

Abstract
In recent years, the rapid analysis of single cells has commonly been performed using flow cytometry and fluorescently-labeled antibodies. However, the issue of spectral overlap of fluorophore emissions has limited the number of simultaneous probes. In contrast, the new CyTOF mass cytometer by DVS Sciences couples a liquid single-cell introduction system to an ICP-MS.1 Rather than fluorophores, chelating polymers containing highly-enriched metal isotopes are coupled to antibodies or other specific probes.2-5 Because of the metal purity and mass resolution of the mass cytometer, there is no "spectral overlap" from neighboring isotopes, and therefore no need for compensation matrices. Additionally, due to the use of lanthanide metals, there is no biological background and therefore no equivalent of autofluorescence. With a mass window spanning atomic mass 103-203, theoretically up to 100 labels could be distinguished simultaneously. Currently, more than 35 channels are available using the chelating reagents available from DVS Sciences, allowing unprecedented dissection of the immunological profile of samples.6-7
Disadvantages to mass cytometry include the strict requirement for a separate metal isotope per probe (no equivalent of forward or side scatter), and the fact that it is a destructive technique (no possibility of sorting recovery). The current configuration of the mass cytometer also has a cell transmission rate of only ~25%, thus requiring a higher input number of cells.
Optimal daily performance of the mass cytometer requires several steps. The basic goal of the optimization is to maximize the measured signal intensity of the desired metal isotopes (M) while minimizing the formation of oxides (M+16) that will decrease the M signal intensity and interfere with any desired signal at M+16. The first step is to warm up the machine so a hot, stable ICP plasma has been established. Second, the settings for current and make-up gas flow rate must be optimized on a daily basis. During sample collection, the maximum cell event rate is limited by detector efficiency and processing speed to 1000 cells/sec. However, depending on the sample quality, a slower cell event rate (300-500 cells/sec) is usually desirable to allow better resolution between cells events and thus maximize intact singlets over doublets and debris. Finally, adequate cleaning of the machine at the end of the day helps minimize background signal due to free metal.
Keywords: Bioengineering, Issue 69, CyTOF, mass cytometry, PBMCs, ICP-MS, multiparametric
Protocol
All cell samples for the CyTOF must be fixed and permeabilized. This enables greater entry of the iridium-containing DNA intercalator, and also prevents cell lysis during the MilliQ water wash and resuspension steps immediately before injecting into the mass cytometer.
1. Start-up of the Mass Cytometer
Open the CyTOF software program. Select Instrument Setup. On the Card Cage tab, go to the lower right corner and click Heater "ON" button. Turn on the heater at least 15 min prior to desired time to allow sufficient time for the spray chamber mantle to reach 200 °C.
Replace the white Norm-Ject 3 ml syringe in the syringe pump with a fresh syringe filled with MilliQ water.
Cleaning nebulizer. Backflush the nebulizer by using the syringe and tubing to pull 5% citranox 2-3 times through both the smaller liquid introduction port and the larger gas introduction port. Repeat backflushing with MilliQ water to remove residual citranox.
Check to confirm that the lights on the front panel of the CyTOF are lit as in Figure 1.
 Figure 1. CyTOF panel lights prior to startup.
Figure 1. CyTOF panel lights prior to startup.
Open argon gas tank valve. This should cause the "ARGON" light on the front to turn on.
Attach the nebulizer to make-up gas inlet. Unscrew the make-up gas tubing connector and remove the black rubber o-ring and the white conical plastic piece. Note the wider "knuckle" on the gas introduction port stem of the nebulizer. Place the connector on the gas introduction port. Place the black o-ring over the end of the gas port, and force the o-ring past the wider part of the port stem. Place the white conical plastic piece on top with the narrow end pointed out, and screw onto the make-up gas tubing.
Attach the nebulizer to the liquid introduction tubing. Carefully insert the small tubing into the opening at the end of the nebulizer. There will be two stops, similar in feel to the stops on a micropipettor. First, press until you meet light resistance; the end of the small tubing should be at the end of the straight section of glass. Second, press harder to force the end of the straight section of glass further into the fitting, and tighten the screw fitting to seal. Insert the end of the nebulizer into the white opening in the heating mantle.
On the RFG Controller tab of the Instrument Setup window, press "Start Plasma". Since the nebulizer is already inserted, press "OK". The software will turn on the chiller, start the flow of gas, and ignite a plasma. The detector voltage will ramp up. There will be a pop-up window saying "Plasma start-up sequence has finished successfully" when finished; press "OK". New lights should be on in the front panel (Figure 2).
 Figure 2. CyTOF panel lights after plasma ignited and start-up successfully completed.
Figure 2. CyTOF panel lights after plasma ignited and start-up successfully completed.
Turning on syringe pump. In the upper right corner, press the green "play" button to start the syringe pump and begin flowing liquid through the tubing and spraying out of the nebulizer. The default syringe settings are for 3 ml of water. In order for sample to be flowing through the sample loop, the syringe pump must be periodically changed when it runs out of water. At the top of the screen, there is a counter indicating how much liquid has flowed out of the syringe pump. The syringe pump will stop when this counter reaches "3.000" or the plunger hits the end of the syringe. When refilling the syringe, you must hit "stop" rather than the blue "pause" button to reset the counter.
2. Daily Calibration of the Mass Cytometer
The mass cytometer will need approximately 20 min to achieve a hot, stable plasma before calibration. The purpose of the tuning steps is to maximize the desired signal of Tm169 or Tb159 while keeping "Gd155" (oxide; La139+O16) signal under 3% of the higher value. Click the Acquisition Settings button to open the Data Acquisition Settings window. In the Analytes tab, click the periodic table to open the isotope selection panel. Select the isotopes in the DVS Sciences tuning solution listed in the manual, then click "save".
Click the Isotopes Per Reading button to open the Mass(es) Per Reading window. Select "Pulses Count", and rescale the y-axis by entering a new number and pressing "enter".
Fill a green 1 ml Norm-Ject syringe with tuning solution. Turn the sample port selector to "LOAD", inject 450 μl of tuning solution, then turn the selector to "INJECT". In the Mass Calibration tab of the Data Acquisition Settings window, press "Run" to monitor which masses are flowing by. The left side of the window is lower mass, while the right side is higher mass. Note: there will already be signals in the TOF 9400-9800 region, due to xenon isotopes in the argon gas (Figure 3). Wait until tuning solution isotopes start appearing as streaks in this display (Figure 4).
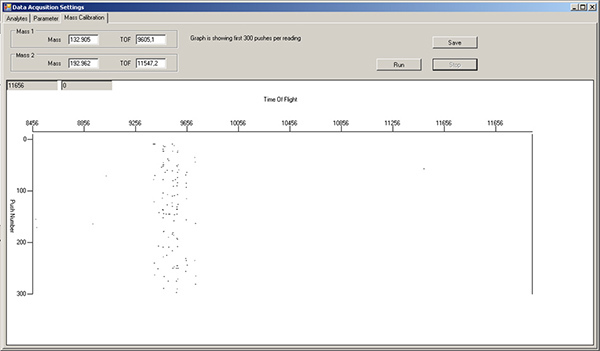 Figure 3. Mass calibration tab window, showing background levels for various isotopes after wash solution and water rinse. The region around TOF 9400-9800 corresponds to xenon isotopes in the argon gas, and should always be present. Click here to view larger figure.
Figure 3. Mass calibration tab window, showing background levels for various isotopes after wash solution and water rinse. The region around TOF 9400-9800 corresponds to xenon isotopes in the argon gas, and should always be present. Click here to view larger figure.
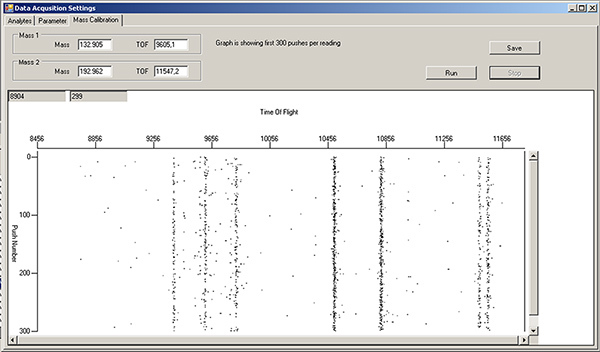 Figure 4. Mass calibration tab window, showing vertical streaks for various isotopes in DVS tuning solution. Click here to view larger figure.
Figure 4. Mass calibration tab window, showing vertical streaks for various isotopes in DVS tuning solution. Click here to view larger figure.
Go back to the Mass(es) Per Reading window and press the green "Play" button in the upper left corner. Each isotope selected in Step 2.1 is represented by a different colored line. The default x-axis is "Time", in sec. Monitor the levels of the different isotopes until they are steady.
Tuning of the current. On the Parameter tab of the Data Acquisition Settings window, select "Current" from the drop-down menu. Let start=0, finish=10, step value=0.5, settling time=200, and press "save". Click back on the Mass(es) Per Reading window and hit play. The x-axis is now "Current". Note where the signals of the isotopes peak (Figure 5), and record. Under the DAC Channels Setup tab of the Instrument Setup window, scroll down to "Current" and enter this new value. Press "Set actual current value", then "save".
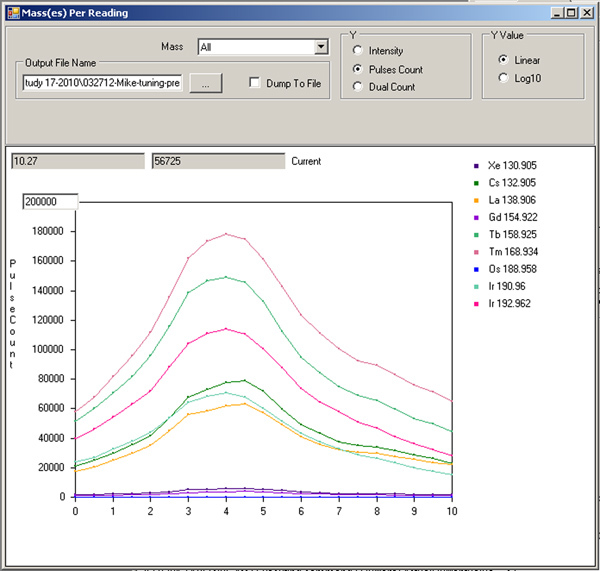 Figure 5. Current tuning profile.
Figure 5. Current tuning profile.
Tuning of the make-up gas. Change drop-down menu to "Make-up gas." Let start=0.55, end=0.95, step value=0.05, settling time=200. Press save. Return to the Mass(es) Per Reading window and hit play. The x-axis is now "Make-up gas" flow rate. Record where the signals of the isotopes peak. Observe the rapid increase of the "Gd155" signal near the end (Figure 6). Return to the DAC Channels Setup tab, scroll down to "Make-up gas", and enter this new value. Press "Set actual current value", then "save".
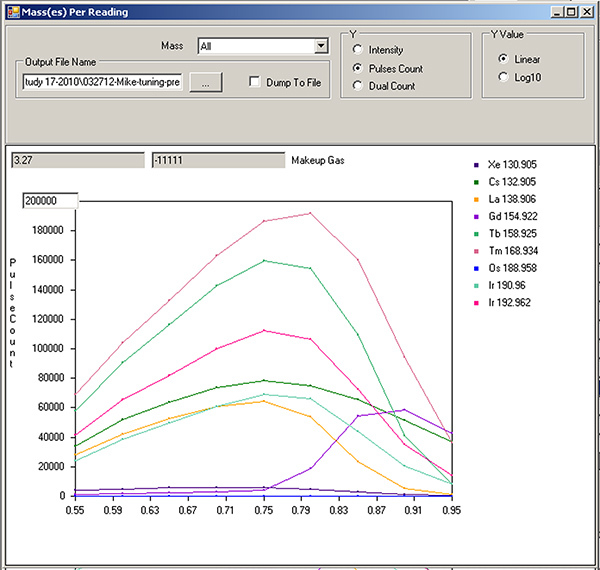 Figure 6. Make-up gas tuning profile.
Figure 6. Make-up gas tuning profile.
Recording ratio of desired isotope vs oxide. Make a second 450 μl injection of tuning solution. On the Parameter tab, select "Time" from the drop-down menu, let start=0, end=10, step value=1, settling time=200. Press "save." Hit "play" in Mass(es) Per Reading window. After the lines have stabilized (Figure 7), use the cursor to read the value in the upper left boxes for Tm169 or Tb159 (higher value) and also the "Gd155" value (<3% of the higher value). These values can also be saved in a tuning file for future reference.
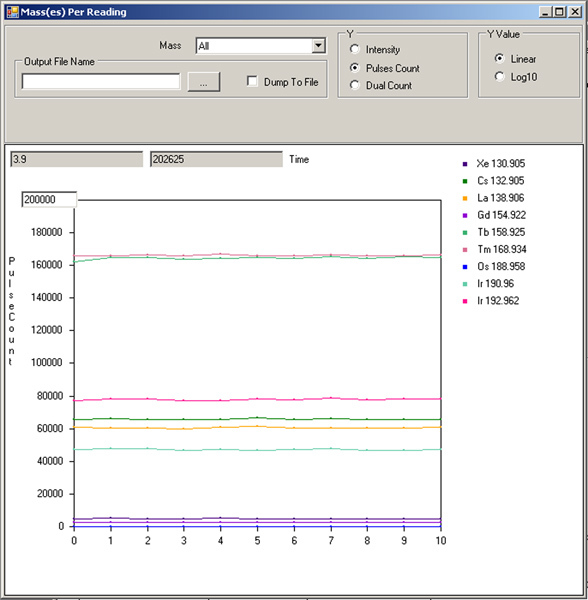 Figure 7. Measurement of tuning solution signal intensities after optimizing Current and Make-up gas.
Figure 7. Measurement of tuning solution signal intensities after optimizing Current and Make-up gas.
Cleaning of sample loop. Inject 3 ml of MilliQ water through the sample loop, followed by 500 μl of DVS Wash Solution. Monitor the progress of the cleaning by pressing "run" in the Mass Calibration tab. Wash until the intensity of the streaks (Figure 4) begins to decrease, then follow by 3 ml of MilliQ water and monitor until down to background levels (Figure 3).
3. Running Samples
Open the Acquisition window. Default settings: Do Analysis = On-The-Fly, Signal to Analyze = Dual, Dual Count Calibration = Data. Noise Reduction is selected. Any of these may be changed if desired. Enter the file name. All files MUST be saved to the E:// drive.
Setting acquisition delays. There will be a slight delay in registering cell events of about 30 sec due to the length of the tubing between the sample loop and the nebulizer. Therefore, the sum total of "Acquisition delay" and "Detector stability delay" should be at least 30 sec. Detector stability delay should be at least 20 sec.
Setting data collection time. "Acquisition time" is the number of seconds you want to run your sample. Filling the entire 450 μl sample loop would require 600 sec to complete. It is suggested that 50-100 sec be added to the end of the run time to minimize sample carry-over.
Resuspension of sample. To minimize build-up of salts in the machine, it is suggested that at least two washes in MilliQ water be done at the end of the cell staining, followed by a final resuspension in MilliQ water. The volume of MilliQ water used to resuspend the cell pellet will vary depending on the number of cells you started with and the cell recovery after the entire staining procedure. A good starting place is 106 (starting) cells/ml. After resuspension, filter through a cell strainer to remove aggregates.
Starting sample run. Enter a new file name for the current sample. Switch the sample loop valve to "LOAD", inject the sample, switch back to "INJECT", then hit "Run" in the Acquisition window.
Registering cell events. Cells will be represented in the snapshot window as horizontal collections of marks in each vertical isotope/marker channel (Figure 8). While the mass cytometer can register 1,000 cells/sec, better resolution between cell events is usually obtained at 300-500 cells/sec. This corresponds to an average of ~1 cell/screen refresh. It may take up to 30 sec before the cell event counter starts to climb. This is due to "On-the-fly" data processing; no cell event information is lost.
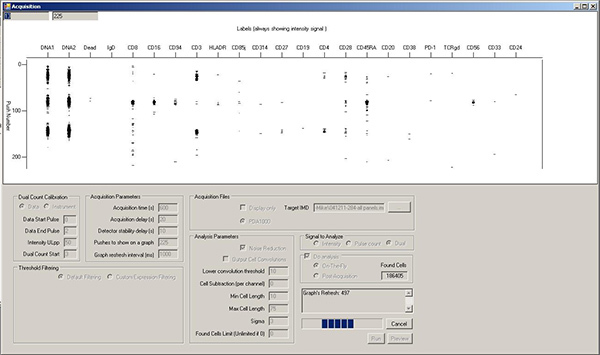 Figure 8. Acquisition window during sample running, showing horizontal rows of spots corresponding to the elements associated with three separate cell events. Click here to view larger figure.
Figure 8. Acquisition window during sample running, showing horizontal rows of spots corresponding to the elements associated with three separate cell events. Click here to view larger figure.
Quality of cell events. Notice any background streaking in each isotope channel, as well as the cell event rate. The mass cytometer recognizes a "cell event" as a certain increase in signal above background, so background signal from free metal or unbound antibody can alter the "cell event" count. Additionally, running cells at too high a concentration may cause an increased number of doublet events. Background can be diminished by additional washes or greater cell dilution. Doublets can be minimized by greater cell dilution, at the cost of increased run time.
Finishing sample data acquisition. When the Acquisition time has been reached, the data collection will finish. There will be a bit of delay before a pop-up window with an "OK" button appears; again, this is due to "On-the-Fly" data processing. Sample acquisition can also be stopped prematurely by hitting the "Stop" button. In this case, all the cell events already registered will be counted and processed into output files.
Injection of at least 500 μl of MilliQ water between each sample also helps minimize sample carryover.
4. Cleaning the Machine After Use
Wash solution. After the final sample is run and the sample loop flushed with water, inject 450 μl of wash solution into the sample loop. Monitor the release of free metal in the Mass Calibration tab of the Data Acquisition Settings window as in Step 2.8 (Figure 9). When the free metal signal begins to decrease, flush with 3 ml of MilliQ water and monitor until background levels are achieved.
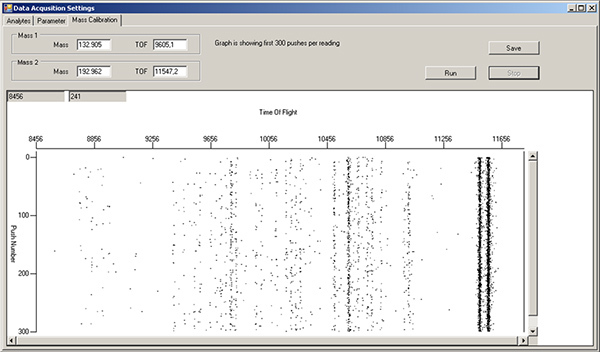 Figure 9. Mass calibration tab window, during post-run cleaning with DVS Wash solution. The vertical streaks in TOF region 9800-11000 are antibody-label isotopes being cleaned out of the machine. The vertical streaks around TOF 11500 correspond to the two Ir intercalator isotopes. Click here to view larger figure.
Figure 9. Mass calibration tab window, during post-run cleaning with DVS Wash solution. The vertical streaks in TOF region 9800-11000 are antibody-label isotopes being cleaned out of the machine. The vertical streaks around TOF 11500 correspond to the two Ir intercalator isotopes. Click here to view larger figure.
5. Shutting Down the Machine
In the RFG Controller tab of the Instrument Setup window, select "Stop Plasma". When the procedure is complete, there will be a pop-up window asking you to remove the nebulizer. Press "OK". In the Card Cage tab, turn off the heater. Close the valve on the argon supply.
Removal of nebulizer. Remove the nebulizer from the spray chamber by carefully pulling backwards. Unscrew the sample introduction tubing from Step 1.6 and place aside. Unscrew the make-up gas introduction connection to loosen slightly, then pull the nebulizer off. Leaving the fitting screwed together will reduce the chances of losing the black rubber o-ring and white conical plastic piece.
Cleaning nebulizer. Backflush both ports of the nebulizer with 5% citranox using the syringe and tubing as in Step 1.3. Store, covered, in 5% citranox until next use.
Representative Results
Following the above protocol should accomplish four things. First, allowing adequate warm-up time for the mass cytometer will produce a hot, stable plasma necessary for optimal signal and minimal oxide formation. Second, adequate washing of the mass cytometer with MilliQ water and DVS Wash solution (Figure 9) will help reduce the levels of metal adsorption to the tubing and other parts of the machine, helping to reduce background during sample acquisition (Figure 8). It will also help remove any cells that might get stuck in the machine and thereby minimizing carryover from sample to sample. Third, while oxide formation cannot be completely prevented, proper tuning of the mass cytometer (Figures 5-7) will help optimize total signal at the desired mass M, while minimizing background at the oxide mass M+16. As seen in Figure 10, improper tuning (magenta) significantly increased oxide formation at M+16 compared to the properly tuned sample (dark blue). This has the effect of slightly decreasing desired signal at M, and significantly increasing undesired background interference at M+16.
Finally, adequate washing of the sample in buffers and finally in MilliQ water should reduce metal/antibody background signal to acceptable levels. The MilliQ water washes and resuspension are critical for maintaining steady Current settings throughout the course of several hours of runtime. Proper dilution of the sample in MilliQ water has to be determined on a daily sample basis. However, diluting to ~106 (starting) cells/ml is a good place to start. Based on the first sample of a set, greater or lesser dilution for subsequent samples can be accommodated. Proper washing and proper dilution will ensure minimal background and the greatest resolution of cells, while balancing the need for speed (Figure 8).
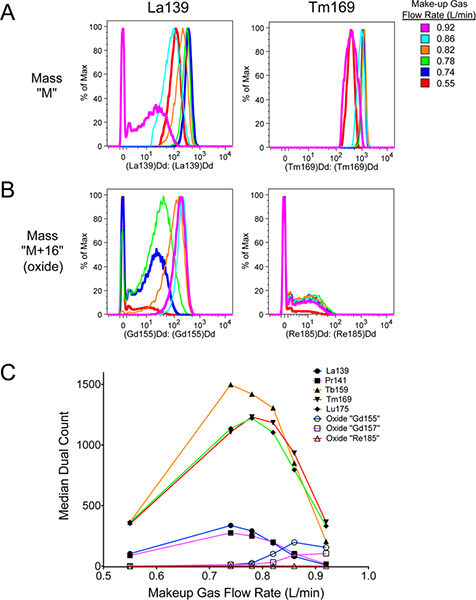 Figure 10. Effect of proper tuning on desired signal, and difference in oxide formation between various lanthanide metals. Polystyrene beads containing natural-abundance La139, Pr141, Tb159, Tm169, and Lu175 from DVS Sciences were run in cell acquisition mode. Separate samples of the beads were run at the indicated Make-up gas flow rates (compare with Figure 6). The data was analyzed using FlowJo v9.4.9 for Mac, including gating out debris from broken beads. The properly tuned sample was at 0.74 L/min. This had higher signal intensities than at lower flow rates, and higher signal at "M" metal masses with less signal at the "M+16" oxide masses than the improperly tuned signal. A. Signal at "M" mass La139 and Tm169. Note that La139 signal intensity is affected more than Tm169 by Make-up gas flow rate. B. Signal at "M+16" oxide masses 155 and 185, corresponding to La139+O16 and Tm169+O16 oxides, respectively. There is no actual "Gd155" or "Re185" present in the beads. The oxide signal in the "Gd155" channel is particularly strong, due to La139 being easily oxidized. This represents one of the highest levels of oxide one can expect to encounter. C. Graph of "M" and "M+16" signal intensities as a function of Make-up gas flow rate. Filled icons represent mass "M", while open icons represent mass "M+16". The color of the lines correspond to the respective mass "M". Notice that La139 and Pr141 are highly affected by Make-up gas flow rate, even reaching a point where more mass "M+16" oxide is present than mass "M". Click here to view larger figure.
Figure 10. Effect of proper tuning on desired signal, and difference in oxide formation between various lanthanide metals. Polystyrene beads containing natural-abundance La139, Pr141, Tb159, Tm169, and Lu175 from DVS Sciences were run in cell acquisition mode. Separate samples of the beads were run at the indicated Make-up gas flow rates (compare with Figure 6). The data was analyzed using FlowJo v9.4.9 for Mac, including gating out debris from broken beads. The properly tuned sample was at 0.74 L/min. This had higher signal intensities than at lower flow rates, and higher signal at "M" metal masses with less signal at the "M+16" oxide masses than the improperly tuned signal. A. Signal at "M" mass La139 and Tm169. Note that La139 signal intensity is affected more than Tm169 by Make-up gas flow rate. B. Signal at "M+16" oxide masses 155 and 185, corresponding to La139+O16 and Tm169+O16 oxides, respectively. There is no actual "Gd155" or "Re185" present in the beads. The oxide signal in the "Gd155" channel is particularly strong, due to La139 being easily oxidized. This represents one of the highest levels of oxide one can expect to encounter. C. Graph of "M" and "M+16" signal intensities as a function of Make-up gas flow rate. Filled icons represent mass "M", while open icons represent mass "M+16". The color of the lines correspond to the respective mass "M". Notice that La139 and Pr141 are highly affected by Make-up gas flow rate, even reaching a point where more mass "M+16" oxide is present than mass "M". Click here to view larger figure.
Discussion
For the last few decades, fluorescence flow cytometry has been a workhorse method for analyzing single cells, both in terms of surface expression and in functional assays. However, the issues of spectral overlap of the fluorescent dyes has limited the number of simultaneous markers. While experiments using more than 12 simultaneous markers have been reported, the amount of compensation necessary makes this technically difficult.
Instead of fluorophores, mass cytometry pioneered by DVS Sciences uses polymers with chelating sites for metals as labels for antibodies.1-3 Due to the purity of the metals and the mass resolution of ICP-MS, there is effectively no "spectral overlap." Since most of the elements used are not biologically relevant, there is also little background to cause "autofluoresence." These facts allow the use of more than 30 simultaneous markers within a single experiment.6,7 There is also a wider dynamic range in signal in ICP-MS than in a typical fluorescence experiment.
However, mass cytometry has limitations. It is a destructive technique: cells cannot be recovered for sorting and further experimentation. The mass cytometer has a strict requirement for the presence of metals: if a metal is not present, a cell will not be detected. This is the primary reason for the use of metal-chelating DNA intercalators to ensure correct percent-of-parent statistics, as all cells will be detected regardless of whether any antibody probes bind.3,6-7 Finally, the current cell transmission efficiency of the machine is ~25%, compared to an efficiency of >90% for a fluorescent flow cytometer. Therefore, a larger starting number of cells is often required, particularly for rare cell populations. However, this is usually balanced out by the fact that a single mass cytometer sample often replaces multiple fluorescent samples. Finally, the maximum cell acquisition rate is much slower than standard fluorescent flow cytometers at ~1,000 cells/sec; optimal rate is often half that. Therefore, each sample takes longer to run.
Several papers have recently been published on the staining of various types of cell samples.3,5-7 The purpose of this article is to provide information on the proper use of a CyTOF mass cytometer for running samples. Maintenance of the nebulizer by backflushing with and storage in 5% citranox will minimized build-up of cell debris and metals, thereby reducing the likelihood of clogs. Maintenance of the machine for optimal performance has two parts. First, the proper tuning (Current, Make-up gas) of the machine on at least a daily basis helps ensure proper measurement. Second, adequate washing with MilliQ water between each sample and with Wash solution at the end of runs to remove built-up organic and inorganic debris will help minimize background in general and carryover between samples. Finally, the quality of sample preparation has a great impact on the quality of data acquisition. Multiple washes in buffer and finally in MilliQ water help remove any metal-background from intercalators or nonspecific antibody binding. Proper dilution of the cell sample will help balance optimal resolution between cell events while minimizing total run time per sample. Proper dilution also helps minimize the likelihood of causing a clog in the nebulizer.
Troubleshooting Common Issues
1. The machine doesn't get past "RFG Prepare" step during start-up
Under the Card Cage tab of the Instrument Set-up window, press "Reset" under RFG. Attempt to start the machine. If this doesn't fix the problem, close the software, reopen, press "Reset" again, and attempt to start. If this still doesn't fix the problem, check the RFG generator breaker switch on the left back corner of the machine. If tripped, reposition to "On", hit "Reset", and try to start. If this still doesn't fix the problem, contact DVS.
2. The machine shuts down while running
Check argon supply. If the argon pressure reaching the machine gets below around 40 psi, the software executes an automatic shutdown.
RFG exceeded 100 W error. This occurs when the power demand for the plasma goes outside the 1,300 +/- 100 W window, and the software executes an automatic shutdown. This is usually caused by uneven spray from the nebulizer, particularly large droplets. This excessively cools the plasma, requiring more energy to maintain the plasma. This can happen because of excessive force used to attach the syringe tubing to the syringe during changing of the syringe pump. This causes a large spray of droplets, cooling the plasma. If this is the case, simply restart the machine.
This can also happen due to large droplets that begin to form when the nebulizer starts to clog. If this error recurs, remove the nebulizer for cleaning, insert a clean nebulizer and restart.
Much less frequently, the thin sample tubing between the sample loop and the nebulizer can clog. This can be diagnosed by anomalously low flow rate of liquid when the tubing is removed from the nebulizer. If this is the case, remove the affected tubing and fittings to be rebuilt with fresh tubing, and replace with a new nebulizer fittings and tubing kit.
3. No isotope streaks (or cell events) visible during cell sample acquisition (Figure 8), or while watching Mass calibration tab window (Figures 3, 4, 9)
Check the syringe pump. This often occurs due to forgetting to hit "play" during the changing of the syringe pump syringe. This can also happen if "pause", rather than "stop", is hit during the syringe change. "Pause" does not reset the volume counter on the syringe pump, and it will stop at 3.000 ml dispensed volume, regardless of whether liquid remains. Finally, the syringe may have run out of water.
Check sample valve. Make sure to load sample while on "Load", and then switch to "Inject" just before starting the sample run.
Depleted or overly dilute sample. You may have run all of your sample; check Acquisition Time calculations (see Step 3.3). Alternatively, you may have miscalculated the dilution factor, and/or lost more cells than expected during sample processing.
Clogged nebulizer. This is usually preceded by an RFG error (see Issue 2b), but a clog can notiecably reduce the cell transmission efficiency prior to causing a shutdown.
Issues 3c and 3d can also be checked by running a sample of the Eu-containing polystyrene calibration beads. They are supplied at approximately 1 million/ml (vortex well before drawing sample!). Therefore, filling the 450 μl sample loop would give approximately 450,000 beads. The CyTOF cell transmission efficiency is machine-dependent, but approximately 15-30%. Therefore, 67,500-135,000 bead events would be expected from that injection. Numbers below that are consistent with a clog in the tubing or nebulizer.
The cell/bead transmission efficiency is something to test occasionally, to note any long-term trends.
Disclosures
The authors both work at the Human Immune Monitoring Center, a service center at Stanford University which charges user fees solely to recover the cost of assays, including mass cytometry.
Acknowledgments
We would like to thank Dr. Evan Newell and Dr. Sean Bendall for feedback. We would also like to thank DVS Sciences and Dr. Sean Bendall for a sample of the Multi-lanthanide-containing polystyrene beads. We are grateful for funding from NIH grant 2 U19 AI057229.
References
- Bandura DR. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma Time-of-Flight mass spectrometry. Anal. Chem. 2009;81:6813–6822. doi: 10.1021/ac901049w. [DOI] [PubMed] [Google Scholar]
- Lou X. Polymer-based elemental tags for sensitive bioassays. Angew. Chem., Int. Ed. 2007;46:6111–6114. doi: 10.1002/anie.200700796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majonis D. Curious results with palladium- and platinum-carrying polymers in mass cytometry bioassays and an unexpected application as a dead cell stain. Biomacromolecules. 2011;12:3997–4010. doi: 10.1021/bm201011t. [DOI] [PubMed] [Google Scholar]
- Leipold MD. Development of mass cytometry methods for bacterial discrimination. Anal. Biochem. 2011;419:1–8. doi: 10.1016/j.ab.2011.07.035. [DOI] [PubMed] [Google Scholar]
- Bendall SC. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newell EW. Cytometry by Time-of-Flight shows combinatorial cytokine expression and virus-specific cell niches within a continuum of CD8+ T cell phenotypes. Immunity. 2012;36:142–152. doi: 10.1016/j.immuni.2012.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]