

Abstract
Using a mouse skin tumor model, we reported previously that cyclooxygenase-2 (COX-2) deficiency reduced papilloma formation. However, this model did not differentiate between the effects of systemic COX-2-deficiency and keratinocyte-specific COX-2 deficiency on tumor formation. To determine whether keratinocyte-specific COX-2 deficiency reduced papilloma formation, v-H-ras-transformed COX-2+/+ and COX-2−/− keratinocytes were grafted onto nude mice and tumor development was compared. Transformed COX-2+/+ and COX-2−/− keratinocytes expressed similar levels of H-ras, epidermal growth factor receptor and phospho-extracellular signal-regulated kinase1/2 in vitro; and COX-2-deficiency did not reduce uninfected or v-H-ras infected keratinocyte replication. In contrast, tumors arising from grafted transformed COX-2+/+ and COX-2−/− keratinocytes expressed similar levels of H-ras, but COX-2 deficiency reduced phospho-extracellular signal-regulated kinase 1/2 and epidermal growth factor receptor levels 50–60% and tumor volume by 80% at 3 weeks. Two factors appeared to account for the reduced papilloma size. First, papillomas derived from COX-2−/− keratinocytes showed about 70% decreased proliferation, as measured by bromodeoxyuridine incorporation, compared with papillomas derived from COX-2+/+ keratinocytes. Second, keratin 1 immunostaining of papillomas indicated that COX-2−/− keratinocytes prematurely initiated terminal differentiation. Differences in the levels of apoptosis and vascularization did not appear to be contributing factors as their levels were similar in tumors derived from COX-2−/− and COX-2+/+ keratinocytes. Overall, the data are in agreement with our previous observations that decreased papilloma number and size on COX-2−/− mice resulted from reduced keratinocyte proliferation and accelerated keratinocyte differentiation. Furthermore, the data indicate that deficiency/inhibition of COX-2 in the initiated keratinocyte is an important determinant of papilloma forming ability.
Introduction
Non-melanoma skin cancers are among the most prevalent cancers affecting humans (1). Because the rising incidence of non-melanoma skin cancers represents a significant impact on human health and medical costs, understanding the molecular mechanisms underlying skin cancer development and developing chemopreventive/chemotherapeutic approaches are active areas of investigation. Prostaglandins (PGs), which are generated by cyclooxygenase-1 and -2 (COX-1 and COX-2), are elevated in most tumor types including skin tumors (for reviews see (2–6)). Mouse models in which COX-1 or COX-2 have been genetically altered or pharmacologically inhibited have shown that the PGs play an important role in the development of both chemical- and ultraviolet (UV) light-induced skin tumors (for reviews see (5,6)). Although both COX-1 and COX-2 can contribute to skin tumor development (7,8), it is COX-2 that is upregulated in most tumor types, including skin tumors (5,6,9). Because COX-2 plays key roles in tumor development, elucidating its cell type–specific location in tumors and its mechanistic contributions to tumor development remains active areas of investigation.
A number of studies have indicated roles for COX-2 and the PGs it generates in skin tumor development and growth. Studies with mice genetically deficient for COX-2 showed decreased PG production and decreased tumor size and multiplicity when either chemical initiation/promotion or UV exposure protocols were used to induce papillomas or squamous cell tumors (7,10). COX-2-deficiency has also been reported to reduce basal-cell carcinoma size and multiplicity in the PTCH1+/− mouse exposed to ionizing radiation (8). Furthermore, transgenic mice overexpressing COX-2 showed increased PG production and increased skin tumor formation following chemical treatment or UV exposure (10–13) suggesting that COX-2 overexpression contributed to the promotion of skin tumor development. Thus, COX-2 deficiency has been demonstrated to decrease mouse skin tumor formation, whereas COX-2 overexpression has been shown to increase both UV and chemically induced mouse skin tumors.
In addition to the effects of COX-2 genetic modification, pharmacological inhibition of COX-2, using COX-2 selective inhibitors, suppressed chemically and UV-induced skin tumor development in mice (14–17) further supporting a role for COX-2-generated PGs in skin tumor development. In addition to the effects of gene disruption and COX-2 inhibitors on skin tumor formation in mice, COX-2 inhibition has also been demonstrated to inhibit the growth of grafted tumors in various animal models. The selective COX-2 inhibitor, meloxicam, suppressed tumor development when a human ovarian cancer cell line was xenotransplanted subcutaneously into nude mice (18), and Ragel et al. (19) demonstrated that celecoxib reduced tumor growth when meningioma cells were transplanted into nude mice.
In the above studies utilizing COX-2-deficient mice or COX-2 selective inhibitors to inhibit tumor development, the results did not differentiate between systemic inhibition of COX-2 in the host and inhibition of COX-2 in the tumor cells. In an attempt to assess the host effects of systemic COX-2 deficiency, Williams et al. (20) transplanted Lewis Lung Carcinoma cells into COX-2+/+ and COX-2−/− mice and observed that host COX-2 deficiency significantly attenuated tumor growth. Although these data support a role for the systemic deficiency of COX-2 in reducing tumor growth, they do not rule out the possibility that COX-2 deficiency in the tumor cell itself can also contribute to reduced tumor growth.
Based on our previous study demonstrating that initiated/promoted COX-2−/− mice developed fewer and smaller papillomas (7), we undertook the present study to investigate the possibility that COX-2 deficiency in the keratinocyte itself could reduce tumor development. As H-ras mutation is a key event in the initiation/promotion skin tumor model (21), we utilized the approach of Dlugosz et al. (22) on grafting primary Harvey ras oncogene (v-H-ras)-transduced COX-2+/+ and COX-2−/− keratinocytes as orthografts onto nude mice and compared the ability of these cells to form tumors in a COX-2+/+ background. Herein, we show that v-H-ras-transformed COX-2−/− keratinocytes, in agreement with our studies utilizing initiated/promoted COX-2−/− mice(7), formed fewer and smaller papillomas compared with v-H-ras-transformed COX-2+/+ keratinocytes. In addition, we demonstrate that papillomas arising from COX-2−/− keratinocytes showed decreased epidermal growth factor receptor (EGFR) and phospho-extracellular signal-regulated kinase 1/2 (p-ERK1/2) levels, as well as decreased keratinocyte proliferation concomitant with accelerated keratinocyte terminal differentiation. Thus, COX-2 deficiency in the keratinocyte itself is sufficient to reduce skin tumor development.
Materials and methods
Animals
COX-2+/+ and COX-2−/− mice (23) were obtained from Taconic Farms (Germantown, NY). The COX-2 mice are maintained on a C57Bl/6-129Ola background as originally described (23). The COX-2+/+ and COX-2−/− mice utilized were generated by mating COX-2+/− X COX-2+/− mice. Athymic nude mice were obtained from the National Cancer Institute (Frederick, MD) and housed in the National Institute of Environmental Health Sciences animal facility under 12-h light/dark cycle and food and water provided ad libitum. All animals were maintained in accordance with Institutional guidelines governing the care and use of experimental animals under an animal-use protocol approved by the National Institute of Environmental Health Sciences Animal Care and Use Committee.
Keratinocyte isolation, culture and v-H-ras infection
The COX-2+/+ and COX-2−/− keratinocytes were obtained from littermates derived by COX-2+/− X COX-2+/− breedings. The keratinocytes were obtained from 1- to 2-day-old pups, and after genotype identification by PCR (24), keratinocytes of similar genotypes were combined. Briefly, the skin was digested overnight in 0.25% trypsin at 4°C to separate the epidermis from the dermis. COX-2+/+ fibroblasts were isolated from the dermis and grown in Ca2+-free Eagle's minimum essential medium (BioWhittaker, Walkersville, MD) supplemented with 10% fetal bovine serum and 100 units/ml penicillin, 100 µg/ml streptomycin, 250ng/ml Fungizone (all from Invitrogen, Carlsbad, CA). Pooled keratinocytes from COX-2+/+ and COX-2−/− neonates were seeded at a density of 3.5 to 4.0×106 cells per 100mm dish, using Eagle's minimum essential medium as described above. After 4h in culture (37°C, 5% CO2), media was removed and replaced with keratinocyte serum-free medium, supplemented with 50 µg/ml bovine pituitary extract, 5ng/ml recombinant human epidermal growth factor, 5 µg/ml gentamycin (all from Invitrogen, Carlsbad, CA). The Ca2+ concentration in the medium was adjusted to 0.05mM. The day the cells were seeded was defined as day 0.
On day 2, keratinocytes in keratinocyte serum-free medium were infected with a 1:10 dilution of replication-defective v-H-ras retrovirus in the presence of 4 µg/ml polybrene (22) for 1h followed by a medium change. For cell growth comparisons, COX-2+/+ and COX-2−/− keratinocytes were seeded at 0.75×106 cells per 35mm dish and infected as described above. Cell counts were obtained in triplicate for control and infected cells on days 2 and 6 for both genotypes with the Beckman Z1 Coulter particle counter. The average cell numbers for the four types of cells, COX-2+/+ control, COX-2+/+ v-H-ras infected, COX-2−/− control and COX-2−/− v-H-ras infected, were compared using the Student’s t-test. A P-value of 0.05 or less was considered significant.
Grafting of v-H-ras-infected keratinocytes onto athymic nude mice
Athymic mice were prepared for grafting by implanting a silicone dome subcutaneously in the dorsal surface skin and allowing it to heal for 2 days (25). The v-H-ras-transformed keratinocytes to be grafted were harvested after 6 days growth in culture, and a slurry containing 3×106 v-H-ras-infected keratinocytes combined with 6×106 COX-2+/+ fibroblasts was applied to the exposed dorsal fascia within the silicone chamber through the port on the apex of the dome (25). The grafting chambers were removed 1 week later and mice were monitored weekly for tumor growth. Tumor dimensions were measured on a weekly basis for 4 weeks and tumor volumes were determined by multiplying tumor length, width and height.
Western blot analysis
For western blot analysis, proteins were isolated from non-infected and v-H-ras-infected COX-2+/+ and COX-2−/− keratinocytes in culture or from grafted papillomas. Cells or tissues were immediately placed in lysis buffer (Cell Signaling Technology, Danvers, MA) containing 1mM phenylmethylsulfonylfluoride. The protein content was determined using the Bicinchoninic acid assay (BCA). (Pierce, Rockford, IL) according to manufacturer’s instructions. Equal amounts (30 µg) of protein were loaded onto 4−12% NuPAGE Bis-Tris Gels (Invitrogen, Carlsbad, CA) and electroblotted onto nitrocellulose membranes. Blots were stained with antibodies to H-ras (anti-mouse, 1:2000, from BD Biosciences, San Diego, CA), COX-1 (1:1000), COX-2 (1:1000) (both from Cayman Chemical, Ann Arbor, MI), p-ERK (1:500, Cell Signaling Technology, Danvers, MA), total ERK (1:1000, Cell Signaling Technology, Danvers, MA) or EGFR (1:1000, Santa Cruz Biotechnology, Santa Cruz, CA). Blots were then incubated with a secondary antibody (anti-rabbit or mouse IgG, horse radish peroxidase-linked antibody from Cell Signaling Technology, Danvers, MA) and signals detected using the enhanced chemiluminescence reagent (GE Healthcare, Piscataway, NJ).
Immunohistochemistry
The preparation of skin tissues for immunohistochemistry has been described previously (7). Following the blocking of endogenous peroxidases with 3% aqueous hydrogen peroxide, sections were subjected to antigen retrieval by heating in antigen unmasking solution according to the manufacturer’s instructions (Vector Laboratories, Burlingame, CA). All antibodies were diluted in 1× automation buffer (Biomeda Corp., Foster City, CA) containing 1% bovine serum albumin (Sigma Chemical Co., St. Louis, MO) and 1% non-fat dry milk. Sections were incubated with various primary antibodies according to the following conditions: COX-1 at 1:1000 dilution, COX-2 at 1:5000 dilution (both antibodies were obtained from Cayman Chemical, Ann Arbor, MI) or keratin 1 at 1:4000 dilution (Covance Research Products, Berkeley, CA) and were incubated overnight at 4°C. For detection of bromodeoxyuridine (BrdU) incorporation, an anti-BrdU antibody was used at 1:800 dilution (Accurate Chemical and Scientific Corp., Westbury, NY) and incubated at room temperature for 30min. Factor VIII primary antibody was used at a 1:500 dilution (Biocare Medical, Concord, CA) and incubated at room temperature for 1h. Antibody binding was detected using Vectastain Elite ABC Kits (Vector Laboratories, Burlingame, CA) according to the manufacturer’s instructions.
BrdU labeling
Mice were euthanized at 14 and 20 days after grafting to compare cell proliferation as measured by BrdU (GE Healthcare, Piscataway, NJ) incorporation. Two hours prior to sacrifice, each mouse received an intraperitoneal injection of a BrdU-labeling solution (10mM BrdU and 1mM FdU in phosphate-buffered saline, 1ml/100g mouse body weight). Tumors were collected and fixed overnight in 10% neutral buffered formalin and then processed by standard histological procedures and paraffin embedded. Tissue sections were prepared for histological examination and immunohistochemical analysis as described above. BrdU incorporation in COX-2+/+ and COX-2−/− tumors was compared by counting BrdU-labeled keratinocytes per 100 basal cells in four papillomas at each time point and genotype. The percentage of BrdU-labeled cells was then determined and a P-value of 0.05 or less was considered statistically significant.
Assessment of vascularization
Angiogenesis in COX-2+/+ and COX-2−/− tumors was evaluated using Factor VIII immunohistochemistry to identify endothelial cells and counting the stained blood vessels. Photos of stained slides were taken from four papillomas for each time point of each genotype and the average number of vessels per mm2 determined. The data from COX-2+/+ and COX-2−/− papillomas were compared using the Student’s t-test and a P-value of 0.05 or less was considered statistically significant.
Apoptosis
Apoptotic cells were detected in paraffin-embedded sections using the Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling (TUNEL) system kit (DeadEndTM, Promega, Madison, WI) according to the manufacturer’s instructions. Four papillomas of each genotype were collected 14 days after grafting and 20 microscope fields were counted for each mouse. The data are expressed as the number of apoptotic cells/10,000 cells.
Results
Characterization of v-H-ras-transformed COX-2+/+ and COX-2−/− keratinocytes
Keratinocytes harvested from the skin of newborn COX-2+/+ and COX-2−/− littermates were infected with a replication-defective v-H-ras retrovirus, and the effects of COX-2 deficiency on the growth of control and infected cells were determined (Figure 1A). Both COX-2+/+ and COX-2−/− keratinocytes exhibited similar cell numbers when infected 2 days after seeding (Figure 1A). However, after 6 days in culture (4 days post infection) uninfected and v-H-ras-infected COX-2−/− keratinocytes displayed a trend toward slower growth compared with COX-2+/+ keratinocytes; but the differences in growth did not reach statistical significance at P < 0.05 after 6 days in culture. Furthermore, both transformed COX-2+/+ and COX-2−/− keratinocytes expressed significantly increased and comparable levels of total H-ras at 4 days post infection (Figure 1B), indicating that COX-2 deficiency did not influence v-H-ras expression. As expected, v-H-ras transfection increased p-ERK1/2 levels in both COX-2+/+ and COX-2−/− keratinocytes, but COX-2 deficiency did not reduce the level of activated ERK1/2 in keratinocytes in vitro. Figure 1B also shows that EGFR levels were similar in COX-2+/+ and COX-2−/− keratinocytes and that EGFR levels in vitro were not significantly altered by v-H-ras infection or by COX-2 deficiency. COX-1 expression in COX-2+/+ and COX-2−/− keratinocytes also was unchanged by v-H-ras-infection or COX-2 deficiency (Figure 1B), and v-H-ras transfection did not alter COX-2 levels in COX-2+/+ cells. Thus, the data indicate that both COX-2+/+ and COX-2−/− keratinocytes were infected equally by v-H-ras and that p-ERK1/2 levels in keratinocytes were elevated by v-H-ras infection independent of COX-2 expression.
Fig. 1.
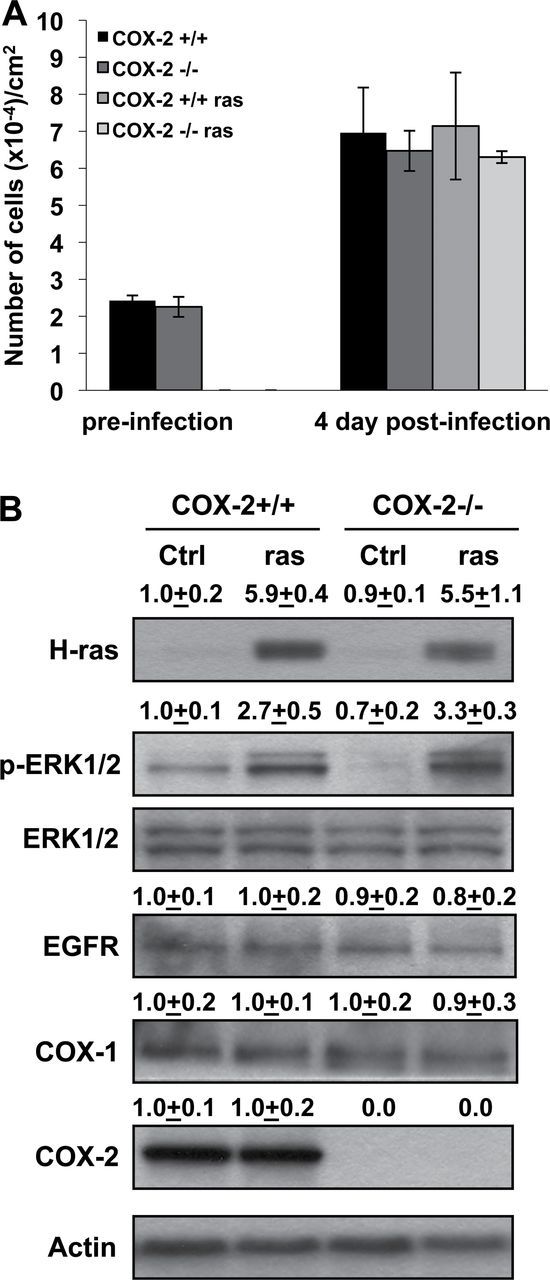
Effects of COX-2 deficiency and v-H-ras transformation on keratinocyte growth and expression of H-ras, pERK1/2, EGFR, COX-1 and COX-2. (A) Keratinocytes were prepared from COX-2+/+ and COX-2−/− 1 to 2-day-old neonates, seeded at 0.75×106 cells per 35mm dish and infected with v-H-ras 2 days after seeding. Cells from three dishes for each genotype plus/minus v-H-ras infection were counted 2 and 6 days after seeding; and the experiment was repeated. Data are reported as mean ± standard error, n = 6 dishes. (B) Cell lysates were prepared 4 days post infection and western blot analysis conducted to determine H-ras, p-ERK1/2, EGFR, COX-1 and COX-2 levels in untransformed and v-H-ras-transformed keratinocytes (30 µg per lane loaded). Actin was used as loading control and all protein levels were determined by densitometry. The data are reported as mean densitometry values ± standard error from three independent blots. Ctrl: control, ras: v-H-ras-transformed keratinocytes.
COX-2 deficiency reduced the growth of papillomas arising from v-H-ras-transformed keratinocytes
Previous work in our laboratory demonstrated that COX-2 deficiency resulted in reduced skin papilloma development using the two-stage mouse skin model (7). To determine whether COX-2 deficiency in the keratinocytes was sufficient to decrease tumor development, v-H-ras-infected COX-2+/+ and COX-2−/− keratinocytes were grafted as cutaneous orthografts onto nude mice and their abilities to form tumors were compared. Figure 2A and 2B show the morphologies and relative sizes of COX-2+/+ and COX-2−/− tumors at 4 weeks post grafting. Tumor volume was significantly diminished in COX-2−/− papillomas (Figure 2C), although lesions formed from both the v-H-ras-transformed COX-2+/+ and COX-2−/− keratinocytes had the typical morphology of squamous cell papillomas, consisting of an inner stromal core surrounded by a stratified squamous epithelium. Tumors arising from both COX-2+/+ and COX-2−/−keratinocytes were apparent by 2 weeks post grafting (Figure 2C), but tumor sizes were decreased in COX-2−/− tumors at all time points over the 4-week course of the study. Furthermore, the tumor incidence of grafted COX-2−/− keratinocytes was decreased compared with grafted COX-2+/+ keratinocytes at all time points (Figure 2D). These data demonstrate that COX-2 deficiency reduces both tumor size and incidence in v-H-ras-infected keratinocytes through a cell-autonomous mechanism.
Fig. 2.
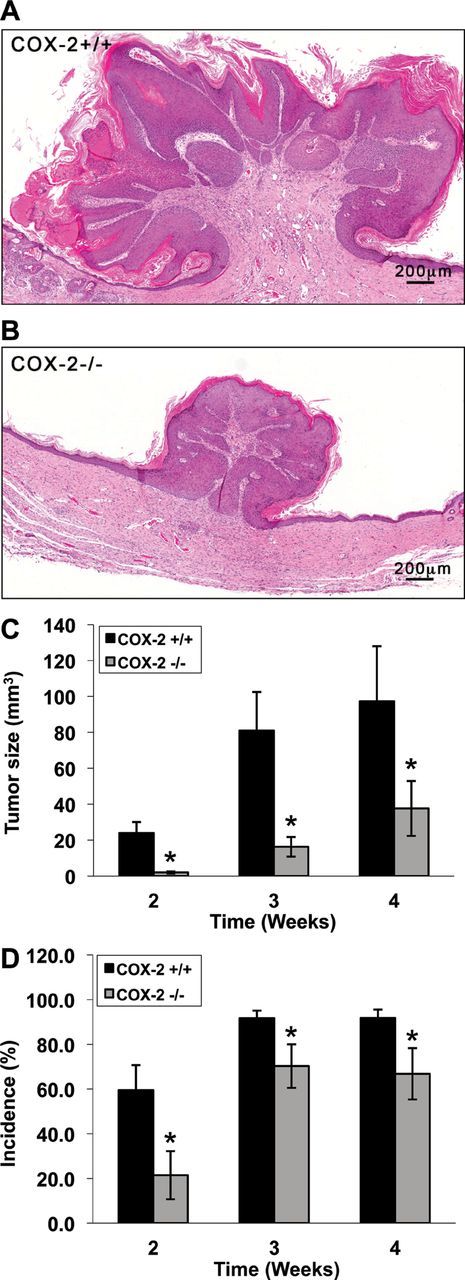
Deficiency of COX-2 in v-H-ras -ransformed keratinocytes causes reduced papilloma growth. (A and B) Hematoxylin and eosin staining of papillomas developed from v-H-ras-transformed COX-2+/+ or COX-2−/− keratinocytes showed that both papilloma genotypes had the typical morphology of squamous cell papillomas, consisting of a stromal core surrounded by a proliferating stratified squamous epithelium, with COX-2−/− tumors being significantly smaller. (C) Papillomas were visible by day 14 after grafting in both genotypes and the sizes were determined by multiplying tumor length, width and height. Size and incidence comparison for COX-2+/+ and COX-2−/− papillomas were made at 2, 3 and 4 weeks post grafting. Data are compiled from 33 mice/genotype from four independent experiments and are reported as mean ± standard error, *P ≤ 0.05. (D) Tumor incidence comparison for COX-2+/+ and COX-2−/− papillomas was made at 2, 3 and 4 weeks after grafting. Data are compiled from 33 mice/genotype from four independent experiments and are reported as mean ± standard error, *P ≤ 0.05.
COX-2 deficiency reduced keratinocyte proliferation and accelerated terminal differentiation in tumors
Because our previous work indicated that COX-2 deficiency caused reduced keratinocyte proliferation and increased differentiation in papillomas (7), the levels of keratinocyte proliferation and differentiation were determined in the tumors of grafted COX-2+/+ and COX-2-deficient keratinocytes. To better compare the locations of replicating and differentiating keratinocytes in the two genotypes, serial papilloma sections were stained for BrdU and keratin 1, a marker for the onset of terminal differentiation in keratinocytes (26). As shown in Figure 3A, when proliferation was assessed by BrdU incorporation, the tumors that developed from COX-2−/− keratinocytes had significantly fewer BrdU-positive cells compared with tumors arising from COX-2+/+ keratinocytes. In addition, the localization of BrdU-positive cells was largely confined to the basal layer in COX-2−/− tumors, whereas BrdU-labeled cells were observed in the basal and suprabasal cells of tumors resulting from COX-2+/+ keratinocytes. Figure 3D shows quantitatively the differences in percentage of BrdU-labeled cells for the two genotypes at days 14 and 20 post grafting. Thus, the data indicate an increase in the number of proliferative cells in tumors arising from COX-2+/+ compared with tumors arising from COX-2−/− keratinocytes.
Fig. 3.
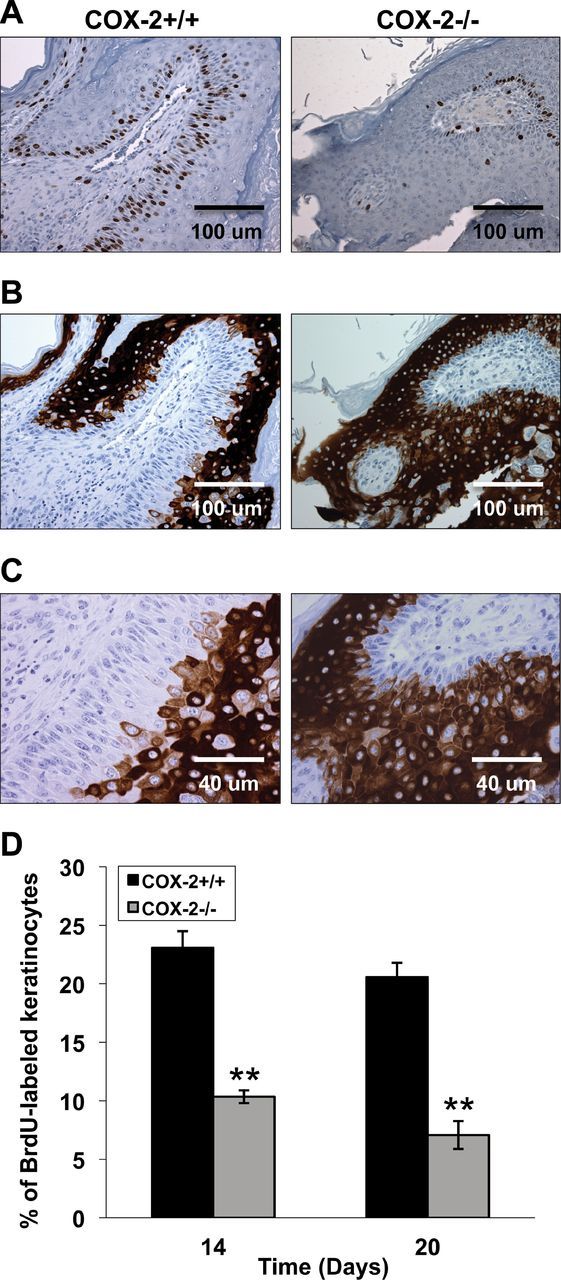
Papillomas arising from mutant v-H-ras-transformed COX-2 −/− keratinocytes exhibit decreased proliferation and increased differentiation. (A) BrdU-labeled cells in the COX-2−/− papillomas mainly resided in the basal cells, whereas in COX-2+/+ papillomas, BrdU-labeled cells are observed both in basal cells and suprabasal cells. (B) The expression of keratin 1 in COX-2+/+ and COX-2−/− papillomas. It should be noted that BrdU incorporation and keratin 1 expression were mutually exclusive both in COX-2+/+ and COX-2−/− papillomas. (C) Keratin 1 expression in COX-2+/+ and COX-2−/− papilloma from (B) above shown at higher magnification is detected about 3–5 cell layers above the basal layer in COX-2+/+ papillomas, whereas keratin 1 expression is generally detected 1–2 cell layers above the basal cells in COX-2−/− papillomas. (D) Comparison of BrdU incorporation at days 14 and 20 was determined by counting the BrdU-labeled cells from four papillomas for each genotype and for each time point. Data were expressed as the percentage of BrdU-labeled keratinocytes per 100 basal cells per papilloma counted. Data are reported as mean ± standard error, n=4 papillomas. **P ≤ 0.001.
The levels of terminal differentiation in the tumors resulting from grafted COX-2+/+ and COX-2-deficient keratinocytes were determined by assessing the expression of keratin 1. As shown in Figure 3B, and at a higher magnification in Figure 3C, keratin 1 expression was primarily detectable 3–5 cell layers above the basal layer in COX-2+/+ tumors, whereas keratin 1 expression was detectable 1–2 cell layers above the basal cells in COX-2−/− tumors (Figure 3B and 3C). These data are in agreement with our previous report that COX-2 deficiency caused the premature onset of terminal differentiation in epidermal keratinocytes (7). The data in Figure 3A and 3B, which are serial sections, indicate that areas of keratinocyte replication and terminal differentiation are essentially mutually exclusive in papillomas resulting from both v-H-ras-transformed COX-2+/+ and COX-2−/− keratinocytes.
COX-2 deficiency reduces p-ERK1/2 and EGFR levels in papillomas, but not H-ras or COX-1 levels
Because papillomas resulting from COX-2-deficient keratinocytes showed decreased keratinocyte proliferation, the levels of H-ras, p-ERK1/2 and EGFR, effectors known to influence cell replication (27,28), in COX-2+/+ and COX-2−/− papillomas were compared. Western blot analysis showed that the levels of H-ras were similar in the tumors that developed from COX-2+/+ and COX-2−/− keratinocytes (Figure 4A). However, the levels of p-ERK1/2 and EGFR were decreased by about 50–60% in tumors arising from COX-2-deficient keratinocytes compared with the tumors derived from COX-2+/+ keratinocytes. The levels of these effectors in tumors arising from COX-2+/+ and COX-2-deficient keratinocytes are in contrast to the similar levels of p-ERK1/2 and EGFR observed in transformed COX-2+/+ and COX-2−/− keratinocytes in culture (Figure 1B). Furthermore, the data in Figure 4A show that COX-1 was expressed at similar levels in the tumors that developed from COX-2+/+ and COX-2−/− keratinocytes. In agreement with our previous study (7), immunostaining for COX-1 in COX-2+/+ and COX-2−/− tumors showed COX-1 expression was detected 2– 4 cell layers above the basal layer in COX-2+/+ tumors and detected 1–2 cell layers above the basal layer in COX-2−/− tumors. In general, COX-1 expression was co-localized with keratin 1 expression in COX-2+/+ and COX-2−/− papillomas (compare Figures 3B and 4C). The data in Figure 4B show that COX-2 on the other hand was primarily localized in the replicating basal-cell region of wild-type tumors (Figure 4B). As expected, COX-2 was not detected by western blotting or immunohistochemistry in tumors that developed from COX-2-deficient keratinocytes (Figure 4A and 4B).
Fig. 4.
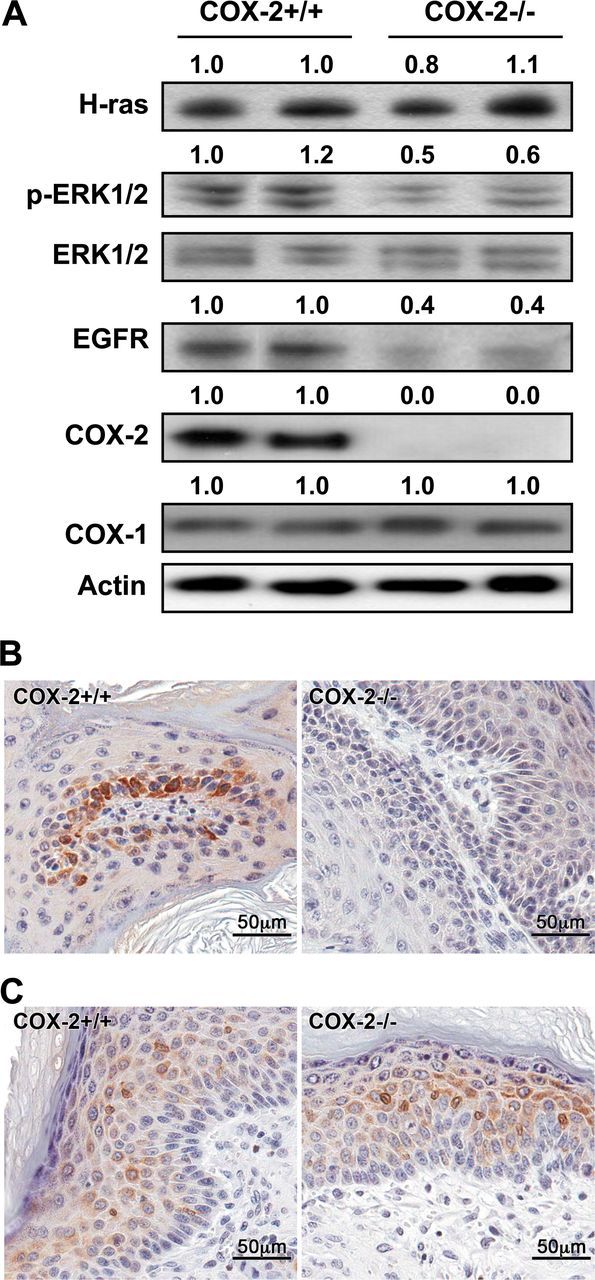
Western blotting and immunohistochemistry of COX-2+/+ and COX-2−/− papillomas. (A) Protein lysates were prepared from two COX-2+/+ papillomas and two COX-2−/− papillomas and analyzed for H-ras, p-ERK1/2, total ERK1/2, EGFR, COX-2 and COX-1 by western blotting (30 µg per lane loaded). The numbers above each lane represent densitometry readings. (B) COX-2 immunostaining of COX-2+/+ papillomas indicated that COX-2 is primarily located in the basal cells and, as expected, COX-2 was not detected in the COX-2−/− papillomas. (C) COX-1 immunostaining indicated that the COX-2+/+ and COX-2−/− papillomas expressed COX-1 in the suprabasal cell layers at comparable levels in agreement with the western blotting results.
COX-2 deficiency did not affect levels of apoptosis or vascularization in papillomas
It has been reported that inhibition of COX-2 increases apoptosis (29,30). However, in agreement with our previous finding (7), the levels of apoptosis in tumors arising from COX-2+/+ or COX-2-deficient keratinocytes were similar (Figure 5A), indicating that apoptosis was not a factor that contributed to the smaller size of tumors resulting from COX-2-deficient keratinocytes. Figure 5B shows actual TUNEL-positive cells in COX-2+/+ and COX-2−/− papillomas.
Fig. 5.
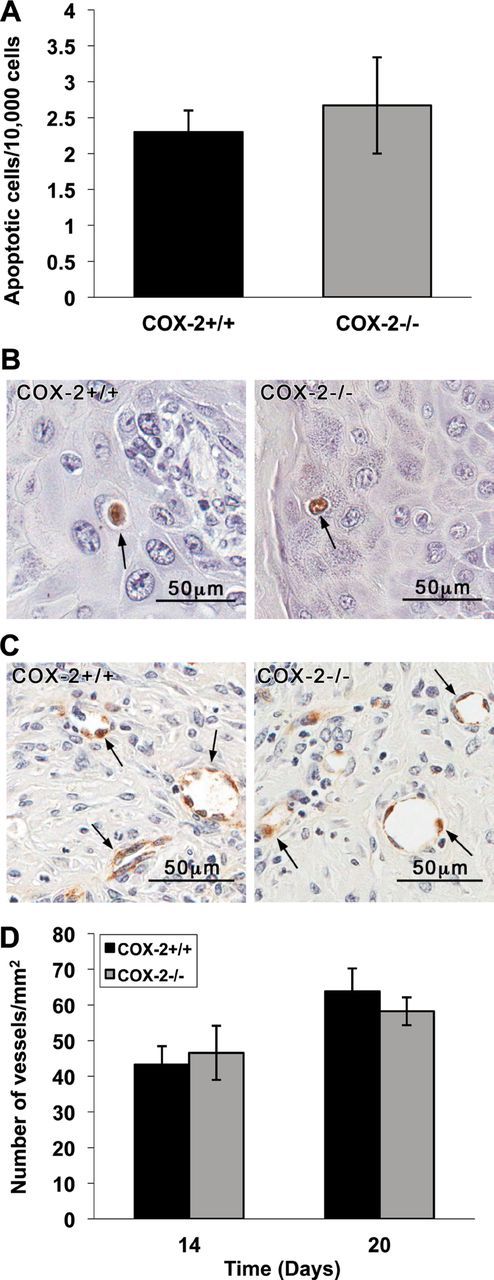
COX-2 deficiency did not affect levels of apoptosis or vascularization in papillomas. (A) Apoptotic cells were detected in paraffin-embedded papilloma sections using the TUNEL assay. Four papillomas of each genotype were collected 14 days after grafting and 20 microscope fields were counted for each tumor. Data are expressed as the number of apoptotic cells/10,000 cells (mean ± standard error, n = 4). Both the COX-2+/+ and COX-2−/− papillomas showed similarly low levels of apoptosis. (B) COX-2+/+ and COX-2−/− papilloma sections showing apoptotic cells stained in the TUNEL assay. (C) Tissue sections of COX-2+/+ and COX-2−/− tumors removed on day 20 after grafting and stained for FactorVIII, a marker of endothelial cells. COX-2+/+ and COX-2−/− papillomas showed similar levels of vascularization in both papilloma genotypes. (D) Quantitative analysis of the number of vessels in four COX-2+/+ and COX-2−/− papillomas at day 14 or 20 post grafting was determined and the number of vessels per mm2 calculated. Data are reported as mean ± standard error, n = 4. Statistical analysis showed no difference in blood vessel numbers between COX-2+/+ and COX-2−/− papillomas.
COX-2 has also been reported to play an important role in tumor angiogenesis (31–33), and therefore, tumors arising from COX-2−/− keratinocytes were compared with tumors arising from COX-2+/+ keratinocytes for possible reduced levels of vascularization. Figure 5C shows tissue sections of COX-2+/+ and COX-2-deficient tumors removed on day 20 after grafting and stained for Factor VIII, a marker of endothelial cells (34). The data in Figure 5D show that COX-2+/+ and COX-2−/− tumors removed on day 14 or 20 contained similar numbers of blood vessels in the stromal region of the tumors. These data indicate that the intrinsic loss of COX-2 in transformed keratinocytes did not alter the level of vascularization during tumor development and that decreased vascularization was not a cause of reduced tumor growth in this model.
Discussion
In the present study, we investigated the contributions of keratinocyte-specific COX-2 deficiency on skin tumor development by utilizing a mouse keratinocyte grafting model (25). The data indicated that v-H-ras-transformed COX-2-deficient keratinocytes displayed delayed tumor onset and formed smaller tumors than transformed COX-2+/+ keratinocytes. These results are in agreement with our previous report in which initiation/promotion of COX-2-deficient mice resulted in delayed skin tumor onset and smaller tumors compared with COX-2+/+ mice (7). Furthermore, as was observed with tumors induced on COX-2+/+ and COX-2-deficient mice, tumors arising from v-H-ras-transformed COX-2−/− keratinocytes displayed reduced keratinocyte replication and accelerated onset of keratinocyte terminal differentiation compared with tumors arising from COX-2+/+ keratinocytes. Tumors arising from transformed COX-2−/− keratinocytes also showed reduced levels of the growth effectors, p-ERK1/2 and EGFR. Thus, the results indicate that keratinocyte-specific deficiency of COX-2 results in similar effects on tumor development and growth as does the systemic deficiency of COX-2 when skin tumors are induced with the initiation/promotion protocol. Overall, the data indicate that cell-autonomous COX-2 deficiency in the keratinocyte is a significant factor in reducing skin tumor development in the mouse.
In an attempt to gain insight into the mechanism(s) by which COX-2 deficiency decreased the size of tumors arising from v-H-ras-transformed COX-2−/− keratinocytes (Figure 2A and 2B), the levels of vascularization and apoptosis in wild-type and COX-2−/− tumors were compared. It is known that blood vessel development is an integral part of tumor growth and studies had linked COX-2 inhibition to decreased angiogenesis (33,35). However, when the number of blood vessels in the stromal tissue of COX-2+/+ and COX-2−/− papillomas was compared, no significant difference was observed (Figure 5C and 5D). COX-2 inhibition has also been associated with increased apoptosis in various types of cancer (36,37). However, when apoptosis was measured in tumor epithelium arising from grafted COX-2−/− keratinocytes, the data indicated that COX-2+/+ and COX-2−/− papillomas had similar levels of apoptosis (Figure 5A). These vascularization and apoptotic data are in agreement with our earlier observations for the tumors that developed on initiated/promoted COX-2-deficient mice (7) and indicate that neither decreased vascularization nor increased apoptosis contributed to the smaller tumor sizes in either model.
Mutated H-ras is a key effector in skin tumor development in the initiation/promotion mouse model (21), and therefore the possible effects of COX-2 deficiency on H-ras expression in v-H-ras-transformed keratinocytes in vitro and in grafted tumors in vivo were determined. When COX-2+/+ and COX-2−/− keratinocytes were transformed by v-H-ras, it was observed that neither the levels of H-ras expression in the keratinocytes in vitro (Figure 1B) nor the levels of H-ras in the resultant tumors (Figure 4A) was affected by COX-2 deficiency. Thus, these results indicated that decreased H-ras expression was not the cause of the reduced growth of tumors arising from COX-2-deficient keratinocytes. However, when the levels of p-ERK and EGFR were determined in the tumors, it was observed that their levels were decreased in COX-2−/− tumors compared with COX-2+/+ tumors (Figure 4A) although their levels in COX-2+/+ and COX-2−/− keratinocytes in vitro were not significantly different (Figure 1B). Although the mechanisms by which COX-2 deficiency decreased EGFR and p-ERK levels in papillomas were not elucidated in the present study, previous studies have indicated that COX-2 deficiency or its inhibition can reduce EGFR and p-ERK levels (38,39).
EGFR is a receptor tyrosine kinase that is known to have a role in skin tumor formation (40,41) and has been demonstrated to activate ERK1/2 and other effectors as tumors develop (42). The decreased EGFR level in COX-2−/− papillomas observed in the present study (Figure 4A) is relevant because grafting v-H-ras-transformed EGFR-deficient keratinocytes onto nude mice(43) resulted in papillomas demonstrating characteristics of the papillomas arising from the transformed COX-2-deficient keratinocytes described herein. For example, EGFR-deficient keratinocytes also produced smaller tumors and the keratinocytes displayed premature differentiation as indicated by keratin 1 and keratin 10 expressions. Thus, the decreased EGFR expression observed in tumors arising from COX-2−/− keratinocytes may, in part, account for the observed characteristics of COX-2−/− tumors.
It is known that the tumor microenvironment plays an important role in tumor growth; but the tumor–microenvironment interaction is a dynamic and complex process that is not clearly understood (44). In the present study, COX-2-deficient keratinocytes grafted onto nude mice resulted in decreased tumor development compared with grafted COX-2+/+ keratinocytes. The decreased ability of COX-2-deficient keratinocytes to grow into tumors may be due in part to differences in their effects on the microenvironment such as reduced production of growth-promoting cytokines or increased production of growth inhibitory factors. In accord with the latter, it has recently been reported that mammary epithelial cell–specific deletion of COX-2 altered the tumor-associated immune response. In this study, Markosyan et al. (45) reported that COX-2 selective deletion in mammary epithelial cells significantly delayed breast tumor development in mice by causing the tumor-associated immune response to shift toward an antitumorigenic type 1 response. However, alternative effects of COX-2 on the microenvironment and tumor growth are also possible. For example, in a study utilizing grafted ductal carcinoma cells, it was reported that COX-2 expression in the carcinoma cells and their growth rates were influenced by the interaction of the tumor cells with fibroblasts in the microenvironment (46). As an additional effect of the tumor microenvironment, the induction of COX-2 in the tumor stroma has been shown to be important for the maintenance of an inflammatory microenvironment that is conducive for the growth of gastrointestinal tumors (47). In addition to the effects of COX-2 expression in epithelial tumor cells versus stromal cells, the epithelial cell type expressing the COX-2 can also affect the microenvironment’s inflammatory response. Using a cell type–specific COX-2 inactivation approach, Ishikawa et al. (48) recently reported that mice with a deficiency of COX-2 in colon epithelial cells did not differ from wild-type mice when a dextran sulfate sodium-induced colitis model was used. However, when mice with endothelial- or myeloid-cell-specific COX-2 deficiency were studied, a reduced response to dextran sulfate sodium -induced colitis was observed. These studies demonstrate that the cell type deficient in COX-2 can differentially influence the microenvironment. Of relevance to the effects of COX-2 deficiency in keratinocytes on tumor growth in the present study, it was observed that EGFR, p-ERK and cell growth were decreased in tumors arising from COX-2−/− keratinocytes compared with tumors arising from COX-2+/+ keratinocytes. Because significant changes in these signaling pathways were not observed in the COX-2−/− keratinocytes in vitro (Figure 1A and 1B), it appears that the tumor microenvironment may cause the changes in their levels in vivo. For example, the effect of COX-2 deficiency in the keratinocytes could cause the microenvironment to produce either lower levels of growth-promoting cytokines or increased levels of factors that inhibit tumor cell growth as described in the studies cited above (44–48). However, it is unclear as to which of these possible responses in the microenvironment are contributing to the reduced tumorigenic growth of COX-2-deficient keratinocytes in the present study.
In addition to the studies investigating the effect of cell-specific COX-2 deficiency on tumorigenesis, the effects of host COX-2 deficiency on transplanted tumor cell growth have also been reported. Williams et al. (20) injected Lewis lung carcinoma cells into COX-2−/− mice and observed that tumor volume was significantly decreased in COX-2−/− mice compared with COX-2+/+ mice. Thus, in combination with our data, the report of Williams et al. (20) suggests that both systemic and tumor cell–specific COX-2 effects may be involved in tumor formation. However, our data showing that COX-2 deficiency in the transformed tumor cell itself decreased tumor formation indicates that the COX-2 status of the keratinocyte itself plays an important role in tumor growth.
In summary, our data demonstrate that cell-specific loss of COX-2 in v-H-ras-transformed keratinocytes was able to significantly attenuate the growth rate and incidence of papilloma formation when the keratinocytes were grafted onto nude mice. Furthermore, the characteristics of the papillomas arising from transformed grafted COX-2−/− keratinocytes agreed well with the characteristics of the papillomas that developed on initiated/promoted COX-2-deficient mice. However, because papilloma development did occur with grafted COX-2−/− keratinocytes, albeit at a slower rate, and because recent studies have indicated the importance of COX-2 on the tumor microenvironment, the deficiency of COX-2 in the grafted keratinocyte may not be the sole cause of reduced tumor formation.
Acknowledgements
This work was supported by the National Institute of Environmental Health Sciences, National Institute of Health, Intramural Program.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations:
- COX
Cyclooxygenase
- PG
prostaglandin
- v-H-ras
Harvey ras oncogene
- ERK
extracellular signal-regulated kinase
- EGFR
epidermal growth factor receptor
- BrdU
bromodeoxyuridine
- TUNEL
Terminal Deoxynucleotidyl Transferase-Mediated dUTP Nick End Labeling
- UV
ultraviolet.
References
- 1. Donaldson M.R. et al. (2011). No end in sight: the skin cancer epidemic continues. Semin. Cutan. Med. Surg. 30 3–5 [DOI] [PubMed] [Google Scholar]
- 2. Dannenberg A.J. et al. (2003). Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell 4 431–436 [DOI] [PubMed] [Google Scholar]
- 3. Hull M.A. (2005). Cyclooxygenase-2: how good is it as a target for cancer chemoprevention? Eur. J. Cancer 41 1854–1863 [DOI] [PubMed] [Google Scholar]
- 4. Doré M. (2011). Cyclooxygenase-2 expression in animal cancers. Vet. Pathol. 48 254–265 [DOI] [PubMed] [Google Scholar]
- 5. Rundhaug J.E. et al. (2010). Molecular mechanisms of mouse skin tumor promotion. Cancers (Basel). 2 436–482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Müller-Decker K. (2011). Cyclooxygenase-dependent signaling is causally linked to non-melanoma skin carcinogenesis: pharmacological, genetic, and clinical evidence. Cancer Metastasis Rev. 30 343–361 [DOI] [PubMed] [Google Scholar]
- 7. Tiano H.F. et al. (2002). Deficiency of either cyclooxygenase (COX)-1 or COX-2 alters epidermal differentiation and reduces mouse skin tumorigenesis. Cancer Res. 62 3395–3401 [PubMed] [Google Scholar]
- 8. Tang J.Y. et al. (2010). Basal cell carcinoma chemoprevention with nonsteroidal anti-inflammatory drugs in genetically predisposed PTCH1+/- humans and mice. Cancer Prev. Res. (Phila). 3 25–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Fischer S.M. (2002). Is cyclooxygenase-2 important in skin carcinogenesis? J. Environ. Pathol. Toxicol. Oncol. 21 183–191 [PubMed] [Google Scholar]
- 10. Fischer S.M. et al. (2007). Cyclooxygenase-2 expression is critical for chronic UV-induced murine skin carcinogenesis. Mol. Carcinog. 46 363–371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Muller-Decker K. et al. (2002). Transgenic cyclooxygenase-2 overexpression sensitizes mouse skin for carcinogenesis. Proc. Natl. Acad. Sci. U.S.A. 99 12483–12488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Müller-Decker K. et al. (2007). The cyclooxygenase-2-mediated prostaglandin signaling is causally related to epithelial carcinogenesis. Mol. Carcinog. 46 705–710 [DOI] [PubMed] [Google Scholar]
- 13. Rundhaug J.E. et al. (2007). A role for cyclooxygenase-2 in ultraviolet light-induced skin carcinogenesis. Mol. Carcinog. 46 692–698 [DOI] [PubMed] [Google Scholar]
- 14. Fischer S.M. et al. (1999). Chemopreventive activity of celecoxib, a specific cyclooxygenase-2 inhibitor, and indomethacin against ultraviolet light-induced skin carcinogenesis. Mol. Carcinog. 25 231–240 [PubMed] [Google Scholar]
- 15. Pentland A.P. et al. (1999). Reduction of UV-induced skin tumors in hairless mice by selective COX-2 inhibition. Carcinogenesis 20 1939–1944 [DOI] [PubMed] [Google Scholar]
- 16. Wilgus T.A. et al. (2003). Inhibition of cutaneous ultraviolet light B-mediated inflammation and tumor formation with topicalcelecoxib treatment. Mol. Carcinog. 38 49–58 [DOI] [PubMed] [Google Scholar]
- 17. Chun K.S. et al. (2006). Inhibition of phorbol ester-induced mouse skin tumor promotion and COX-2 expression by celecoxib: C/EBP as a potential molecular target. Cancer Res. Treat. 38 152–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xin B. et al. (2007). Inhibitory effect of meloxicam, a selective cyclooxygenase-2 inhibitor, and ciglitazone, a peroxisomeproliferator-activated receptor gamma ligand, on the growth of human ovarian cancers. Cancer 110 791–800 [DOI] [PubMed] [Google Scholar]
- 19. Ragel B.T. et al. (2007). Celecoxib inhibits meningioma tumor growth in a mouse xenograft model. Cancer 109 588–597 [DOI] [PubMed] [Google Scholar]
- 20. Williams C.S. et al. (2000). Host cyclooxygenase-2 modulates carcinoma growth. J. Clin. Invest. 105 1589–1594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yuspa S.H. (1994). The pathogenesis of squamous cell cancer: lessons learned from studies of skin carcinogenesis–thirty-third G. H. A. Clowes Memorial Award Lecture. Cancer Res. 54 1178–1189 [PubMed] [Google Scholar]
- 22. Dlugosz A.A. et al. (1995). Isolation and utilization of epidermal keratinocytes for oncogene research. Meth. Enzymol. 254 3–20 [DOI] [PubMed] [Google Scholar]
- 23. Morham S.G. et al. (1995). Prostaglandin synthase 2 gene disruption causes severe renal pathology in the mouse. Cell 83 473–482 [DOI] [PubMed] [Google Scholar]
- 24. Loftin C.D. et al. (2001). Failure of ductus arteriosus closure and remodeling in neonatal mice deficient in cyclooxygenase-1 and cyclooxygenase-2. Proc. Natl. Acad. Sci. U.S.A. 98 1059–1064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roop D.R. et al. (1986). An activated Harvey ras oncogene produces benign tumours on mouse epidermal tissue. Nature 323 822–824 [DOI] [PubMed] [Google Scholar]
- 26. Zhu S. et al. (1999). C/EBPbeta modulates the early events of keratinocyte differentiation involving growth arrest and keratin 1 and keratin 10 expression. Mol. Cell. Biol. 19 7181–7190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Roberts P.J. et al. (2007). Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26 3291–3310 [DOI] [PubMed] [Google Scholar]
- 28. Pastore S. et al. (2008). The epidermal growth factor receptor system in skin repair and inflammation. J. Invest. Dermatol. 128 1365–1374 [DOI] [PubMed] [Google Scholar]
- 29. Moeckel G.W. et al. (2003). COX2 activity promotes organic osmolyte accumulation and adaptation of renal medullary interstitial cells to hypertonic stress. J. Biol. Chem. 278 19352–19357 [DOI] [PubMed] [Google Scholar]
- 30. Papadimitriou A. et al. (2007). Anti-apoptotic effects of arachidonic acid and prostaglandin E2 in pancreatic beta-cells. Cell. Physiol. Biochem. 20 607–616 [DOI] [PubMed] [Google Scholar]
- 31. Gately S. (2000). The contributions of cyclooxygenase-2 to tumor angiogenesis. Cancer Metastasis Rev. 19 19–27 [DOI] [PubMed] [Google Scholar]
- 32. Seno H. et al. (2002). Cyclooxygenase 2- and prostaglandin E(2) receptor EP(2)-dependent angiogenesis in Apc(Delta716) mouse intestinal polyps. Cancer Res. 62 506–511 [PubMed] [Google Scholar]
- 33. Chang S.H. et al. (2004). Role of prostaglandin E2-dependent angiogenic switch in cyclooxygenase 2-induced breast cancer progression. Proc. Natl. Acad. Sci. U.S.A. 101 591–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Vacca A. et al. (2001). Bone marrow angiogenesis in patients with active multiple myeloma. Semin. Oncol. 28 543–550 [DOI] [PubMed] [Google Scholar]
- 35. Masferrer J.L. et al. (1999). COX-2 inhibitors. A new class of antiangiogenic agents. Ann. N. Y. Acad. Sci. 889 84–86 [DOI] [PubMed] [Google Scholar]
- 36. Gee J. et al. (2006). Selective cyclooxygenase-2 inhibitors inhibit growth and induce apoptosis of bladder cancer. Oncol. Rep. 15 471–477 [PubMed] [Google Scholar]
- 37. Kern M.A. et al. (2006). Cyclooxygenase-2 inhibition induces apoptosis signaling via death receptors and mitochondria in hepatocellular carcinoma. Cancer Res. 66 7059–7066 [DOI] [PubMed] [Google Scholar]
- 38. Li G. et al. (2009). Cyclooxygenase-2 prevents fas-induced liver injury through up-regulation of epidermal growth factor receptor. Hepatology 50 834–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agarwal S. et al. (2009). Inhibition of 12-LOX and COX-2 reduces the proliferation of human epidermoid carcinoma cells (A431) by modulating the ERK and PI3K-Akt signalling pathways. Exp. Dermatol. 18 939–946 [DOI] [PubMed] [Google Scholar]
- 40. Dlugosz A.A. et al. (1997). Targeted disruption of the epidermal growth factor receptor impairs growth of squamous papillomas expressing the v-ras(Ha) oncogene but does not block in vitro keratinocyte responses to oncogenic ras. Cancer Res. 57 3180–3188 [PubMed] [Google Scholar]
- 41. El-Abaseri T.B. et al. (2005). Chemoprevention of UV light-induced skin tumorigenesis by inhibition of the epidermal growth factor receptor. Cancer Res. 65 3958–3965 [DOI] [PubMed] [Google Scholar]
- 42. Normanno N. et al. (2006). Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 366 2–16 [DOI] [PubMed] [Google Scholar]
- 43. Hansen L.A. et al. (2000). The epidermal growth factor receptor is required to maintain the proliferative population in the basal compartment of epidermal tumors. Cancer Res. 60 3328–3332 [PubMed] [Google Scholar]
- 44. Hanahan D. et al. (2011). Hallmarks of cancer: the next generation. Cell 144 646–674 [DOI] [PubMed] [Google Scholar]
- 45. Markosyan N. et al. (2011). Deletion of cyclooxygenase 2 in mouse mammary epithelial cells delays breast cancer onset through augmentation of type 1 immune responses in tumors. Carcinogenesis 32 1441–1449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Hu M. et al. (2009). Role of COX-2 in epithelial-stromal cell interactions and progression of ductal carcinoma in situ of the breast. Proc. Natl. Acad. Sci. U.S.A. 106 3372–3377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Oshima H. et al. (2012). The inflammatory network in the gastrointestinal tumor microenvironment: lessons from mouse models. J. Gastroenterol. 47 97–106 [DOI] [PubMed] [Google Scholar]
- 48. Ishikawa T.O. et al. (2011). Cox-2 deletion in myeloid and endothelial cells, but not in epithelial cells, exacerbates murine colitis. Carcinogenesis 32 417–426 [DOI] [PMC free article] [PubMed] [Google Scholar]