

Abstract
The p53 tumor suppressor is a critical component of the cellular response to stress. As it can inhibit cell growth, p53 is mutated or functionally inactivated in most tumors. A multitude of protein–protein interactions with transcriptional cofactors are central to p53-dependent responses. In its activated state, p53 is extensively modified in both the N- and C-terminal regions of the protein. These modifications, especially phosphorylation of serine and threonine residues in the N-terminal transactivation domain, affect p53 stability and activity by modulating the affinity of protein–protein interactions. Here, we review recent findings from in vitro and in vivo studies on the role of p53 N-terminal phosphorylation. These modifications can either positively or negatively affect p53 and add a second layer of complex regulation to the divergent interactions of the p53 transactivation domain.
Introduction
The p53 tumor suppressor protein is a key component of the cellular stress response. p53 is activated by DNA damage, hypoxia, heat shock and other stresses and, depending on the cellular context and the nature of the stress, regulates the cellular responses of DNA repair, cell cycle arrest, senescence and apoptosis. The stress response enacted by p53 derives primarily from its function as a transcription factor. p53 activates or represses the transcription of a large number of genes, including PUMA, CDKN1A (p21) and MDM2 (1), in part through sequence-specific interaction with DNA. It serves as a critical monitor of genome stability; as such, it is mutated in approximately half of all human tumors (2). Mutant p53 facilitates tumor formation both through dominant-negative inhibition of wild-type p53 and gain-of-function roles (recently reviewed in ref. 3). Furthermore, in the majority of tumors retaining wild-type p53, it may be functionally impaired by misregulation, such as through overexpression of its repressor Mdm2 (4).
p53 is a multi-domain protein (Figure 1). At its N terminus is the transactivation domain (TAD), important for interaction with transcriptional coactivators and corepressors. The TAD is composed of two homologous subdomains, TAD1 (residues 1–40) and TAD2 (residues 41–61), which both contain conserved Φ-X-X-Φ-Φ sequence motifs (Φ = hydrophobic and X = any amino acid) common to many proteins regulating transcription. The TAD is followed by a proline-rich region (residues 63–97) and then by the highly conserved DNA-binding domain (residues 102–292) that exhibits sequence-specific DNA binding (5), a linker region with an embedded nuclear localization signal (residues 301–323), the tetramerization domain (residues 323–356) and the mainly disordered C-terminal regulatory domain (REG, residues 363–393). This last domain is highly basic, contains two additional nuclear localization signals and is a locus for important protein–protein interactions regulating p53 activity.
Fig. 1.

Domain structure of p53 showing known sites of posttranslational modification. Documented sites of p53 posttranslational modification are shown; known modifying (black arrow) or unmodifying (red arrow) enzyme(s) are indicated above the modification. Circle: serine (yellow) or threonine (orange) phosphorylation; hexagon: ubiquitin or ubiquitin-like modification; square: acetylation; oval: N-acetylglucosamine; octagon: poly ADP-ribosylation; pentagon: arginine (gray) or lysine mono- (lavender) or di- (violet) methylation; star: nitration.
p53 is normally a short-lived protein, maintained at low levels in unstressed mammalian cells. Following stress, p53 becomes stabilized and activated through extensive posttranslational modification (Figure 1), including: phosphorylation, acetylation, methylation, ubiquitination, neddylation, sumoylation, poly ADP-ribosylation, nitration and addition of N-acetylglucosamine (6–8). Phosphorylation is largely clustered in the TAD, linker and REG domain, whereas acetylation occurs on lysine residues in the DNA-binding domain, linker region and REG domain. Several of these lysines may also be methylated (9,10), neddylated (11) or ubiquitinated (12,13). Phosphorylation in the TAD generally results in p53 stabilization and activation: alanine substitution of seven phosphorylated serine and theonine residues in TAD1 leads to a stark loss of p53 transcriptional effects (14). Ubiquitination is associated with p53 destabilization and degradation, monomethylation is generally repressive of p53 activity and acetylation is activating, although exceptions are known (15).
As indicated by the large number of reported interacting proteins and regulating posttranslational modifications (Figure 1), the control of p53 function is extremely complex. As the roles of p53 acetylation and ubiquitination have been reviewed recently (16–19), we will focus here on TAD phosphorylation. These modifications can critically affect p53 complex formation, either positively or negatively. For example, phosphorylation of Thr55 in TAD2 mediates nuclear export of p53 by increasing its interaction with CRM1 (20). Similarly, following resolution of stress signaling, SMAR1 binds to p53 to suppress its activity during recovery. This binding is enhanced by Ser15 phosphorylation and leads to increased interaction with Mdm2, decreased DNA binding and deacetylation of the p53 REG domain (21). Phosphorylation of Ser6 and Ser9 is required for the interaction of p53 with Smad proteins, which is critical for p53 involvement in transforming growth factor β signaling (TGF-beta) (22). Among all interactors, the effects of phosphorylation on the interactions with Mdm2, the p62 subunit of general transcription factor IIH (TFIIH) and CREB-binding protein (CBP)/p300 have been studied in the greatest detail. Interestingly, these interactions have opposite effects on p53 activity and represent very different modes of binding. Here, we will examine the effects of posttranslational modification regulation of p53 observed in knock-in mouse models and explore how these effects can be understood by analysis of p53 complexes.
Studies in vivo: knock-in Mice
The development of mouse models containing knock-in mutations of TAD phosphorylation sites has helped elucidate the role of these modifications in regulating p53 activity. Homozygous mice containing alanine mutants of Ser18 (human Ser15), Ser23 (human Ser20) or both have been generated, as have mice containing mutations of Thr21 (human Thr18) and Ser23 to aspartic acid (Figure 2A) (23–29). In addition, the human p53 knock-in (HUPKI) mouse containing a mutation of Ser46 to alanine has been studied (30). In general, each of these knock-in mutations leads to a defect associated with p53-dependent signaling. The effects of single mutations are less severe than those of double mutations. The TAD1 mutations result in more severe defects than the TAD2 mutation.
Fig. 2.

Phenotypes of p53 knock-in mice. (A) Sequence of human and murine p53, showing sites of mutation in p53 knock-in mouse models. (B) Summary of phenotypic observations for various knock-in mice.
p53S18A/S18A mice have shorter life spans than wild-type mice, with a median of 81 weeks compared with 98 weeks (23). p53S23A/S23A mice also exhibit a reduced life span, with a median of 63 weeks (28). In cells from p53S18A/S18A and p53S18A,S23A/S18A,S23A knock-in mice, the stabilization of p53 after stress was not significantly affected, although p53 induction after exposure to ionizing radiation (IR) in p53S23A/S23A thymocytes was modestly reduced due to a shorter protein half-life (Figure 2B) (23,26–28). All three knock-in mutant mice displayed some defect in p53-dependent stress responses. Following exposure to IR, p53-dependent apoptosis in thymocytes from p53S18A/S18A mice was impaired, as was UV-induced G1/S arrest in mouse embryonic fibroblasts (MEFs) (26). Although G1/S arrest following exposure to IR was unaffected in p53S23A/S23A MEFs, apoptosis in thymocytes was moderately reduced (28). In contrast, cells from p53S18A,S23A/S18A,S23A mice were severely compromised in p53-dependent apoptosis (27), with overall levels of apoptosis in γ-irradiated thymocytes similar to those in p53−/− thymocytes. In comparison, knock-in mice containing mutations of residues Leu25 and Trp26 (Leu22 and Trp23 in humans) to glutamine and serine, respectively (p53L25Q,W26S/L25Q,W26S; Figure 2A), have impaired G1/S cell cycle arrest and very low rates of apoptosis after DNA damage (31–33). These mutations abrogate p53 transactivaton by 90–95% (reviewed in ref. 34); as detailed below, they abolish hydrophobic interactions central to the interaction of p53 with transcriptional coactivators.
The impairment in p53-induced stress responses correlates with specific effects on the induction of p53 target genes. For example, the increased expression of Cdkn1a, Perp, Sfn and Tnf after IR was significantly impaired in p53S18A/S18A thymocytes, whereas Mdm2, Noxa, Bax, Apaf1 and Wig1 induction was unaffected (26). Decreased histone acetylation at the Cdkn1a promoter was observed in thymocytes from p53S18A/S18A mice compared with wild-type mice, whereas histone acetylation at the Mdm2 promoter was unaffected (26). Furthermore, the REG domain of p53 from p53S18A/S18A MEFs showed less acetylation following exposure to UV than in wild-type MEFs. The induction of p53 target genes in p53S18A,S23A/S18A,S23A thymocytes was more dramatically decreased, with changes similar to those in p53−/− cells after IR (27). For comparison, p53L25Q,W26S/L25Q,W26S mice were defective in transactivation of most p53 target genes, including Cdkn1a, Noxa and Puma, although Bax expression was unaffected (31–33). The similarity in the phenotypes of posttranslational modification knock-in mice and the p53L25Q,W26S/L25Q,W26S knock-in mice demonstrates that these modifications are critical in regulating the stability and activity of p53 after stress.
Although born at the expected ratio, knock-in mice containing a single p53 allele with mutation of Thr21 and Ser23 to aspartic acid (p53T21D,S23D/−), which mimics constitutively phosphorylated p53, exhibited premature aging and a significantly reduced life span of only 6 weeks (35). Two copies of the mutated allele resulted in embryonic lethality. Cells from p53T21D,S23D/− mice showed increased p53-dependent transcription and apoptosis in the untreated state as compared with p53+/− cells, but this activity was unaffected by DNA damage and was lower than that observed in p53+/− cells after damage. Thus, although the aspartic acid mutation imperfectly mimics phosphorylation, the results in p53T21,S23D/− mice are concordant with other knock-in mice in demonstrating the importance of phosphorylation in modulating p53 function.
Recent studies have demonstrated involvement of p53 in regulating cellular metabolism. p53 can promote oxidative phosphorylation, inhibit glycolysis and regulate several mitochondrial and non-mitochondrial genes involved in metabolism (recently reviewed in ref. 36). Consistent with these activities, insulin and fasting blood triglyceride levels were increased in p53S18A/S18A mice (24). Additionally, 24-week-old mutant mice showed a modest increase in body weight as compared with wild-type mice and exhibited glucose intolerance and insulin resistance. Crossing p53S18A/S18A mice with Atm−/− mice resulted in reduced embryonic viability and decreased weight of surviving offspring as compared with Atm−/− mice (25). Finally, tests of motor coordination demonstrated a gender-specific effect of the Ser18 mutation. These studies demonstrate the importance of p53 phosphorylation not only for its DNA damage response functions but also for its role in regulating energy pathways in the cell.
Concordant with the functional defects in p53 signaling, p53S18A/S18A, p53S23A/S23A and p53S18A,S23A/S18A,S23A knock-in mice were prone to development of spontaneous tumors at 1–2 years of age (23,27,28). Unlike p53−/− mice, which develop thymic lymphomas, the knock-in mice predominantly developed B-cell lymphomas (23,26,37). Thus, the loss of p53 phosphorylation has specific functional consequences that overlap with, but do not fully recapitulate, protein loss.
Mutation of Ser46 to alanine was generated in the HUPKI mouse model, as this residue is not conserved in mice (Figure 2A). Since the signaling pathway that leads to its phosphorylation is conserved, the mouse model adds important understanding regarding this site of modification in a physiological setting. Unlike the TAD1 knock-in mice, MEFs and thymocytes from p53S46A/S46A HUPKI mice showed decreased p53 levels after exposure to UV and IR, respectively, as compared with the corresponding wild-type HUPKI mouse cells (30). This result is interesting, as Mdm2 binds exclusively within TAD1. The decreased stabilization of p53S46A/S46A in thymocytes was accompanied by a moderate decrease in transactivation of p53 target genes and a modest reduction in p53-dependent apoptosis. In contrast, most p53 targets were not affected by the mutation following exposure of MEFs to UV, although Noxa and Perp expression was significantly reduced as compared with wild-type HUPKI MEFs. In p53 knock-in mice containing mutation of Phe53 and Phe54 to glutamine and serine, respectively, expression of target genes in MEFs was generally similar to wild-type mice after doxorubicin treatment and only a small reduction in p53-dependent apoptosis was observed in the small intestine after exposure to IR (32). Thus, the moderate cell type and stress-specific effects of Ser46 mutation are comparable with loss of protein interaction within TAD2.
Structural features of p53 TAD complexes and effects of phosphorylation
The p53 TAD is phosphorylated by a number of activated kinases and is critical for the many protein–protein interactions that either modulate the stability and subcellular localization of p53 or effect its function as a transcription factor. When unbound, the TAD is unstructured (38), but it adopts a helical conformation upon complex formation. Intrinsic disorder is a common feature of ‘hub’ proteins such as p53 that interact with a large number of binding partners (39,40). Rather than forming an extended random coil, recent studies indicate that the free TAD is in a partially compact collapsed state, with transient elements of helical secondary structure (41). The partially folded nascent helical structures lower the energetic barrier to induced-fit binding but concomitantly enable promiscuous binding (42).
Range of bound structures.
To date, structures of p53 TAD in complex with six different partner proteins have been reported. In all cases, the complexes contain portions of the TAD bound to an isolated domain of the interacting protein. As depicted in Figure 3A, the structures exhibit considerable diversity: one or both subdomains can bind individually or the two can bind simultaneously. The helix lengths and locations also vary among the complexes (Figure 3A), although in all cases, a Φ-X-X-Φ-Φ motif is included. These helices are amphipathic, leading to a mixture of hydrophobic and electrostatic interactions with the binding proteins.
Fig. 3.

Effect of phosphorylation on p53 complexes. (A) Sequence of p53 TAD showing regions of helical structure in the different complexes. The helical boundaries are taken from the description in the header of the Protein Data Bank structure file. The Φ-X-X-Φ-Φ motif is shown for TAD1 and TAD2, and the dominant modifications in each domain are marked. (B) Changes in the affinity of p53 TAD complexes for domains of CBP/p300. The general increase in affinity for modified forms of p53 is shown; changes are summarized from (60,62).
The first structure reported was the complex of p53 with the N-terminal domain of human Mdm2 (Figure 4A) (43). The minimal Mdm2-binding region resides fully within TAD1, which forms a helix encompassing residues 19–25 (Figure 3A). In cells, complex formation results in ubiquitination of the p53 REG domain by the C-terminal E3 ligase domain of Mdm2 (Figure 1), leading to nuclear export and degradation of p53. Since Mdm2 is a transcriptional target of p53, these two proteins form a negative feedback loop that controls p53 levels in the absence of stress and during the return to homeostasis following stress. In the complex, three highly conserved hydrophobic residues (Phe19, Trp23 and Leu26) align along one face of the TAD1 helix and are packed deeply in a hydrophobic cleft of Mdm2 (43). This results in a relatively strong complex (K d ∼ 100 nM) stabilized primarily by the hydrophobic effect. Similar structures have been determined for the interaction of TAD1 with MdmX, a homolog of Mdm2 that also negatively regulates p53 (44,45).
Fig. 4.
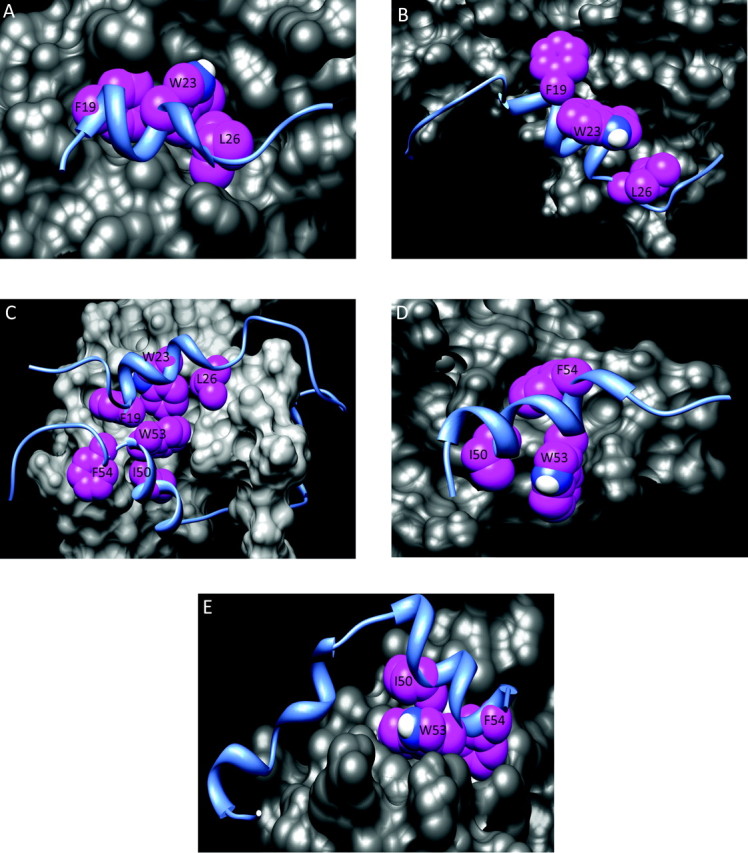
Hydrophobic interactions in p53 complexes. In each structure, p53 is shown as a blue ribbon and the binding partner is shown as gray surfaces. Hydrophobic residues from p53 are highlighted as magenta spheres. (A) Mdm2 (PDB: 1YCQ); (B) p300 Taz2 (PDB: 2K8F); (C) CBP NCBD (PDB: 2L14); (D) Tfb1 (PDB: 2GS0); (E) RPA70 (PDB: 2B3G) PDB, Protein Data Bank.
In contrast, the interaction of p53 with CBP or p300, two homologous histone acetyltransferases, facilitates its transcriptional activator activity. By binding the TAD, these proteins are recruited to p53 binding sites in chromatin near the promoters of target genes, resulting in modification of proximal histones and chromatin unwinding (46–48). Additionally, CBP and p300 acetylate lysine residues in the REG domain, which further stabilizes and activates p53 (Figure 1). Inhibition of binding by competitor proteins or downregulation of CBP/p300 by small interfering RNA represses p53-mediated transcriptional activation and reduces local histone acetylation at promoters of p53 target genes (47,49–51). Furthermore, catalytically inactive deletion mutants of p300 dominantly inhibit p53-dependent apoptosis and G1 arrest (52,53).
CBP and p300 are composed of eight distinct domains, five of which have been shown to interact with the p53 TAD in vitro: Taz1(CH1), IHD, KIX, Taz2(CH3) and the nuclear receptor coactivator-binding domain (NCBD, also referred to as IBiD) (52,54–58). The affinities of p53 for these isolated domains range from 0.02 to 10 μM (59–62). It should be noted that the binding affinities vary between studies, probably because of differences in the specific method used to measure the affinity and buffer conditions. We and others have shown that several of these domains interact with both TAD1 and TAD2 individually (61,63,64). For interaction of isolated TAD1 or TAD2 with the Taz2 domain of p300, we found that both subdomains bound with equal affinity to the same face of Taz2 (63,65). In contrast, the NCBD and KIX domains each have two distinct binding sites, allowing for the simultaneous binding of TAD1 and TAD2 in the context of the intact TAD (66).
To date, the structures of only two p53-CBP/p300 complexes have been determined: TAD1 with p300 Taz2 and a contiguous TAD1/2-containing peptide with CBP NCBD (Figure 3A). The former complex differs considerably from the TAD1-Mdm2 complex (Figure 4) (65). First, TAD1 forms a longer helix (residues 15–27 versus 19–25; Figure 3A). Second, the side chains of Phe19, Trp23 and Leu26, which are buried in the TAD1-Mdm2 complex, are significantly exposed to solvent in the TAD1-Taz2 complex (especially Trp23 and Leu26). For the TAD1-Taz2 complex, the major stabilizing hydrophobic interactions come from burial of p53 Leu22 and Leu25; individual alanine substitutions of these residues led to complete loss of binding (65). The TAD1-Taz2 complex is further stabilized by electrostatic interactions, including salt bridges involving p53 Glu11 and Glu17 and hydrogen bonds with Ser15, Thr18 and Asp21.
In the TAD-NCBD complex, the p53 TAD wraps around the CBP NCBD, allowing the helical portions of TAD1 and TAD2 to bind simultaneously at proximal sites (Figure 4C) (67). The non-helical portions of the TAD peptide are less well defined, as demonstrated by the range of conformations in the Nuclear Magnetic Resonance spectroscopy (NMR) models (67). In addition to Phe19, Trp23 and Leu26 of TAD1, Ile50, Trp53 and Phe54 of TAD2 are buried with minimal solvent exposure in a non-polar pocket of the NCBD. While these hydrophobic interactions provide the major stabilization of the complex, some salt bridges and hydrogen bonds may form at the periphery.
Binding of p53 TAD2 to the pleckstrin homology (PH) domain of the p62 subunit of TFIIH (and its yeast homolog Tfb1) facilitates p53-dependent activation of transcription (68). In the structure of TAD2 bound to Tfb1, the three hydrophobic motif residues (Ile50, Trp53 and Phe54) each bind in a non-polar pocket of the PH domain (Figure 4D). Consequently, as with both the Taz2 and NCBD complexes, binding is primarily stabilized by hydrophobic interactions and supplemented by salt bridges, hydrogen bonds and cation/aromatic interactions.
The fifth complex depicted in Figure 3A is that of TAD2 bound to the N-terminal domain of the largest subunit of the heterotrimeric replication protein A (RPA) complex, RPA70N (69). This is an example of a separate class of p53-interacting proteins that recognize the p53 TAD as a single-stranded DNA mimetic. Other members of this class include the respective C-terminal DNA-binding domains of positive cofactor 4 (PC4), BRCA2, and the human mitochondrial single-stranded DNA binding protein (HmtSSB). In complex with RPA70N or BRCA2, TAD2 binds to the oligonucleotide/oligosaccharide-binding folds of the cofactors, directly competing with single-stranded DNA (69,70), whereas both subdomains bind to HmtSSB (71). Binding of PC4 to the TAD, DNA-binding domain and REG domains of p53 promotes the transcriptional activation of p53 by increasing binding to its DNA response element (72,73). In contrast, interactions with RPA70N and BRCA2 repress the transcriptional activity of p53, possibly by preventing contact with other cofactors. As shown in Figure 4E, the hydrophobic consensus residues of TAD2 bind as a cluster in a hydrophobic pocket of RPA70N. A second region of helical structure in TAD2 formed by residues 36–44 also contributes to binding. For this class of binding proteins, electrostatic interactions play a significant role in complex stabilization. While mutation of hydrophobic residues in p53 TAD reduced its affinity for BRCA2 and PC4 by 5- to 10-fold, substitution of the acidic residues completely eliminated complex formation (70,73). This reflects a unique type of interaction with this class of cofactors in which acidic residues in TAD compete with negatively charged phosphate groups of single-stranded DNA.
Effects of phosphorylation.
The importance of TAD phosphorylation in the regulation of p53 function has led to numerous in vitro studies examining the effects of p53 phosphorylations on interactions with its binding partners. These typically utilize biophysical techniques such as fluorescence depolarization, isothermal titration calorimetry or NMR chemical shift perturbation studies. Due to the −2e formal charge under physiological conditions, phosphorylation can be thought of as adding an electrostatic-based component to the binding energy. For the interaction with Mdm2, which is primarily stabilized by the hydrophobic effect, phosphorylation prevents complex formation. In contrast, TAD phosphorylation enhances binding to CBP/p300 and p62. Thus, phosphorylation couples relief of negative regulation with enhancement of transcriptional activation. To this end, the structures of p53 complexes have facilitated understanding the detailed steric and physiochemical bases of the effects of phosphorylation.
As described above, in the absence of cellular stress, most of the serines and threonines of the p53 TAD are unphosphorylated. In particular, the absence of phosphorylation of Thr18 allows tight binding of Mdm2 to suppress p53 activity by enhancing nuclear export and proteosomal degradation. Once a cell experiences a stress, the concentration of p53 rapidly rises to stimulate the appropriate response: e.g. cell cycle arrest or apoptosis. One measure of the speed of response is that phosphorylation of Ser15, a marker of p53 activation, is detectable within 30 min after exposure to IR (74). This rapid change necessitates a switch-like inhibition of the interaction with Mdm2. As in vitro experiments have shown, the binding affinity of the TAD1-Mdm2 complex can be reduced 5- to 25-fold solely by phosphorylation of Thr18 (60–62). Mutational studies have demonstrated that the effect results from electrostatic repulsion of negatively charged pThr18 by a proximal patch of acidic and aromatic residues on the surface of the Mdm2 domain (Figure 5A) (75,76).
Fig. 5.
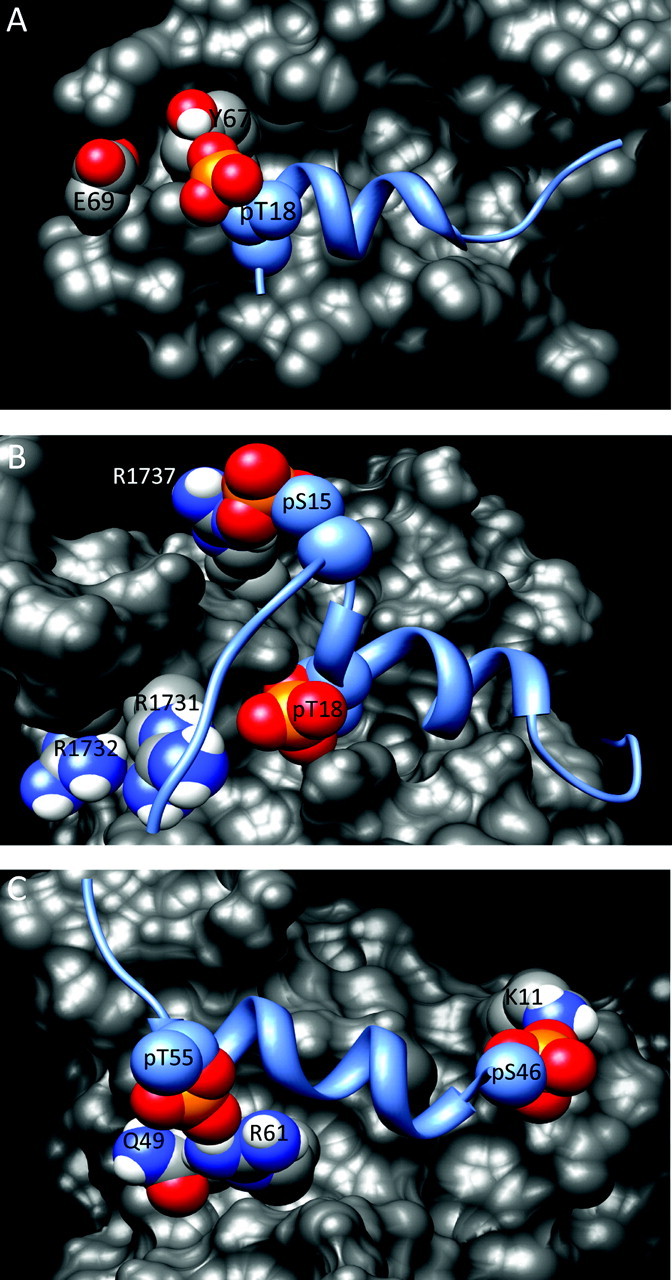
Models of phosphorylated p53 in complexes. The models shown are derived from the structures of the unmodified complexes with phosphate groups added on the specified residues. In each panel, p53 is shown as a blue ribbon and the binding partner is shown as a gray surface. The oxygen and phosphorus atoms of the phosphate group are in red and orange, respectively. Basic residues of each binding partner that could interact with phosphorylated sites on p53 are indicated. In (A), Glu69 of Mdm2, one of the residues mutated in the study by Brown et al. (69), is not indicated as it is obscured by Asp68 in this view.
In contrast, the interactions of p53 with its positive cofactors generally start out weak and increase in affinity with increasing phosphorylation. This allows for a nuanced response in which the interactions of p53 with different subgroups of cofactors change over time. This phenomenon has been best demonstrated for the interaction with CBP/p300 (59–64). As shown in Figure 3B, the strength of the effect depends on the location of the phosphorylation within the TAD sequence and varies for the different domains of CBP/p300. Single phosphorylation of Ser15, Thr18, Ser20, Ser33, Ser37 or Ser46 generally increases the binding affinities to the Taz1, Taz2 and KIX domains by 2- to 7-fold, whereas the effects on the NCBD are smaller and phosphorylation of Thr18 inversely leads to an ∼2-fold decrease in binding affinity (60,62–64). With the exception of the KIX domain, phosphorylation of Thr55 does not significantly affect binding to any of the CBP/p300 domains, consistent with the observation that this modification primarily occurs in unstressed cells (20,77). Following triphosphorylation of p53 at specific sites in TAD1, the binding of the TAD to CBP/p300 domains is comparable in strength to that of unmodified TAD1 with Mdm2 (59–64). For example, triphosphorylation of Ser15, Thr18 and Ser20 results in a 20-fold increase in binding to the Taz2 and KIX domains, and triphosphorylation of Ser33, Ser37 and Ser46 results in a nearly 30-fold increase in binding to the Taz1 and Taz2 domains (63,67). This enhanced binding to CBP/p300 enables p53 to compete successfully with other transcription factors for the limited number of these cofactors in the cell.
Figure 5B depicts the hypothetical location of the pSer15 and pThr18 phosphate groups in the TAD1-Taz2 complex, presuming the conformation of the complex remains the same as with unmodified TAD1 (65). This model suggests that salt bridges could form between pSer15 and Arg1737 of Taz2 and between pThr18 and the cluster of Arg1731 and Arg1732. Although the latter interaction was confirmed with alanine mutants, the binding affinity of TAD1-pSer15 was unaffected by mutation of Arg1737 (63). As Ser15 is in a more flexible region of the peptide than Thr18, the phosphorylated residue could shift to bind a different amino acid of Taz2 or, alternately, the entire TAD1 could bind in a different orientation. Three observations support these possibilities (63,65): (i) the affinity of diphosphorylated TAD1 is intermediate between the affinities of the corresponding monophosphorylated forms; (ii) changes in amide 1H and 15N chemical shifts measured by NMR indicate conformational changes in Taz2 when Thr18 is phosphorylated; (iii) differences in the temperature dependence of the heat capacity suggest that stabilization of the mono- and diphosphorylated forms arises primarily from hydrophobic and electrostatic interactions, respectively. Determination of the NMR solution structures of the complexes with phosphorylated p53 is hampered by the difficulty and expense of producing isotopically labeled phosphopeptides.
In contrast to the TAD1-Taz2 complex, analysis of the structure of unmodified TAD bound to the NCBD (Figure 4C) failed to suggest proximal positively charged residues that could form stabilizing salt bridges, an observation consistent with the relatively weak effect of phosphorylation on binding (Figure 3B). Additionally, negatively charged residues on the surface of the NCBD that could explain the reduced affinity upon Thr18 phosphorylation were not observed. Therefore, the moderate changes in affinity that result from p53 phosphorylation likely reflect a structural rearrangement of the TAD-NCBD complex.
Modification of TAD2 enhances the binding of p53 to p62, with monophosphorylation of Ser46 or Thr55 each increasing the affinity of TAD2 for p62 6-fold and the diphosphorylated form binding 32-fold tighter (78). As in the TAD1-Taz2 complex, the structure of the unmodified complex suggests positively charged residues that could form stabilizing interactions with the phosphorylated residues. In the Tfb1 complex, pSer46 of TAD2 is adjacent to Lys11 of Tfb1 and pThr55 is proximal to Arg61 (Figure 5C). Interaction of the former was confirmed by mutagenesis of the corresponding residue in p62 (78). In contrast, the residue in p62 that corresponds to Arg61 in Tfb1 is Gln66, which lacks a formal charge. Instead, pThr55 may interact with Lys54 of p62, which corresponds to Gln49 in Tfb1 and is adjacent to Arg61 (Figure 5C). The physiological implication of the increased binding of pThr55 is unclear, as this modification is associated with repression of p53.
Less is known about the effects of p53 phosphorylation on interactions with proteins that bind p53 as a single-stranded DNA mimetic. For the interaction of the TAD with PC4, hepta-phosphorylated p53 increased the affinity to the net positively charged PC4, consistent with the ability of this cofactor to activate p53 (73). In contrast, UV exposure abrogates the interaction of p53 with RPA, suggesting that phosphorylation may reduce the affinity of the complex (79). Based on the negative regulatory role of BRCA2 binding to p53, phosphorylation may also repress this interaction. The structure of the complex of p53 TAD with RPA70N (Figure 4E) does not suggest any formally charged residues that could interact with phosphorylated residues in TAD2.
The effects (loss or gain) on p53 transcriptional activity observed in the knock-in mice are consistent with the role of these sites on binding of cofactors. Phosphorylation generally increases the affinity of p53 for its positive regulators; serine to alanine mutations that block phosphorylation decreased p53 transcriptional activity, probably due in part to the decreased affinity of the mutant p53 for critical cofactors. Additionally, the increased defects in p53-dependent stress responses in p53S18A,S23A/S18A,S23A mice as compared with p53S18A/S18A and p53S23A/S23A knock-in mice are consistent with the additive effects of p53 TAD1 phosphorylation on binding.
Concluding remarks
One of the interesting questions raised by these studies is why p53 contains two similar acidic transactivation subdomains. Although both contain Φ-X-X-Φ-Φ motifs, the surrounding residues differ. TAD1 contains seven modifiable serine or threonine residues, whereas TAD2 contains only two. TAD1 contains seven acidic residues (18% of the total domain) with an estimated pI of 3.71 in the unmodified state; TAD2 contains nine acidic residues (35% of the total domain) with an estimated pI of 3.10. Thus, in the unstressed state, TAD2 presents a greater density of charged residues than TAD1; following p53-activating stress, however, the two subdomains become approximately equally charged. The nine phosphorylation sites within the full TAD are modified by enzymes involved in different signaling pathways. Moreover, the same site can be modified by mutliple kinases. For example, Ser15 can be phosphorylated by kinases that respond to DNA damage, nutrient deprivation, hormone stimulation and hypoxia. Specific stresses result in distinct patterns of phosphorylation over time (80). Differences in the charge of the subdomain and pattern of modification give rise to specificity in the protein–protein interactions in which the subdomains participate. Although many of the domains of CBP/p300 interact with both TAD1 and TAD2, Mdm2 only binds to TAD1, whereas RPA70 and p62 only bind to TAD2 (69,78). Thus, the two subdomains provide extra flexibility for p53 to respond to different stress signals and to mediate the multiple responses required by the specific stress in a cell- or tissue-type-dependent manner. This is exemplified by ΔNp53 (an isoform of p53 in which translation is initiated at Met40 such that it contains only TAD2), which has specific functions in stem cells, embryonic development and following endoplasmic reticulum stress (81–83).
The different functional effects of Ser15 and Ser46 phosphorylation also demonstrate the differences between the TAD subdomains. When aligning TAD1 and TAD2 by their Φ-X-X-Φ-Φ motifs, Ser15 correlates to Ser46; phosphorylation of these two residues might be expected to exhibit similar regulation and function. With the sole exception of the adenosine monophosphate-activated protein kinase (AMPK), which can directly phosphorylate both, the two sites are specifically modified by several different kinases, none of which has been shown to directly phosphorylate the other site. In addition, distinct phosphatases remove the modifications. One recent study demonstrated that the amount of p53 phosphorylated at Ser15 bound to DNA was similar following treatment of U2OS osteosarcoma cells with actinomycin D to induce cell cycle arrest or etoposide to induce apoptosis; in contrast, DNA-bound p53 phosphorylated on Ser46 increased 5-fold after etoposide treatment as compared with actinomycin D treatment (84). Thus, the regulation and functional outcome of phosphorylation of these two sites are quite different, indicating that they play unique roles.
Another aspect of the presence of two TAD subdomains of p53 is that it allows the formation of ternary complexes, such as those observed between p53, CBP/p300 and Mdm2 (61,63). Although these form on a single molecule of p53 using isolated domains in vitro, the sizes of the intact proteins would likely sterically preclude the same from occurring in vivo. However, as p53 forms a tetramer in the nucleus, it is possible that Mdm2 could bind one monomer of p53 while CBP/p300 binds either subdomain on a separate monomer. Such a ternary complex may represent an intermediate state early after stress. In experiments with nutlin-3a, an inhibitor of the TAD1-Mdm2 interaction, CBP/p300-dependent acetylation of the REG domain was observed in the absence of TAD1 phosphorylation (85,86). These results suggest that upon removal of the inhibitory effect of Mdm2, p53 is able to accumulate to a level comparable with that following stress, which allows critical protein–protein interactions to form in the absence of phosphorylation.
The stability and activity of p53 are very tightly controlled by phosphorylation. Within the TAD, phosphorylation can have either a positive or negative effect on p53 stability, activity or both. The same trends are observed in the REG domain, in which phosphorylation, methylation, acetylation and ubiquitination are all found. As the same lysine residues can be methylated, acetylated and ubiquitinated, the complexity of p53 regulation quickly rises. In addition, N-terminal modifications can affect C-terminal ones. For example, phosphorylation of the TAD promotes acetylation by CBP/p300 and negatively regulates ubiquitination and sumoylation in the REG domain (87). As illustrated here, p53 posttranslational modifications can modulate protein–protein interactions; additionally, they can affect p53 tetramerization, which has direct implications for both stability and activity. Intriguingly, p53 polymorphisms have recently been shown to also affect modification, with p53 Arg72 showing enhanced phosphorylation of Ser6 and Ser20 as compared with p53 Pro72 (88).
Clearly, the regulation of p53 protein level is critical. It must be stabilized in response to stress to protect the cell, as evidenced by the hightumor rate among p53−/− mice. However, following removal of the stress and clearance of the resulting damage, p53 levels must return to steady-state levels: Mdm2−/− mice are embryonically lethal and overactive p53 leads to an accelerated aging phenotype. p53 messenger RNA levels are generally static; thus, the primary regulation of p53 stability and activity is through the modulation of protein interactions by posttranslational modification.
Although much has been done to identify the sites and effects of p53 posttranslational modifications, there are gaps in our understanding. Most of the research on the modulation of protein–protein interactions by phosphorylation has necessarily been done in vitro; in the future, experiments should be performed to analyze these complexes in cells as well. In addition, more effort should be devoted to unraveling the interplay between different sites of modification, such as how modification on one site affects the modification of a second site. Signaling cascades among N-terminal phosphorylation sites have been studied following different stresses (80), but the interplay of modifications in the REG domain of p53 and how they are affected by N-terminal phosphorylation have received less focus. Finally, it will be critical to explore the effect of posttranslational modification on new functions of p53, including metabolism. Combined these studies will lead to a new era in the understanding of the complex layers of p53 regulation.
Funding
All authors are supported by the Intramural Research Program of the National Cancer Institute, National Institutes of Health, USA.
Acknowledgments
Conflict of Interest Statement: None declared.
Glossary
Abbreviations
- CBP
CREB-binding protein
- HUPKI
human p53 knock-in
- IR
ionizing radiation
- MEF
mouse embryonic fibroblast
- NCBD
nuclear receptor coactivator-binding domain
- NMR
nuclear magnetic resonance spectroscopy
- REG
regulatory domain
- RPA
replication protein A
- TAD
transactivation domain
References
- 1.Beckerman R, et al. Transcriptional regulation by p53. Cold Spring Harb. Perspect. Biol. 2010;2:a000935. doi: 10.1101/cshperspect.a000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Petitjean A, et al. Impact of mutant p53 functional properties on TP53 mutation patterns and tumor phenotype: lessons from recent developments in the IARC TP53 database. Hum. Mutat. 2007;28:622–629. doi: 10.1002/humu.20495. [DOI] [PubMed] [Google Scholar]
- 3.Goh AM, et al. The role of mutant p53 in human cancer. J. Pathol. 2011;223:116–126. doi: 10.1002/path.2784. [DOI] [PubMed] [Google Scholar]
- 4.Woods DB, et al. Regulation of p53 function. Exp. Cell Res. 2001;264:56–66. doi: 10.1006/excr.2000.5141. [DOI] [PubMed] [Google Scholar]
- 5.El-Deiry WS, et al. Definition of a consensus binding site for p53. Nat. Genet. 1992;1:45–49. doi: 10.1038/ng0492-45. [DOI] [PubMed] [Google Scholar]
- 6.Anderson CW, et al. Signaling to the p53 tumor suppressor through pathways activated by genotoxic and non-genotoxic stresses. In: Bradshaw RA, Dennis EA, editors. Handbook of Cell Signaling. Amsterdam, The Netherlands: Elsevier; 2009. pp. 2185–2203. [Google Scholar]
- 7.Meek DW, et al. Posttranslational modification of p53: cooperative integrators of function. Cold Spring Harb. Perspect. Biol. 2009;1:a000950. doi: 10.1101/cshperspect.a000950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yakovlev VA, et al. Nitration of the tumor suppressor protein p53 at tyrosine 327 promotes p53 oligomerization and activation. Biochemistry. 2010;49:5331–5339. doi: 10.1021/bi100564w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chuikov S, et al. Regulation of p53 activity through lysine methylation. Nature. 2004;432:353–360. doi: 10.1038/nature03117. [DOI] [PubMed] [Google Scholar]
- 10.Huang J, et al. Repression of p53 activity by Smyd2-mediated methylation. Nature. 2006;444:629–632. doi: 10.1038/nature05287. [DOI] [PubMed] [Google Scholar]
- 11.Xirodimas DP, et al. Mdm2-mediated NEDD8 conjugation of p53 inhibits its transcriptional activity. Cell. 2004;118:83–97. doi: 10.1016/j.cell.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 12.Chan WM, et al. Ubiquitination of p53 at multiple sites in the DNA-binding domain. Mol. Cancer Res. 2006;4:15–25. doi: 10.1158/1541-7786.MCR-05-0097. [DOI] [PubMed] [Google Scholar]
- 13.Rodriguez MS, et al. Multiple C-terminal lysine residues target p53 for ubiquitin-proteasome-mediated degradation. Mol. Cell. Biol. 2000;20:8458–8467. doi: 10.1128/mcb.20.22.8458-8467.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ohki R, et al. Dissecting functional roles of p53 N-terminal transactivation domains by microarray expression analysis. Cancer Sci. 2007;98:189–200. doi: 10.1111/j.1349-7006.2006.00375.x. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 15.Berger SL. Keeping p53 in check: a high-stakes balancing act. Cell. 2010;142:17–19. doi: 10.1016/j.cell.2010.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brooks CL, et al. The impact of acetylation and deacetylation on the p53 pathway. Protein Cell. 2011;2:456–462. doi: 10.1007/s13238-011-1063-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dai C, et al. p53 post-translational modification: deregulated in tumorigenesis. Trends Mol. Med. 2010;16:528–536. doi: 10.1016/j.molmed.2010.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brooks CL, et al. p53 regulation by ubiquitin. FEBS Lett. 2011;585:2803–2809. doi: 10.1016/j.febslet.2011.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hock A, et al. Regulation of the p53 pathway by ubiquitin and related proteins. Int. J. Biochem. Cell Biol. 2010;42:1618–1621. doi: 10.1016/j.biocel.2010.06.011. [DOI] [PubMed] [Google Scholar]
- 20.Cai X, et al. Inhibition of Thr-55 phosphorylation restores p53 nuclear localization and sensitizes cancer cells to DNA damage. Proc. Natl Acad. Sci. USA. 2008;105:16958–16963. doi: 10.1073/pnas.0804608105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pavithra L, et al. SMAR1 forms a ternary complex with p53-MDM2 and negatively regulates p53-mediated transcription. J. Mol. Biol. 2009;388:691–702. doi: 10.1016/j.jmb.2009.03.033. [DOI] [PubMed] [Google Scholar]
- 22.Cordenonsi M, et al. Integration of TGF-beta and Ras/MAPK signaling through p53 phosphorylation. Science. 2007;315:840–843. doi: 10.1126/science.1135961. [DOI] [PubMed] [Google Scholar]
- 23.Armata HL, et al. The ataxia telangiectasia-mutated target site Ser18 is required for p53-mediated tumor suppression. Cancer Res. 2007;67:11696–11703. doi: 10.1158/0008-5472.CAN-07-1610. [DOI] [PubMed] [Google Scholar]
- 24.Armata HL, et al. Requirement of the ATM/p53 tumor suppressor pathway for glucose homeostasis. Mol. Cell. Biol. 2010;30:5787–5794. doi: 10.1128/MCB.00347-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Armata HL, et al. Loss of p53 Ser18 and Atm results in embryonic lethality without cooperation in tumorigenesis. PLoS One. 2011;6:e24813. doi: 10.1371/journal.pone.0024813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chao C, et al. Cell type- and promoter-specific roles of Ser18 phosphorylation in regulating p53 responses. J. Biol. Chem. 2003;278:41028–41033. doi: 10.1074/jbc.M306938200. [DOI] [PubMed] [Google Scholar]
- 27.Chao C, et al. Ser18 and 23 phosphorylation is required for p53-dependent apoptosis and tumor suppression. EMBO J. 2006;25:2615–2622. doi: 10.1038/sj.emboj.7601167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.MacPherson D, et al. Defective apoptosis and B-cell lymphomas in mice with p53 point mutation at Ser 23. EMBO J. 2004;23:3689–3699. doi: 10.1038/sj.emboj.7600363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sluss HK, et al. Phosphorylation of serine 18 regulates distinct p53 functions in mice. Mol. Cell. Biol. 2004;24:976–984. doi: 10.1128/MCB.24.3.976-984.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Feng L, et al. Ser46 phosphorylation regulates p53-dependent apoptosis and replicative senescence. Cell Cycle. 2006;5:2812–2819. doi: 10.4161/cc.5.23.3526. [DOI] [PubMed] [Google Scholar]
- 31.Johnson TM, et al. The p53QS transactivation-deficient mutant shows stress-specific apoptotic activity and induces embryonic lethality. Nat. Genet. 2005;37:145–152. doi: 10.1038/ng1498. [DOI] [PubMed] [Google Scholar]
- 32.Brady CA, et al. Distinct p53 transcriptional programs dictate acute DNA-damage responses and tumor suppression. Cell. 2011;145:571–583. doi: 10.1016/j.cell.2011.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gaidarenko O, et al. Transcription activity is required for p53-dependent tumor suppression. Oncogene. 2009;28:4397–4401. doi: 10.1038/onc.2009.290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson TM, et al. p53QS: an old mutant teaches us new tricks. Cell Cycle. 2005;4:731–734. doi: 10.4161/cc.4.6.1696. [DOI] [PubMed] [Google Scholar]
- 35.Liu D, et al. Puma is required for p53-induced depletion of adult stem cells. Nat. Cell Biol. 2010;12:993–998. doi: 10.1038/ncb2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang PY, et al. p53: exercise capacity and metabolism. Curr. Opin. Oncol. 2012;24:76–82. doi: 10.1097/CCO.0b013e32834de1d8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Donehower LA, et al. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature. 1992;356:215–221. doi: 10.1038/356215a0. [DOI] [PubMed] [Google Scholar]
- 38.Vise P, et al. Identifying long-range structure in the intrinsically unstructured transactivation domain of p53. Proteins. 2007;67:526–530. doi: 10.1002/prot.21364. [DOI] [PubMed] [Google Scholar]
- 39.Gsponer J, et al. The rules of disorder or why disorder rules. Prog. Biophys. Mol. Biol. 2009;99:94–103. doi: 10.1016/j.pbiomolbio.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 40.Oldfield CJ, et al. Flexible nets: disorder and induced fit in the associations of p53 and 14-3-3 with their partners. BMC Genomics. 2008;9(suppl. 1):S1. doi: 10.1186/1471-2164-9-S1-S1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Daughdrill GW, et al. Understanding the structural ensembles of a highly extended disordered protein. Mol. Biosyst. 2012;8:308–319. doi: 10.1039/c1mb05243h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hazy E, et al. Limitations of induced folding in molecular recognition by intrinsically disordered proteins. Chemphyschem. 2009;10:1415–1419. doi: 10.1002/cphc.200900205. [DOI] [PubMed] [Google Scholar]
- 43.Kussie PH, et al. Structure of the MDM2 oncoprotein bound to the p53 tumor suppressor transactivation domain. Science. 1996;274:948–953. doi: 10.1126/science.274.5289.948. [DOI] [PubMed] [Google Scholar]
- 44.Popowicz GM, et al. Structure of the human Mdmx protein bound to the p53 tumor suppressor transactivation domain. Cell Cycle. 2008;7:2441–2443. doi: 10.4161/cc.6365. [DOI] [PubMed] [Google Scholar]
- 45.Popowicz GM, et al. Molecular basis for the inhibition of p53 by Mdmx. Cell Cycle. 2007;6:2386–2392. doi: 10.4161/cc.6.19.4740. [DOI] [PubMed] [Google Scholar]
- 46.Barlev NA, et al. Acetylation of p53 activates transcription through recruitment of coactivators/histone acetyltransferases. Mol. Cell. 2001;8:1243–1254. doi: 10.1016/s1097-2765(01)00414-2. [DOI] [PubMed] [Google Scholar]
- 47.Liu G, et al. The activation domains, the proline-rich domain, and the C-terminal basic domain in p53 are necessary for acetylation of histones on the proximal p21 promoter and interaction with p300/CREB-binding protein. J. Biol. Chem. 2003;278:17557–17565. doi: 10.1074/jbc.M210696200. [DOI] [PubMed] [Google Scholar]
- 48.Espinosa JM, et al. Transcriptional regulation by p53 through intrinsic DNA/chromatin binding and site-directed cofactor recruitment. Mol. Cell. 2001;8:57–69. doi: 10.1016/s1097-2765(01)00283-0. [DOI] [PubMed] [Google Scholar]
- 49.Hsu CH, et al. HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J. 2004;23:2269–2280. doi: 10.1038/sj.emboj.7600239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Luo J, et al. Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell. 2001;107:137–148. doi: 10.1016/s0092-8674(01)00524-4. [DOI] [PubMed] [Google Scholar]
- 51.Vaziri H, et al. hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell. 2001;107:149–159. doi: 10.1016/s0092-8674(01)00527-x. [DOI] [PubMed] [Google Scholar]
- 52.Avantaggiati ML, et al. Recruitment of p300/CBP in p53-dependent signal pathways. Cell. 1997;89:1175–1184. doi: 10.1016/s0092-8674(00)80304-9. [DOI] [PubMed] [Google Scholar]
- 53.Scolnick DM, et al. CREB-binding protein and p300/CBP-associated factor are transcriptional coactivators of the p53 tumor suppressor protein. Cancer Res. 1997;57:3693–3696. [PubMed] [Google Scholar]
- 54.Dornan D, et al. DNA-dependent acetylation of p53 by the transcription coactivator p300. J. Biol. Chem. 2003;278:13431–13441. doi: 10.1074/jbc.M211460200. [DOI] [PubMed] [Google Scholar]
- 55.Grossman SR, et al. p300/MDM2 complexes participate in MDM2-mediated p53 degradation. Mol. Cell. 1998;2:405–415. doi: 10.1016/s1097-2765(00)80140-9. [DOI] [PubMed] [Google Scholar]
- 56.Gu W, et al. Synergistic activation of transcription by CBP and p53. Nature. 1997;387:819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 57.Livengood JA, et al. p53 Transcriptional activity is mediated through the SRC1-interacting domain of CBP/p300. J. Biol. Chem. 2002;277:9054–9061. doi: 10.1074/jbc.M108870200. [DOI] [PubMed] [Google Scholar]
- 58.Van Orden K, et al. Binding of p53 to the KIX domain of CREB binding protein. A potential link to human T-cell leukemia virus, type I-associated leukemogenesis. J. Biol. Chem. 1999;274:26321–26328. doi: 10.1074/jbc.274.37.26321. [DOI] [PubMed] [Google Scholar]
- 59.Teufel DP, et al. Four domains of p300 each bind tightly to a sequence spanning both transactivation subdomains of p53. Proc. Natl Acad. Sci. USA. 2007;104:7009–7014. doi: 10.1073/pnas.0702010104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lee CW, et al. Graded enhancement of p53 binding to CREB-binding protein (CBP) by multisite phosphorylation. Proc. Natl Acad. Sci. USA. 2010;107:19290–19295. doi: 10.1073/pnas.1013078107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ferreon JC, et al. Cooperative regulation of p53 by modulation of ternary complex formation with CBP/p300 and HDM2. Proc. Natl Acad. Sci. USA. 2009;106:6591–6596. doi: 10.1073/pnas.0811023106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Teufel DP, et al. Regulation by phosphorylation of the relative affinities of the N-terminal transactivation domains of p53 for p300 domains and Mdm2. Oncogene. 2009;28:2112–2118. doi: 10.1038/onc.2009.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Jenkins LM, et al. Two distinct motifs within the p53 transactivation domain bind to the Taz2 domain of p300 and are differentially affected by phosphorylation. Biochemistry. 2009;48:1244–1255. doi: 10.1021/bi801716h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Polley S, et al. Differential recognition of phosphorylated transactivation domains of p53 by different p300 domains. J. Mol. Biol. 2008;376:8–12. doi: 10.1016/j.jmb.2007.11.082. [DOI] [PubMed] [Google Scholar]
- 65.Feng H, et al. Structural basis for p300 Taz2-p53 TAD1 binding and modulation by phosphorylation. Structure. 2009;17:202–210. doi: 10.1016/j.str.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Lee CW, et al. Mapping the interactions of the p53 transactivation domain with the KIX domain of CBP. Biochemistry. 2009;48:2115–2124. doi: 10.1021/bi802055v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee CW, et al. Structure of the p53 transactivation domain in complex with the nuclear receptor coactivator binding domain of CREB binding protein. Biochemistry. 2010;49:9964–9971. doi: 10.1021/bi1012996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Blau J, et al. Three functional classes of transcriptional activation domain. Mol. Cell. Biol. 1996;16:2044–2055. doi: 10.1128/mcb.16.5.2044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bochkareva E, et al. Single-stranded DNA mimicry in the p53 transactivation domain interaction with replication protein A. Proc. Natl Acad. Sci. USA. 2005;102:15412–15417. doi: 10.1073/pnas.0504614102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rajagopalan S, et al. Mapping the physical and functional interactions between the tumor suppressors p53 and BRCA2. Proc. Natl Acad. Sci. USA. 2010;107:8587–8592. doi: 10.1073/pnas.1003689107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wong TS, et al. Physical and functional interactions between human mitochondrial single-stranded DNA-binding protein and tumour suppressor p53. Nucleic Acids Res. 2009;37:568–581. doi: 10.1093/nar/gkn974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Debnath S, et al. Peptide-protein interactions suggest that acetylation of lysines 381 and 382 of p53 is important for positive coactivator 4-p53 interaction. J. Biol. Chem. 2011;286:25076–25087. doi: 10.1074/jbc.M110.205328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rajagopalan S, et al. Interaction between the transactivation domain of p53 and PC4 exemplifies acidic activation domains as single-stranded DNA mimics. J. Biol. Chem. 2009;284:21728–21737. doi: 10.1074/jbc.M109.006429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Al Rashid ST, et al. Protein-protein interactions occur between p53 phosphoforms and ATM and 53BP1 at sites of exogenous DNA damage. Radiat. Res. 2011;175:588–598. doi: 10.1667/RR2084.1. [DOI] [PubMed] [Google Scholar]
- 75.Brown CJ, et al. The electrostatic surface of MDM2 modulates the specificity of its interaction with phosphorylated and unphosphorylated p53 peptides. Cell Cycle. 2008;7:608–610. doi: 10.4161/cc.7.5.5488. [DOI] [PubMed] [Google Scholar]
- 76.Lee HJ, et al. Modulation of the p53-MDM2 interaction by phosphorylation of Thr18: a computational study. Cell Cycle. 2007;6:2604–2611. doi: 10.4161/cc.6.21.4923. [DOI] [PubMed] [Google Scholar]
- 77.Shouse GP, et al. Serine 15 phosphorylation of p53 directs its interaction with B56gamma and the tumor suppressor activity of B56gamma-specific protein phosphatase 2A. Mol. Cell. Biol. 2008;28:448–456. doi: 10.1128/MCB.00983-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Di Lello P, et al. Structure of the Tfb1/p53 complex: insights into the interaction between the p62/Tfb1 subunit of TFIIH and the activation domain of p53. Mol. Cell. 2006;22:731–740. doi: 10.1016/j.molcel.2006.05.007. [DOI] [PubMed] [Google Scholar]
- 79.Abramova NA, et al. Interaction between replication protein A and p53 is disrupted after UV damage in a DNA repair-dependent manner. Proc. Natl Acad. Sci. USA. 1997;94:7186–7191. doi: 10.1073/pnas.94.14.7186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Saito S, et al. Phosphorylation site interdependence of human p53 post-translational modifications in response to stress. J. Biol. Chem. 2003;278:37536–37544. doi: 10.1074/jbc.M305135200. [DOI] [PubMed] [Google Scholar]
- 81.Bourougaa K, et al. Endoplasmic reticulum stress induces G2 cell-cycle arrest via mRNA translation of the p53 isoform p53/47. Mol. Cell. 2010;38:78–88. doi: 10.1016/j.molcel.2010.01.041. [DOI] [PubMed] [Google Scholar]
- 82.Melis JP, et al. Genotoxic exposure: novel cause of selection for a functional DeltaN-p53 isoform. Oncogene. 2011;30:1764–1772. doi: 10.1038/onc.2010.552. [DOI] [PubMed] [Google Scholar]
- 83.Ungewitter E, et al. Delta40p53 controls the switch from pluripotency to differentiation by regulating IGF signaling in ESCs. Genes Dev. 2010;24:2408–2419. doi: 10.1101/gad.1987810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Smeenk L, et al. Role of p53 serine 46 in p53 target gene regulation. PLoS One. 2011;6:e17574. doi: 10.1371/journal.pone.0017574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kumamoto K, et al. Nutlin-3a activates p53 to both down-regulate inhibitor of growth 2 and up-regulate mir-34a, mir-34b, and mir-34c expression, and induce senescence. Cancer Res. 2008;68:3193–3203. doi: 10.1158/0008-5472.CAN-07-2780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Thompson T, et al. Phosphorylation of p53 on key serines is dispensable for transcriptional activation and apoptosis. J. Biol. Chem. 2004;279:53015–53022. doi: 10.1074/jbc.M410233200. [DOI] [PubMed] [Google Scholar]
- 87.Lin JY, et al. Association of Ubc9, an E2 ligase for SUMO conjugation, with p53 is regulated by phosphorylation of p53. FEBS Lett. 2004;573:15–18. doi: 10.1016/j.febslet.2004.07.059. [DOI] [PubMed] [Google Scholar]
- 88.Ozeki C, et al. Cancer susceptibility polymorphism of p53 at codon 72 affects phosphorylation and degradation of p53 protein. J. Biol. Chem. 2011;286:18251–18260. doi: 10.1074/jbc.M110.208587. [DOI] [PMC free article] [PubMed] [Google Scholar]