

Abstract
Lung cancer is the leading cause of cancer-related death, with non-small cell lung cancer (NSCLC) being the predominant form of the disease. Most lung cancer is caused by the accumulation of genomic alterations due to tobacco exposure. To uncover its mutational landscape, we performed whole-exome sequencing in 31 NSCLCs and their matched normal tissue samples. We identified both common and unique mutation spectra and pathway activation in lung adenocarcinomas and squamous cell carcinomas, two major histologies in NSCLC. In addition to identifying previously known lung cancer genes (TP53, KRAS, EGFR, CDKN2A and RB1), the analysis revealed many genes not previously implicated in this malignancy. Notably, a novel gene CSMD3 was identified as the second most frequently mutated gene (next to TP53) in lung cancer. We further demonstrated that loss of CSMD3 results in increased proliferation of airway epithelial cells. The study provides unprecedented insights into mutational processes, cellular pathways and gene networks associated with lung cancer. Of potential immediate clinical relevance, several highly mutated genes identified in our study are promising druggable targets in cancer therapy including ALK, CTNNA3, DCC, MLL3, PCDHIIX, PIK3C2B, PIK3CG and ROCK2.
Introduction
Lung cancer is the leading cause of cancer-related death in the USA. In 2011, there will be an estimated 220 000 cases of lung cancer diagnosed, and only 15% of those patients are expected to survive for >5 years (1). Although active cigarette smoking causes most lung cancer deaths, 10–15% of patients diagnosed with lung cancer have never smoked. Prolonged exposure to carcinogens found in tobacco smoke and other environmental carcinogens that interact with various genetic susceptibility and/or resistance factors contribute to the accumulation of genomic alterations, including nucleotide substitutions, small insertions and deletions, copy number variations and chromosomal rearrangements, in human lung cancers.
Several tumor suppressor genes are inactivated in many cases of lung cancer including TP53 (2), RB1 (3,4) and PTEN (5), whereas activating mutations are found in EGFR (6) and KRAS (7) in some cases. Recent DNA sequencing of 623 genes with known or potential relationships to cancer revealed >1000 somatic mutations in lung adenocarcinomas (ADC), a major subtype of non-small cell lung cancer (NSCLC). The newly identified genes include tumor suppressor genes (NF1, ATM and APC) along with tyrosine kinase genes (ephrin receptor genes, ERBB4, KDR, FGFR4 and NTRK genes) that may function as proto-oncogenes (8). More recently, the genomes of a small-cell cancer line NCI-H209 and a male Caucasian diagnosed with ADC were sequenced (9,10). Although a broad spectrum of mutations was observed in these whole-genome sequencing studies, the large majority of mutations are predicted to be passenger mutations. It is not yet clear whether the somatic alterations identified in these two whole-genome sequencing samples will be found recurrently in human lung cancers. The identification of recurrent driver mutations will require the sequencing of a large number of samples.
Next-generation sequencing technologies have revolutionized cancer genomics research by providing an unbiased and comprehensive method of detecting somatic cancer genome alterations (11). Characterization of these cancer genome hallmarks will lead to new therapeutic treatments and personalized medicine. To identify a more comprehensive landscape of somatic mutations in lung cancer, we carried out whole-exome sequencing and subsequent Sanger sequencing validation in 31 NSCLC patients. Our study identified both common and unique mutation spectra and pathway activation in ADC and squamous cell carcinomas (SCC). This data provides new insights into lung carcinogenesis and the potential molecular basis for directing the treatment of lung cancer.
Materials and methods
Lung tumor histology
Thirty-one pairs of lung tumors and matched normal tissues were analyzed, including 16 ADC, 12 SCC, 1 adenosquamous carcinoma, 1 bronchioloalveolar carcinoma (BAC) and 1 ADC with BAC (Supplementary Table 1, available at Carcinogenesis Online). Five samples were from never smokers and the others were from smokers. Five patients had at least one first-degree relative with lung cancer, and the other 26 patients did not have a family history of lung cancer. The study protocols were approved by Roswell Park Cancer Institute and Mayo Clinic Institute Review Boards.
DNA library preparation
Tumor tissues were microdissected to identify areas of adequate tumor cellularity (>70%) for DNA extraction. Paired-end libraries were prepared following the manufacturer's protocol (Illumina and Agilent). Briefly, 3 μg of genomic DNA was fragmented to 150–200 bp using the Covaris E210 sonicator. The ends were repaired, and an ‘A’ base was added to the 3′ ends. Paired-end DNA adaptors (Illumina) with a single ‘T’ base overhang at the 3′ end were ligated and the resulting constructs were purified using AMPure SPRI beads from Agencourt. The adapter-modified DNA fragments were enriched by four cycles of PCR using PE 1.0 forward and PE 2.0 reverse (Illumina) primers. The concentration and size distribution of the libraries were determined on an Agilent Bioanalyzer DNA 1000 chip.
Capture of target genome
Exonic sequences from tumor (purity >70%) and normal DNAs were enriched using Agilent's SureSelect technology for targeted exon capture, targeting 38 Mb of sequence from 212 911 exons and their flanking regions in ∼20 000 genes. Five hundred nanograms of the prepped library was incubated with whole exon biotinylated DNA capture baits supplied in the kit for 24 h at 65°C. The captured DNA hybrids were recovered using Dynabeads MyOne Streptavidin T1 from Dynal. The DNA was eluted from the beads and desalted using Qiagen MinElute PCR purification columns. The purified capture products were then amplified using the SureSelect GA PCR primers (Agilent) for 12 cycles.
Massively parallel sequencing
Sequencing was carried out for the captured libraries with the Illumina Genome Analyzer IIx platform (GAIIx) using 75-bp paired-end reads (three samples) and with HiSeq 2000 using 100-bp paired-end reads. Libraries were loaded onto paired-end flow cells at concentrations of 6–8 pM (GAIIx) or 4–5 pM (HiSeq 2000) to generate cluster densities of 250 000–350 000/tile (GAIIx) or 300 000–500 000/mm2 (HiSeq 2000) following Illumina's standard protocol using the Illumina cluster station and Paired-End Cluster Kit version 4 (GAIIx) or the Illumina cBot and HiSeq Paired-End Cluster Kit version 1 (HiSeq 2000). To achieve a high level of sensitivity and accuracy for detecting all the mutations in the whole exome, each sample was sequenced at the depth of, on average, 123X (Supplementary Table 2, available at Carcinogenesis Online). Image analysis and base calling were carried out by Illumina software (CASAVA) with default parameters. All the sequence runs used in the data analysis passed quality controls with error rates of <2% and Eland alignment rates of >80%.
Read mapping and alignment and variant analysis
We recently developed an in-house analysis pipeline for cancer genome sequencing data. This pipeline essentially includes (i) mapping and alignment, (ii) sequence nucleotide variations (SNVs) and insertions and deletions (indels) discovery and (iii) SNVs and indels filtering and annotation. Briefly, once the raw sequence data was created, the output short reads were aligned to a reference genome (NCBI human genome assembly build 36) using the Burrows-Wheeler Aligner (12). Each alignment was assigned a mapping quality score by Burrows-Wheeler Aligner, which is the Phred-scaled probability that the alignment is incorrect. The PCR duplicates were detected and removed by Picard (http://picard.sourceforge.net). After alignment, we used the SomaticSniper (http://genome.wustl.edu/software/somaticsniper) and VarScan (13) to call SNV for each chromosomal position. SNVs and indels filtering and annotation were performed using ANNOVAR (http://www.openbioinformatics.org/annovar/). We defined high-quality SNVs as those detected both by SomaticSniper with Somatic Score 60 and single-nucleotide polymorphism (SNP) mapping quality 60 and by VarScan with somatic P value 0.05. To minimize false positives, we also set minimum coverage as 8× in normal and 15× in tumor, minimum reads of variant allele as 4 and minimum proportion of variant allele as 15% in tumor. We also filter by minimum number of 2 reads supporting the variant allele per strand. For indels called from VarScan, we further remove them with any support reads from its matched normal control.
Pathway and protein interaction network analyses
Direct interaction protein networks were generated separately for the frequently mutated genes in common between ADC and SCC, the genes only mutated in ADC and the genes only mutated in SCC. The networks were generated using the GeneGo MetaCore software and include only those proteins that are products of the relevant genes and that are annotated to have direct interactions with other such proteins according to their databases. The gene selected as input and the network construction are as follows. For the commonly mutated genes between ADC and SCC, we included all those genes with 3 or more non-silent mutations of at least two subtypes as well as those genes with a critical mutation (nonsense, frameshift and splicing) as defined above in each subtype and which belongs to one of the top 25 scoring pathways based on the critical mutation definitions (219 genes). We created direct interaction networks for the protein products of these genes. We similarly created direct interaction protein network for SCC-only mutated genes (in two or more samples) and genes of the frequently mutated pathways for which mutations were observed only in SCC (158 genes) and for the ADC-only mutated genes (106 genes) and genes of the frequently mutated pathways for which mutations were observed only in ADC. In the figures, the proteins are arranged according to their annotated cellular localization.
Cell culture and short-hairpin RNA knockdown
Beas-2b cells were cultured in complete BEGM with supplements (Lonza, Basel, Switzerland) on tissue culture dishes coated with 10 μg/ml fibronectin, 30 μg/ml bovine collagen type I and 10 μg/ml bovine serum albumin. Cells were transduced with lentiviral short-hairpin RNA vectors based on the pLKO.1 vector and designed to specifically target human CSMD3 transcript (Open Biosystems, Huntsville, AL). Empty vector or vector knocking down CSMD3 transcript were first packaged in 293T cells (Orbigen, San Diego, CA) with helper plasmids and then transduced into A549 cells with 8 μg/ml polybrene (Sigma, St Louis, MO). Media were replaced 24 h after transduction, and cells were split 1:4 48 h after transduction. At 72 h post-transduction, cells harboring lentiviral constructs were selected with 1 μg/ml puromycin for 2–4 days, until mock-infected cells were dead.
MTS proliferation assay
Cells were seeded onto 12-well tissue culture dishes at a density of 5000 cells per well. Cells were assayed for viable cell numbers in triplicate by adding 100 μl per well of the MTS-based CellTiter 96 AQueous One Solution Cell Proliferation reagent (Promega, Madison, WI) and measuring A490 on a plate reader over 8 days in culture.
Full description of methods and any associated references are presented in Supplementary data.
Results
Sequencing and variant detection
We conducted exome sequencing of 31 primary lung tumors and their matched normal controls. On average, we generated 14.4 Gb of sequence per sample to a mean depth of 123-fold exon coverage (Supplementary Table 2, available at Carcinogenesis Online). In total, 89.2% of the 212 911 exons on the genome were covered with more than one sequencing read. To eliminate common germline mutations, we removed any potential somatic mutations that were observed in dbSNP130 (http://www.ncbi.nlm.nih.gov/projects/SNP/), 1000 Genomes Projects (http://www.1000genomes.org/), Hapmap exomes (14) or 200 Danish exomes (15). To reduce false-positive rates, we adopted comprehensive filtering criteria to identify high-confidence tumor-specific SNVs. As a result, we identified 7201 high-confidence somatic mutations in the 31 samples sequenced, including 2633 in protein-coding regions (Supplementary Tables 3 and 4, available at Carcinogenesis Online).
To evaluate false-positive rates of the identified high-confidence somatic SNVs, we selected all the nonsense, splicing mutations and exonic indels and a random subset of somatic substitutions causing missense mutations, totaling 481 SNVs, for Sanger sequencing validation. The Sanger sequencing yielded an ∼90% validation rate of the SNVs (i.e. a false-positive rate of 10%) that passed our comprehensive filtering criteria (Supplementary Table 5, available at Carcinogenesis Online). It is worth noting that the other types of mutations such as intronic and untranslated regions variants will have approximately similar validation rates since they were filtered by the same criteria. We further validated additional somatic SNPs using the NimbleGen AccuSNP™ genotyping platform. Based on the HapMap sample NA07022, the newly launched platform achieves 99.9% (SE, 0.01) genotyping accuracy with a false call rate as low as 0.09% (0.01%) and no call rate of 0.35% (0.07%) (Supplementary Table 6, available at Carcinogenesis Online). SNPs (13 860) that were submitted to the AccuSNP™ genotyping platform were predicted directly from either software VarScan (13) or SomaticSniper and were not subjected to our filtering criteria. As a result, 665 new somatic SNPs were identified with the AccuSNP™ platform (Supplementary Table 7, available at Carcinogenesis Online). We found that 75.5% of the validated somatic SNPs are contained in our 7201 high-confidence SNVs. This number can be translated into a <25% false-negative rate when using the filtering criteria adopted in our study while keeping the false-positive rate as low as 10%.
Overview of somatic mutation profiles
The 7201 SNVs are distributed in various genomic regions including exonic (44.8%), splicing (2.1%), ncRNA (14.4%), 5′-untranslated regions (1.0%), 3′-untranslated regions (1.8%), intronic (23.4%), upstream (0.9%), downstream (0.8%) and intergenic regions (10.7%) (Table I and Supplementary Table 3, available at Carcinogenesis Online). Among them, 2271 substitutions caused amino acid changes (missense), 194 nonsense mutations led to truncated proteins, 1 led to abnormal protein extension and 151 occurred at splicing sites. There were 754 silent (synonymous) substitutions in protein-coding regions. We also observed 67 insertions and deletions (indels) causing exonic frameshifts and 2 causing non-frameshifts, ranging from 1 to 19 bp in length (Supplementary Table 8, available at Carcinogenesis Online). Nonsense, frameshift indels and splicing mutations generally lead to inactivation of the protein products. To evaluate missense mutations, we used four algorithms to make a consensus prediction to identify putative driver mutations. These functional prediction algorithms are based on phylogenetics, structure biology, bioinformatics or population genetics and were trained on different sets of data; each has its own strength and weakness. Of 2271 missense mutations, 397 (17.5%) were predicted by all the four algorithms and 772 (34.0%) were predicted by at least three of them to affect protein function (Supplementary Table 9, available at Carcinogenesis Online).
Table I.
Summary of somatic sequence mutations in the lung cancer exome study
| Total SNVs | 232.3 |
| Exonic | 104.1 (44.8%) |
| Splicing | 4.9 (2.1%) |
| ncRNA | 33.5 (14.4%) |
| UTR5 | 2.4 (1.0%) |
| UTR3 | 4.3 (1.8%) |
| Intronic | 54.3 (23.4%) |
| Upstream | 2.2 (0.9%) |
| Downstream | 1.9 (0.8%) |
| Intergenic | 24.8 (10.7%) |
| SNVs in protein-coding regions | |
| Frameshift deletion | 1.7 (1.6%) |
| Frameshift insertion | 0.5 (0.5%) |
| Nonframeshift deletion | 0.1 (0.1%) |
| Nonframeshift insertion | 0.0 (0.0%) |
| Nonsense (truncated) | 6.3 (6.0%) |
| Nonsense (extension) | 0.0 (0.0%) |
| Nonsynonymous | 73.3 (70.4%) |
| Synonymous | 24.3 (23.4%) |
These exonic SNVs and mutations occurring in splicing sites are distributed in 2170 genes. Comparison of the identified 2170 mutated genes with the Catalogue of Somatic Mutations in Cancer (COSMIC) database (http://www.sanger.ac.uk/genetics/CGP/cosmic/) revealed that 1443 of them have been previously reported in the literature or COSMIC database to be somatically mutated in human tumors. The exome sequencing confirmed 23 point mutations and 5 frameshift deletions previously reported in the COSMIC database (Supplementary Table 10, available at Carcinogenesis Online). These included 10 TP53 and 2 RB1 somatic mutations that have been observed multiple times in different types of tumors as reported by the COSMIC. Identical somatic mutations in other important cancer genes such as KRAS, EGFR, PTEN, PIK3R1, APC and SLC37A3 were also found in our tumor samples. Our data further highlight the importance of these mutations in lung tumorigenesis.
We observed on average, 104 somatic SNVs in exonic regions per exome with the most frequently altered exome having 424 SNVs. No somatic mutations in exonic regions were identified in two samples. On average, SCC had 154 somatic, exonic SNVs, whereas only 86 somatic SNVs were found in ADC (Supplementary Figure 1, available at Carcinogenesis Online). The observed ratio of non-synonymous to synonymous change is significantly higher in ADC than in SCC (3.6 versus 2.8) (P vaule = 0.05) (Supplementary Figure 2, available at Carcinogenesis Online). Among 7201 SNVs identified by exome sequencing, somatic variants occurred predominantly at G•C base pairs (63.8% in ADC and 68.8% in SCC) with the most prevalent changes being G•C to A•T (24.3% in ADC and 26.8% in SCC) and G•C to T•A transversions (26.8% in ADC and 28.2% in SCC). As compared with germline mutation patterns, G•C to T•A transversions (7.8 versus 26.8% in ADC and 7.8 versus 28.2% in SCC) and A•T to T•A transitions (6.0 versus 11.4% in ADC and 6.1 versus 0.12.3% in SCC) were strongly enriched in somatic mutations (Supplementary Figure 3, available at Carcinogenesis Online).
As expected, the number of somatic SNVs identified in never smokers were much less than in smokers that have an average of 130 exonic somatic SNVs per genome (Supplementary Figure 1, available at Carcinogenesis Online). In smokers, G•C to T•A transversions (29.4%) were the commonest change observed, followed by G•C to A•T (24.5%) and A•T to G•C (14.8%) transversions. Although similar trends were observed in never smokers, G•C to T•A transversions in never smokers are significantly fewer than in smokers (P = 0.002) (Supplementary Figure 3, available at Carcinogenesis Online). These results are largely consistent with previously documented tobacco exposure-related mutation signatures (8–10).
Somatic mutations in ADC and SCC
At the gene level, we first looked to see if mutated genes have been previously reported in lung cancer. As compared with a recent sequencing study (8) that identified 24 significantly mutated genes in lung ADC, 12 of them were also mutated in our samples, including TP53, KRAS, EGFR, LRP1B, NF1, ATM, APC, EPHA3, PTPRD, EBRR4, KDR and RB1. Eleven of 12 genes that are not identified in our study have <4% of mutation frequencies reported in the original samples. Then, we looked for somatic alternations enriched in both ADC and SCC samples. As expected, TP53 was the most frequently mutated gene in lung tumors. Six 1-bp frameshift deletions and five substitutions causing amino acid changes in TP53 were identified in 10 of 31 samples, including 3 ADC and 7 SCC. In addition to TP53, we found 51 more genes harboring at least three recurrent mutations, of which 10 significantly exceed their background mutation rate (P < 0.001) (Table II and Supplementary Table 11, available at Carcinogenesis Online). These significantly mutated genes are LRRC7, SLC7A13, PCDH11X, CSMD3, DNAH3, CD1B, CACNA2D1, KEAP1, PIK3C2B and CTNNA3 (Figure 1). All the 10 significantly mutated genes have not been reported in recent sequencing studies (8–10). It is not surprisingly since the previous studies were focused on either a limited set of genes or a single sample. Particularly, seven nonsynonymous mutations (G700W, R899T, N954D, T1054K, T1104K, T2735S and D3135Y) and one mutation occurring splicing site in CSMD3 were observed in 6 of 31 samples, whereas another member of CSMD gene family, CSMD2, bears four nonsynonymous mutations in 4 of 31 samples. Six of seven missense mutations in CSMD3 are located in either its CUB or sushi domains (Supplementary Figure 4, available at Carcinogenesis Online). Pathways enriched in the set of frequently mutated genes found in both subtypes include the Role of SUMO in p53 regulation, c-jun N-terminal kinase pathway, DNA damage-induced response pathways as well as hepatocyte growth factor and transforming growth factor-beta signaling pathways (Table II and Supplementary Table 14, available at Carcinogenesis Online). The DNA damage control and c-jun N-terminal kinase signaling pathways had significantly high ratios of non-silent mutations to silent mutations across our samples (Supplementary Tables 15 and 16, available at Carcinogenesis Online).
Table II.
Highly mutated genes and their associated pathways in lung cancer
| Set | Regulatory process or pathway | Representative altered genes | Adjusted P values |
| Common | DNA damage control | TP53, MYO5A,POT1, DAXX, RB1, NBN | 2E-14 |
| c-jun N-terminal kinase signaling | TP53, DAXX, PLCB1, PIK3CG, ATF2 | 3E-21 | |
| Role of SUMO in p53 regulation | TP53, DAXX | 5E-20 | |
| Hepatocyte growth factor signaling in pancreatic cancer | TP53, HGF | 2E-18 | |
| Transforming growth factor-beta signaling | TP53, ATF2 | 5E-12 | |
| ADC | Mitogen-activated protein kinase signaling | KRAS and NOX4 | 3E-04 |
| SCC | Invasion | DDI1, PROC, MMP16, HSPA6, PCCB, LCT and AOX1 | 2E-03 |
| Small GTPase signaling | WNK1, PLCXD3, PREX2, FMN2, DAB1 and CD36 | 5E-03 | |
| ECM remodeling | LAMB1 and MMP16 | 7E-04 |
Fig. 1.

Significantly mutated genes in lung cancer. Red circles represent genes mutated in both ADC and SCC; blue circles represent genes mutated only in ADC and green circles represent genes mutated only in SCC. P values represent the evidence of a gene having higher mutation rate than its background mutation rate. The new Poisson model for mutational process was described in Supplementary data.
We also observed histology-specific mutated genes. For ADC, we identified 43 such genes mutated in at least two ADC samples (Table II and Supplementary Table 12, available at Carcinogenesis Online). We then determined if this group of genes is significantly altered in the ADC samples. We found that the ratios of nonsynonymous to synonymous mutation are significantly higher than the expectations across ADC samples, suggesting that they are not random subset of genes with passenger mutations (P value = 1.09 × 10−3). As expected, KRAS point mutations in Codon 12 from lysine to valine were present in two lung ADC. Three genes (ZNF717, RAB6B and DCAF12L2) are significantly mutated in ADC. At the individual mutation level, 30 of 43 ADC-specific genes bear mutations that are predicted to affect protein function (Supplementary Tables 9 and 12, available at Carcinogenesis Online). We found that genes involved in mitogen-activated protein kinase signaling were preferentially mutated in ADC (Table II and Supplementary Table 14, available at Carcinogenesis Online).
Similarly, we identified 64 SCC-specific genes mutated in at least two samples (Table II and Supplementary Table 13, available at Carcinogenesis Online). This group of genes is significantly altered in specific SCC samples (P = 1.11 × 10−8). Three new genes ARMCX2, COL27A1 and APBB1IP are significantly mutated in SCC. Mitochondrial dysfunction has been linked to tumorigenesis. Interestingly, we found SCC-specific somatic mutations in NDUFA13 (GRIM19), PCCB and SDHB, all of which play essential roles in mitochondrial energy production (16–18). Other processes enriched in the SCC specifically mutated genes included ECM remodeling, invasion and small GTPases signaling (Table II and Supplementary Table 14, available at Carcinogenesis Online).
Somatic mutations in other types of lung tumors
Four lung ADC from never smokers were sequenced in our study. Only 1, 4, 7 and 10 somatic SNVs occurring in exons were identified in these four never smokers (Supplementary Table 4, available at Carcinogenesis Online). For the first never smoker with lung cancer, we observed only one nonsynonymous substitution in ANO9, which is a p53-inducible protein. In the second never smoker, three somatic SNVs causing nonsynonymous substitutions in VEGFA, VPS37B and RBM39 and one 4-bp deletion causing frameshift in SETD2 were validated. SETD2 protein is a histone methyltransferase that is specific for lysine-36 of histone H3, and methylation of this residue is associated with active chromatin. Among seven exonic SNVs identified in the third case, four are missense mutations (INADL, FAM129A, CLCN7 and OR2C3), one is a single-base deletion causing a frameshift in MARK1 and two are silent substitutions. MARK1 is a kinase regulating microtubule-associated protein–microtubule affinity. The fourth case bears a 2-bp deletion in TP53 and four missense mutations in SLC25A12, AGTR1, RPAP1 and DNAH2.
In addition to ADC and SCC, we also sequenced one BAC which is one of four histologically distinct subtypes of lung ADC. It possesses unique clinical and pathological features, and prognoses, and responds to different treatments. We identified one SNV causing truncation of PIK3R1 protein and one in a CCHCR1 splicing site. Depletion of PIK3R1 induces cell cycle arrest and apoptosis in colorectal cancer cells (19). In addition, five nonsynonymous substitutions were identified in AHCTF1, EGFR, GSTA3, SLC7A13 and SHROOM2. We identified one recurrent mutation in EGFR in one of five BACs that were sequenced in whole genomes (data not shown).
Protein interaction networks
We used protein interaction networks in order to examine possible interrelationships between individual genes, which are most likely to be relevant for the mutation profiles that we observed. Among the genes commonly mutated in multiple subtypes, there was one primary cluster (21 nodes) of interacting proteins as well as four smaller clusters (2 or 3 nodes). The hub of the primary cluster was p53 with three proteins upstream of p53, but most of the proteins downstream of p53. Thus, p53, in addition to being the most frequently mutated gene, has the greatest frequency of mutated interacting proteins as well, with most of them being downstream. The ATF2 and RB1 protein products also had several interacting proteins (seven in ATF2 and five in RB1) (Figure 2A and Supplementary Figure 5, available at Carcinogenesis Online). In contrast, the corresponding ADC direct interaction protein network for ADC-only mutated genes had only a few interactions (Figure 2B). SLIT2 and SLIT3 have activation effects on ROBO2. CALDAG-GEFI activates KRAS, which activates A-Raf-1 and RalGDS. Neuroreglin-1 activates ErbB. The MAPK14 protein, p38-alpha, acts on MAP2 and HNF-1-alpha acts on two other genes. The network for the SCC-specific genes contains >50 nodes including 10 genes having five of more interactions (e.g. 10 in p73 and 9 in SMAD4). Key divergence hubs (upstream proteins that act on many proteins) include PKC-beta and SMAD4. PTEN is a major convergence hub (a protein that is affected by many other proteins), while p73 is a major hub with both converging interactions, including PKC-beta, and diverging interactions, including PTEN (Figure 2C).
Fig. 2.
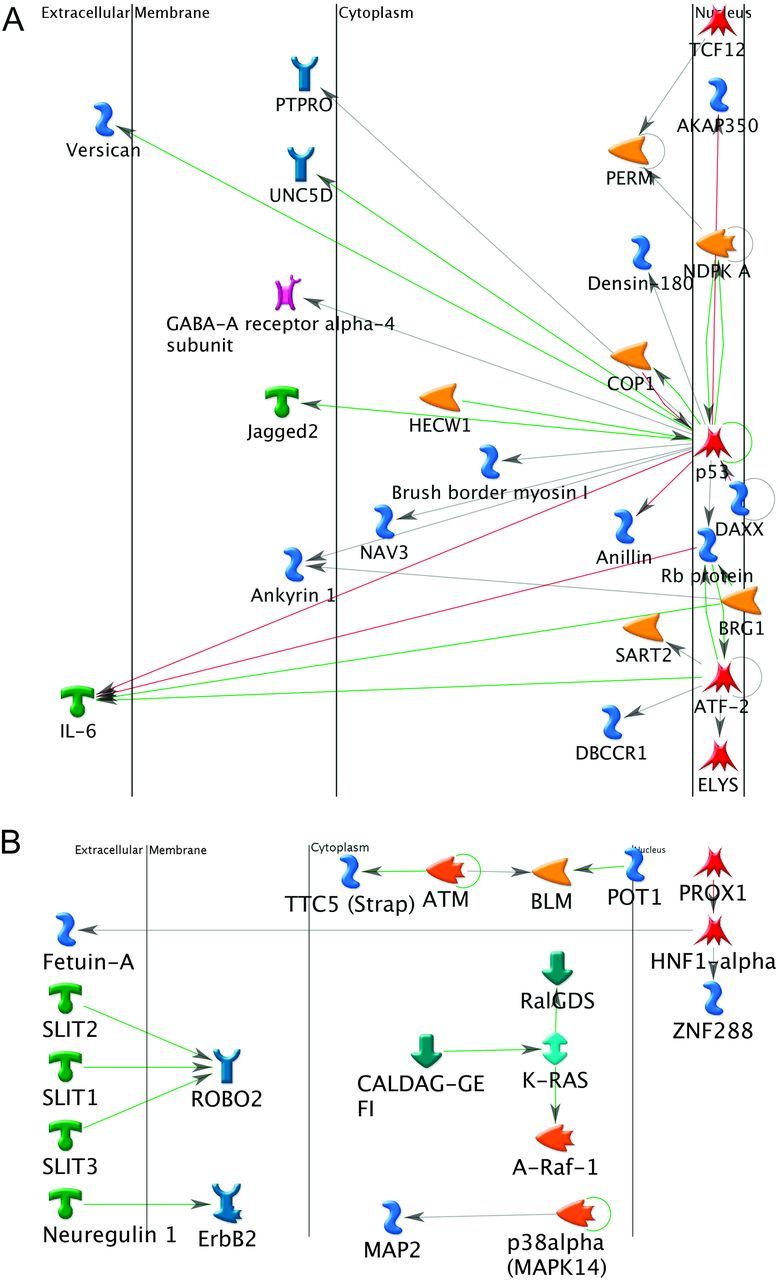
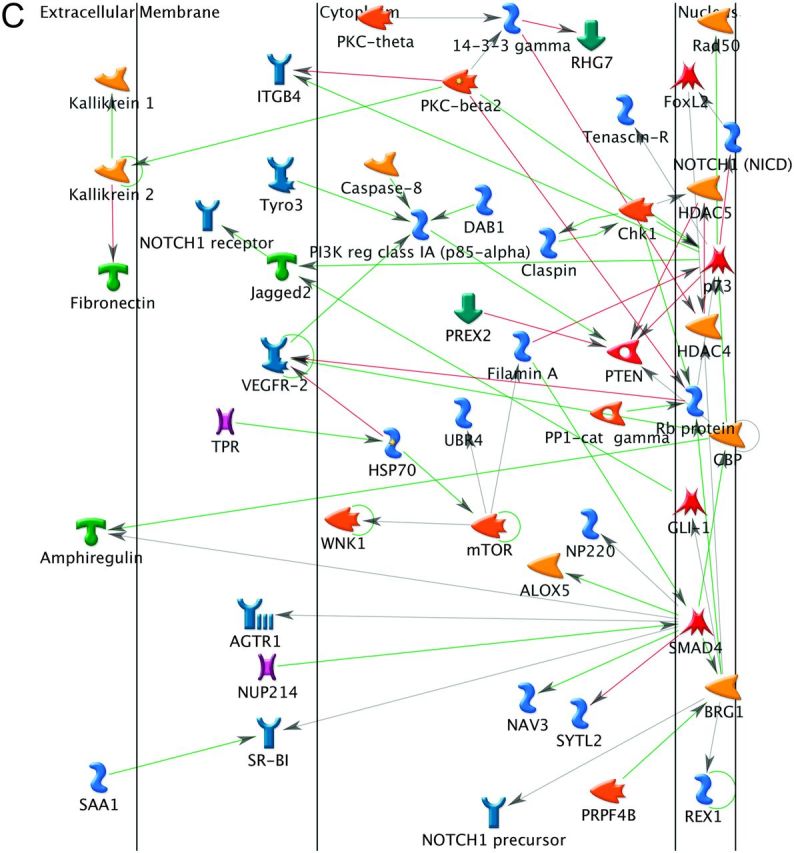
Direct interaction networks. (A) Frequently mutated genes observed in both ADC and SCC. (B) ADC-specific genes. (C) SCC-specific genes. Only clusters with visible interactions (not genes of the same complex) are shown. The direct of action is arranged to be mostly from left to right. The color of the edges or connecting lines indicate the type of regulation (activation: green, inhibition: red and unspecified: gray). Arrows indicated the direction of the interaction with the arrow pointing to the gene being acted on. The meaning of the symbols in the network is presented in Supplementary data.
Loss of CSMD3 results in increased proliferation of airway epithelial cells
Our discovery of frequent and recurrent CSMD3 mutations in lung cancer and the lack of prior knowledge regarding the function of this gene led us to pursue in vitro functional characterization of CSMD3. Upon stable infection of immortalized airway epithelial Beas-2b cells with viral short-hairpin RNA vectors targeting CSMD3, we were able to achieve a range of CSMD3 knockdown efficiencies, up to ∼90% knockdown of endogenous levels with sh3 (Figure 3A). These stable cell lines were subjected to an MTS-based growth curve analysis to evaluate the effect of loss of CSMD3 expression on proliferative rate. Airway epithelial cells with loss of CSMD3 transcript accumulation demonstrated increased rates of growth (Figure 3B). Cells with the sh1 vector, which demonstrated no significant knockdown of CSMD3, grew to an average absorbance reading of 0.933 at Day 6, whereas sh2 and sh3 grew to average absorbance readings of 1.36 and 1.65, respectively. Differences in growth after 6 days in culture were statistically significant with sh2 and sh3 (P = 0.003 and P = 0.000013, respectively). These results suggest that loss of CSMD3 function by somatic mutation may contribute to the oncogenic transformation of airway epithelial cells.
Fig. 3.
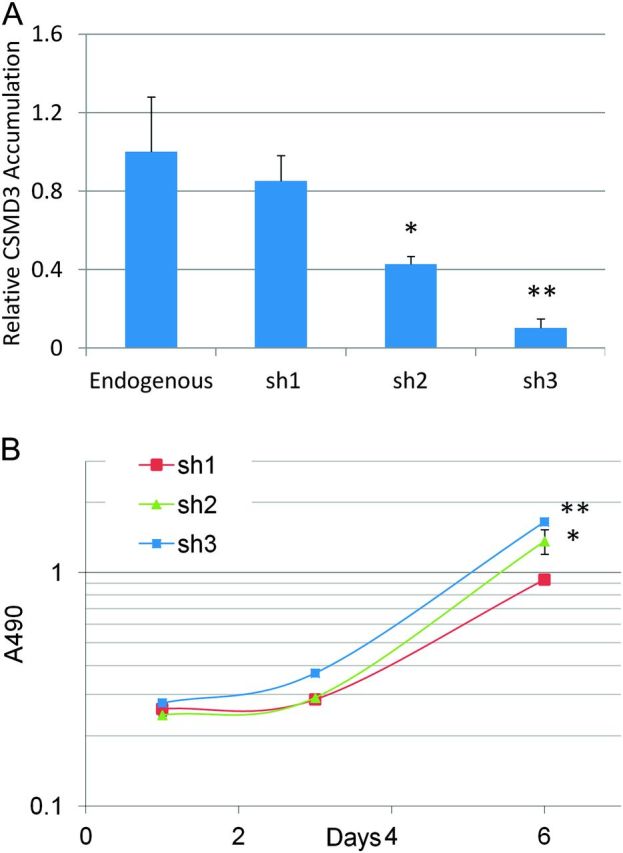
Knockdown of CSMD3 in airway epithelial cells. (A) Real-time PCR quantitation of CSMD3 transcript levels relative to endogenous levels for pooled populations of cells infection with short-hairpin RNA vectors targeting CSMD3 and selection. All data is normalized to actin signal for each sample. *P = 0.008, **P = 0.001. P values were obtained using two-tailed Student's t-test and represent significance of difference from endogenous levels. Error bars represent standard error from the mean. Data points represent triplicate assays. (B) Growth curves of Beas-2b cells with CSMD3 knockdown vectors. Cell viability was determined over 8 days in culture by MTS assay. *P = 0.003, **P = 0.000013. P values were obtained using two-tailed Student's t-test and represent significance of difference in Day 6 values from sh1. Error bars represent 1 SD from the mean.
Discussion
Lung cancer is caused by the accumulation of genomic alterations. Our study represents the latest efforts to characterize genomic alternations in NSCLC using the large-scale exome sequencing. As expected, smokers have much higher mutation rates than never smokers. G•C to T•A transversions were the most common substitution in smokers with lung cancer, consistent with the previously documented tobacco exposure-related mutation signatures (8–10). However, the lung cancers of smokers are highly heterogeneous and the number of somatic mutations ranges widely from no mutations to 414 SNVs in exonic regions. This is probably due to difference in inherited genetic backgrounds and DNA mismatch repair defects. Our study revealed a broad mutation spectrum in NSCLC. Comparison of our list of somatic mutated genes with the COSMIC database revealed that 727 new mutated genes found in our exome study have not yet been reported in the literature or COSMIC database. These data highlight the complexity of lung cancer genomes and strong carcinogenic effects of tobacco exposure on mutagenesis. A multiplicity of partially redundant mutations may exist in genetically complex lung tumors.
Lung ADC and SCC are two major histologies of NSCLC, accounting for ∼60% of all lung cancers. We identified a total of 52 genes commonly mutated in both ADC and SCC samples. The newly identified genes include LRRC7, SLC7A13, PCDH11X, CSMD3, DNAH3, CD1B, CACNA2D1, KEAP1, PIK3C2B, CTNNA3, ALK, BAI3, CDH18, MLL3, DCC, FAT4, FLNC, NAV3, PAPPA2, MYO5A, PIK3CG, PHKA1, ROCK2 and SALL1. Each of these somatic genes is mutated in 11–36% of lung cancer samples. Notably, CSMD3 was identified as the second most frequently mutated gene in lung cancer. CSMD3 encodes a transmembrane protein with CUB and sushi multiple domains. The role of CSMD3 in carcinogenesis has not yet been studies so far. Our functional evaluation suggests that loss of CSMD3 results in increased proliferation of airway epithelial cells. The identification of a second major cancer gene in NSCLC will further define the pathogenesis and genetic architecture of this tumor type. Understanding the contribution of CSMD3 mutation to tumor initiation as well as disease progression and outcome is important future areas of lung cancer research.
In addition to commonly mutated genes, we also demonstrated distinct mutational signatures and signaling pathways between ADC and SCC subtypes. We identified 43 ADC-specific genes mutated exclusively in ADC samples we studied. Three genes are involved in nitric oxide synthesis, including NOS1, NOX4 and QSOX1. NO-induced signaling pathways play important roles in cancer cell apoptosis and survival. Similarly, we identified 64 SCC-specific genes mutated in at least 2 of 12 samples. Three mutated genes NDUFA13 (GRIM19), PCCB and SDHB are potentially involved in mitochondrial dysfunction during SCC carcinogenesis. One of SCC-specific gene, GRIN2A, was recently identified as the most frequently mutated gene in melanoma (20). GRIN2A encodes a glutamate receptor subunit N-methyl-d-aspartate, which is one of the ionotropic glutamate receptors (iGluRs). We identified additional four iGluRs including GRIA1, GRIA4, GRID1 and GRIN2D that were mutated in 3 of 12 SCC samples but not in ADC. NAGR1, a regulator of N-methyl-d-aspartate receptors, was also mutated in one SCC. Furthermore, another metabotropic glutamate receptor (mGluRs) GRM8 and its downstream effector, PLCB4, involved in glutamate signaling pathway were mutated in the SCC samples. These results imply that the glutamate signaling pathway may be highly significant in squamous cell lung carcinogenesis.
Although there are substantial overlaps between the mutation profile of our experiment and mutation profiles of lung and other tissues reported elsewhere (Supplementary Tables 14–16, available at Carcinogenesis Online), we observed some differences in pathway alterations between ADC and SCC. SCC has a higher frequency of mutation than ADC and a more highly connected network of mutated genes (Supplementary Table 16 and Figure 3, available at Carcinogenesis Online). The SCC subtype has more enrichment associated with cell adhesion, cytoskeleton, transforming growth factor and WNT remodeling. PTEN and p53 form a positive feedback loop in normal cells (21,22). We observed many mutations in TP53 and its downstream genes and PTEN and its upstream genes (Figure 3). Mechanisms that keep this positive feedback loop in check may be disrupted in lung cancer and, in particular, in SCC. There are some connections among mutated genes that appear to be more common in ADC. This subtype may have more mutations involving RAS signaling and Slit-Robo signaling.
Of potential immediate clinical relevance, this study has identified a number of druggable targets of relevance in cancer therapy (Supplementary Table 17a, available at Carcinogenesis Online). Among them, 77 drugs per drug target combination are specifically used for cancer and are Food and Drug Administration approved or are or have been used in a clinical trial. Thirty-two of the 77 are already being used in lung cancer chemotherapy (Supplementary Table 17b and c, available at Carcinogenesis Online). Some findings recapitulate earlier findings in other tumors, where agents are already in clinical testing. These include GPLD1, GRM5, NOS1, SCN2A, AGTR1, KEAP1, NOTCH1, MTOR, HDAC4, PARP, EPHA7, HGF, KRAS, MAGE, ADAM and the MAP kinases. For example, edelfosine is currently in cancer clinical trial, targeting NOS1 and SCN2A.
Novel genes of significant interest as cancer drug targets include ALK. In addition to crizotinib, an inhibitor of lung cancers harboring the EML4-ALK translocation which is expected to be available for lung cancer therapy in the next few months, there are other ALK kinase inhibitors in the clinic including ASP3026, LDK378, AF802 and AP26113. Although the PI3Kinase inhibitors in the clinic only target PIK3CA, which rarely harbors mutations in lung cancer, other PI3 kinases PIK3C2B and PIK3CG have been found to be frequently mutated in this study and are targets for drug therapy, using either isoform-specific or pan PI3 kinase inhibitors.
In addition, MLL3 has been described in leukemia and lymphomas. This gene encodes a nuclear protein with histone methylase activity and is a target for epigenetic therapy. DCC, a candidate tumor suppressor gene in colon cancer, has been a target for gene therapy approaches. ROCK2 (rho kinase) inhibitors have been studied as a target for cardiovascular disease, however, the pivotal role of ROCK in mediating apoptosis, suggest a role in cancer therapy. While cancer therapeutics has focused on targeting the WNT/beta catenin pathway, alpha T-catenin (CTNNA3) stabilizes cellular adherence, and disruption could enhance tumor metastasis. Thus, CTNNA3 is an attractive drug target for lung cancer. PCDHIIX has been found to be a tumor suppressor gene and is an interesting target, as cancer therapeutics develops approaches to targeting mutated/deleted tumor suppressors.
Although this study represents one of the largest sequencing efforts so far, it would be interesting to screen a large number of independent tumors for somatic mutations in the candidate genes that emerge from our initial screening set of 31 samples. Such a screen serves to determine if these candidate genes are recurrently mutated in lung cancer. Therefore, we performed in silico screening of somatic mutations in independent sets of lung tumors from The Cancer Genome Atlas (TCGA, http://cancergenome.nih.gov/). As of 30 September 2011, we downloaded 195 pairs of lung tumor samples from TCGA (PYL, unpublished). We used the same analysis pipeline to call and filter somatic variants in these samples. As a result, 50 of 52 most frequently mutated genes identified in our 31 samples (Supplementary Table 11, available at Carcinogenesis Online) were also mutated in TCGA samples; the other two not observed in TCGA samples are due to differences in gene annotation between human genome build 36 and 37. Twenty-six of 52 samples have frequency of somatic mutations >5% (data not shown). TP53 and CSMD3 were mutated in 42.6 and 21.5% the TCGA samples, which are comparable with those identified in our screen (32.3 and 19.4%). These data demonstrated that many of the frequently mutated genes identified in our screen are recurrently mutated in lung tumors.
Finally, several caveats for our findings should be acknowledged. First, the lung tumors exhibited extremely complex genomic alterations including point mutations, small insertion and deletion, copy number variation and large chromosomal rearrangements (9,10). Our exome sequencing study has characterized point mutations and small insertions and deletions. Copy number and structural variant analysis should be included in the future investigation. Second, due to the nature of next-generation sequencing technology, some protein-coding regions cannot be captured in library preparation or by sequencing because of regions of high GC content. Only somatic mutations are called when both tumors and their matched normal samples simultaneously have sufficient coverages at those coding regions. For example, mutations in STK11 found to be frequently mutated in the previous study (8) were not identified in our study. Third, the identification of a second major cancer gene in NSCLC will further define the pathogenesis and genetic architecture of this tumor type. Our functional study provided initial evidence of CSMD3 contributing to lung tumorigenesis. Understanding the contribution of CSMD3 mutation to tumor initiation as well as disease progression and outcome are important future areas of lung cancer research.
Supplementary material
Supplementary Tables 1–17 and Figures 1–5 can be found at http://carcin.oxfordjournals.org/
Funding
This work was supported by National Institutes of Health grants R01CA129533 (MY), R01CA113793 (MY), R01CA134682 (MY, PL), R01CA80127 (PY), R01CA84354 (PY); the Advancing a Healthier Wisconsin Chemoprevention Program (PI: MY); and the Mayo Foundation Fund (PY).
Supplementary Material
Acknowledgments
We thank Bruce W. Eckloff and Eric D. Wieben of the Sequencing Core at the Mayo Clinic for technical assistance, and Haris Vikis and Jay Tichelaar for reading and commenting on the manuscript.
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- ADC
adenocarcinomas
- BAC
bronchioloalveolar carcinoma
- NSCLC
non-small cell lung cancer
- SNVs
sequence nucleotide variations
- SCC
squamous cell carcinomas
References
- 1.Siegel R, et al. Cancer statistics, 2011: the impact of eliminating socioeconomic and racial disparities on premature cancer deaths. CA Cancer J. Clin. 2011;61:212–236. doi: 10.3322/caac.20121. [DOI] [PubMed] [Google Scholar]
- 2.Wistuba II, et al. Molecular genetics of small cell lung carcinoma. Semin. Oncol. 2001;28:3–13. [PubMed] [Google Scholar]
- 3.Horowitz JM, et al. Frequent inactivation of the retinoblastoma anti-oncogene is restricted to a subset of human tumor cells. Proc. Natl Acad. Sci. USA. 1990;87:2775–2779. doi: 10.1073/pnas.87.7.2775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mori N, et al. Variable mutations of the RB gene in small-cell lung carcinoma. Oncogene. 1990;5:1713–1717. [PubMed] [Google Scholar]
- 5.Yokomizo A, et al. PTEN/MMAC1 mutations identified in small cell, but not in non-small cell lung cancers. Oncogene. 1998;17:475–479. doi: 10.1038/sj.onc.1201956. [DOI] [PubMed] [Google Scholar]
- 6.Franklin WA, et al. Epidermal growth factor receptor family in lung cancer and premalignancy. Semin. Oncol. 2002;29:3–14. doi: 10.1053/sonc.2002.31520. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki Y, et al. Detection of ras gene mutations in human lung cancers by single-strand conformation polymorphism analysis of polymerase chain reaction products. Oncogene. 1990;5:1037–1043. [PubMed] [Google Scholar]
- 8.Ding L, et al. Somatic mutations affect key pathways in lung adenocarcinoma. Nature. 2008;455:1069–1075. doi: 10.1038/nature07423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee W, et al. The mutation spectrum revealed by paired genome sequences from a lung cancer patient. Nature. 2010;465:473–477. doi: 10.1038/nature09004. [DOI] [PubMed] [Google Scholar]
- 10.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Meyerson M, et al. Advances in understanding cancer genomes through second-generation sequencing. Nat. Rev. Genet. 2010;11:685–696. doi: 10.1038/nrg2841. [DOI] [PubMed] [Google Scholar]
- 12.Li H, et al. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Koboldt DC, et al. VarScan: variant detection in massively parallel sequencing of individual and pooled samples. Bioinformatics. 2009;25:2283–2285. doi: 10.1093/bioinformatics/btp373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ng SB, et al. Targeted capture and massively parallel sequencing of 12 human exomes. Nature. 2009;461:272–276. doi: 10.1038/nature08250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, et al. Resequencing of 200 human exomes identifies an excess of low-frequency non-synonymous coding variants. Nat. Genet. 2010;42:969–972. doi: 10.1038/ng.680. [DOI] [PubMed] [Google Scholar]
- 16.Oishi Y, et al. Mutation analysis of the SDHB and SDHD genes in pheochromocytomas and paragangliomas: identification of a novel nonsense mutation (Q168X) in the SDHB gene. Endocr. J. 2010;57:745–750. doi: 10.1507/endocrj.k10e-023. [DOI] [PubMed] [Google Scholar]
- 17.Lu H, et al. GRIM-19 is essential for maintenance of mitochondrial membrane potential. Mol. Biol. Cell. 2008;19:1893–1902. doi: 10.1091/mbc.E07-07-0683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Muro S, et al. Effect of PCCB gene mutations on the heteromeric and homomeric assembly of propionyl-CoA carboxylase. Mol. Genet. Metab. 2001;74:476–483. doi: 10.1006/mgme.2001.3254. [DOI] [PubMed] [Google Scholar]
- 19.Sun Y, et al. Depletion of PI3K p.85alpha induces cell cycle arrest and apoptosis in colorectal cancer cells. Oncol. Rep. 2009;22:1435–1441. [PubMed] [Google Scholar]
- 20.Wei X, et al. Exome sequencing identifies GRIN2A as frequently mutated in melanoma. Nat. Genet. 2011;43:442–446. doi: 10.1038/ng.810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine A, et al. The P53 pathway: what questions remain to be explored? Cell Death Differ. 2006;13:1027–1036. doi: 10.1038/sj.cdd.4401910. [DOI] [PubMed] [Google Scholar]
- 22.Harris S, et al. The p53 pathway: positive and negative feedback loops. Oncogene. 2005;24:2899–2908. doi: 10.1038/sj.onc.1208615. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.