

Abstract
Actin reorganization has been shown to be important for lymphocyte activation in response to antigenic stimulation. However, methods for quantitative analysis of actin dynamics in live lymphocytes are still underdeveloped. In this study, we describe new methods to examine the actin dynamics in B cells induced by antigenic stimulation. Using the A20 B cell line expressing GFP–actin, we analyzed in real time the redistribution of F-actin and the lateral mobility of actin flow in the surface of B cells in response to soluble and/or membrane associated antigens. Using fluorescently labeled G-actin, we identified the subcellular location and quantified the level of de novo actin polymerization sites in primary B cells. Using A20 B cells expressing G-actin fused with the photoconvertible protein mEos, we examined the kinetics of actin polymerization and depolymerization at the same time. Our studies present a set of methods that are capable of quantitatively analyzing the role of actin dynamics in lymphocyte activation.
Keywords: Actin reorganization, BCR activation, Soluble and membrane antigens, Actin polymerization, Actin depolymerization
1. Introduction
The actin cytoskeleton is essential for the formation of immunological synapses between T cells and antigen presenting cells [1–3]. Actin reorganization in T cells is triggered and regulated by both adhesion molecules and T cell receptor (TCR) [4]. T cells also undergo spreading and contraction during synapse formation, which is associated with a centripetal actin flow [5]. In addition to its well-known role in antigen presentation to T cells, data from previous studies suggest that actin reorganization also plays a crucial role in the activation of B cells. Antigen induced B cell receptor (BCR) activation is important for B cell function. BCR activation was shown to induce global actin depolymerization, followed by actin polymerization. Depolymerization enhanced BCR signaling, while blocking depolymerization reduced the BCR signaling [6]. Recent reports show that depolymerization of the actin cytoskeleton inhibits surface BCR clustering in response to membrane-tethered antigens [7], but increases the lateral diffusion rate of surface BCRs and induces signaling in the absence of antigen [8]. An ezrinand actin-defined network was suggested to influence steady-state BCR diffusion by creating boundaries that restrict BCR diffusion; thus, the membrane skeleton could control signaling by influencing BCR dynamics [9]. Antigen binding to the BCR-induced kinetically and spatially controlled actin reorganization. This actin reorganization is required for B-cell spreading and contraction and controls BCR clustering in response to antigen. The stimulatory kinase Btk and the inhibitory phosphatase SHIP-1 have opposite regulatory roles in actin reorganization, B-cell spreading and contraction, surface BCR clustering, and B-cell surface signaling [10]. Previous research has also shown that actin is important for the presentation of antigens by B cells. Cytochalasin D treatment of F6 B lymphoma cells dramatically reduced the degradation of the invariant chain and delayed the appearance of stable forms of class II molecules, which reduced the efficiency of antigen presentation [11]. Also, Cytochalasin D treatment of CH27 cells decreases the internalization of BCR and blocks the movement of BCR from early endosomes to late endosomes [12].
Actin reorganization in lymphocytes has been traditionally studied by phalloidin staining and with labeled antibodies targeting to actin. FRAP analysis of membrane-proximal regions of interest (ROIs) in LPS/IL-4-activated B cells or IL-2-activated T cells that had been transfected with actin–GFP showed that Ag receptor-induced cell spreading is associated with increased actin dynamics [13]. Lifeact, a 17-amino-acid peptide that labels F-actin in eukaryotic cells, allows visualization of actin dynamics in nontransfectable cells without interfering with actin dynamics caused by GFP tags in vitro and in vivo [14].
Tracking of actin dynamics and reorganization in live cells is rarely performed after BCR activation in response to soluble antigens. However, the predominant form of antigen in vivo has been suggested to be attached to membrane surfaces, such that B cell membranes interact with other membranes to form BCR clusters and an immunological synapse [15]. Because of constraints due to mimicking of membrane-bound antigens and the required microscopy techniques, live cell tracking of actin dynamics and reorganization is rarely performed after BCR activation in response to membrane-bound antigens. It is important to quantify the actin dynamics in this physiological closed system. Experiments with G-actin incorporation indentify the de novo actin polymerization sites and dynamics, and can exclude the background caused by GFP–actin cell lines. Because of the tough environment caused by polymerization buffer, research on G-actin incorporation has been limited to splenic B cells. Actin depolymerization tracking is still a challenge because of the background levels of G-actin. Photo-convertible fluorescent proteins create a brighter signal and lower background, suitable for tracking the actin depolymerization events [16]. In this study, we tracked in real time the actin reorganization in GFP–actin mouse B lymphoma cells (A20) in response to soluble antigens by confocal microscopy and quantified actin flow speed in response to membrane antigens using total internal reflection fluorescence (TIRF) microscopy. De novo actin polymerization sites and dynamics were examined in primary B cells by TIRF microscopy. Finally actin polymerization and depolymerization was investigated using photo-convertible fluorescent actin (mEos-actin) expressed in A20 cells by confocal microscopy.
2. Materials and methods
2.1. Mice and cells
Wild type (wt) (CBA/CaJ), 6–10 weeks old mice (Jackson Laboratories, Bar Harbor, ME) were used. To isolate splenic B cells, mononuclear cells were subjected using Ficoll (Sigma–Aldrich, St Louis, MO) density-gradient centrifugation, treated with anti-Thy1.2 monoclonal antibodies (BD Biosciences, San Jose, CA) and guinea pig complement (Rockland Immunobiochemicals, Gilbertsville, PA) to remove T cells, and panned for 1 h to remove monocytes. B cell lymphoma A20 IIA1.6 cells (H-2d, IgG2a+, FcγRIIB−) were cultured in DMEM supplemented with 10% FBS.
2.2. Generation of A20 cells expressing GFP–G-actin and mEos-G-actin
B cell lymphoma A20 IIA1.6 cells were cultured at 37 °C in DMEM supplemented with 10% FBS. The DNA construct encoding the eGFP fusion protein of actin (eGFP–actin) or mEos fusion protein of actin (mEos-actin) was introduced into A20 B cells by electroporation using the Nucleofection kit V from Amaxa (Gaithersburg, MD). Transfected A20 cells were select with G418 (1 mg/ml) in DMEM supplemented with 10% FBS for two weeks and sorted by Flow Aria, and then maintained with G418 (0.5 mg/ml) in DMEM supplemented with 10% FBS.
2.3. Preparation of mono-biotinylated Fab’ antibody
Mono-biotinylated Fab’-anti-mouse IgM+G antibodies (mBFab’-anti-Ig) were generated from F(ab’)2 fragments (Jackson ImmunoResearch, West Grove, PA) using a published protocol [17]. The disulfide bond that links the two Fab’ was reduced using 20 mM 2-mercaptoethylamine, and the reduced cysteine was biotinylated by maleimide activated biotin (Thermo Scientific, Odessa, TA). Fab’ was further purified using Amicon Ultra Centrifugal Filter (Millipore, Temecula, CA). Titration of one biotin per Fab’ was confirmed by a biotin quantification kit from Thermo Scientific. Fab’ was labeled with Alexa Fluor 546 using a kit from Invitrogen (Carlsbad, CA).
2.4. Immunofluorescence by confocal microscopy
For live cell imaging, A20 cells that express a GFP fusion of actin were incubated with AF546-mB-Fab’-anti-Ig (10 μg/ml) at room temperature for 10 min to label the BCR, washed, and incubated with streptavidin (1 μg/ml) to activate the BCR. Images were acquired 3 s between each frame using a Zeiss 7 Live-DUO confocal microscope.
2.5. Preparation of antigen-tethered planar lipid bilayers
The planar lipid bilayer was prepared as described previously [18–19]. Liposomes were made by sonication of 1,2-Dioleoyl-sn-Glycero-3-phosphocholine and 1,2-Dioleoyl-sn-Glycero-3-phosphoethanola-mine-cap-biotin (Avanti Polar Lipids, Alabaster, AL) in a 100:1 M ratio in PBS at a lipid concentration of 5 mM. Aggregated liposomes were removed by ultracentrifugation and filtering. Coverslip chambers (Lab-Tek Nalge Nunc, Rochester, NY) were incubated with the liposomes (0.05 mM) for 10 min. After extensive washes, the coated coverslip chambers was incubated with 1 μg/ml streptavidin (Jackson ImmunoResearch), followed by 2 μg/ml AF546-mB-Fab’-anti-Ig and 8 μg/ml mB-Fab’-anti-Ig antibody.
2.6. Total internal reflection microcopy and image analysis
The surface dynamics and organization of the BCR and other molecules were analyzed using a TIRFm (TE2000U, Nikon). Images were acquired using a 100 × NA 1.49 Apochromat TIRF objective lens (Nikon). For live cell imaging, time-lapse imaging started upon the addition of B cells onto the lipid bilayers tethered with AF546-mB-anti-Ig and continued for 5–10 min at 37 °C. GFP/AF488, AF546, interference refection images (IRM) were acquired sequentially at each time point. B cell contact area was determined using MATLAB software. Total fluorescence intensities of AF546-mB-anti-Ig in the contact zone and relative fluorescence and IRM intensity along a line across cells were quantified using Andor iQ software (Andor Technology, Belfast, UK). Background fluorescence, such as antigen-tethered lipid bilayers in the absence of B cells was subtracted. For each set of data with statistics, more than 20 individual cells from two or three independent experiments were analyzed.
2.7. Tracking speed and direction of actin flow
Spatio-temporal image correlation spectroscopy [20] was used to detect the directed movement of actin from the analysis of fluorescence image time series. The technique involves calculating the spatial–temporal correlation function of fluorescence intensity fluctuations as a function of the time-lag between successive images. For diffusive motion of the fluorescent particles, the correlation function exhibits a peak that decreases in amplitude and broadens as a function of time-lag. However, for fluorescent proteins exhibiting directional motion, the correlation peak shifts opposite to the direction of motion. The correlation functions were calculated using Fourier transforms and the velocity values were extracted using the STICS algorithm implemented in MATLAB and freely available from the Cell Migration Gateway.
Velocity maps were generated by selecting a grid of overlapping, 16× 16 pixel (1.6×1.6 microns) square regions (with an overlap of 8 pixels) that spanned the region of interest. The images were corrected for immobile fraction on each sub-region time stack within this grid. Image series were collected with time intervals of x seconds; which is slower than the diffusive motion of proteins (in the millisecond range).
2.8. Analysis of actin nucleation sites
Actin nucleation sites were labeled as previously described [21]. For B cells that were activated by membrane tethered mB-Fab’-anti-Ig, B cells were incubated with the membrane-tethered antigen in the presence of AF488-G-actin and 0.025% saponin. Time lapse images were acquired using a TIRF microscope.
2.9. Photo-convertible activation
Before photoactivation, A20-mEos-actin cells stimulated by 10 μg/ml F (ab’)2 -anti-Ig soluble antigens at 37 °C were scanned two times with 488 nm argon laser (10%) and a white light laser turned at 560 nm (50%) under a 63 × oil objective lens of a Leica SP5X laser scanning confocal microscope. The cells were then activated with the 405 nm diode laser (10% power) at time = 2.735 s. The cells were scanned a total of 54 times. The average intensity values for each of the ROIs were recorded by Leica LAS AF software.
3. Results
3.1. Multivalent soluble antigens induce the redistribution of surface BCRs and the actin cytoskeleton
To study the dynamics of actin reorganization induced by antigens, we first used A20 lymphoma B cells that express GFP–actin. B cells were activated by Alexa fluor 546-conjugated, mono-biotinylated Fab’ fragments of goat-anti-mouse IgG+M (AF546-mB-Fab’-anti-Ig) plus streptavidin as a multivalent soluble antigen. The reorganization of the actin cytoskeleton and surface BCRs in B cells activated by the soluble antigen was examined using confocal fluorescence microscopy (CFm). Time lapse imaging showed that in the absence of streptavidin-mediated cross-linking (−XL), surface BCRs distributed evenly on the cell surface, while some of them slowly internalized over time (Fig. 1a–f). Upon cross-linking by streptavidin (+XL), surface BCRs rapidly formed small clusters first (Fig. 1g and h), and the small clusters then merged into a centralized cluster at one pole of the cell (Fig. 1i–l and Video S1). In response to BCR cross-linking, the actin cytoskeleton underwent a dramatic redistribution, from an even distribution at the cell periphery in unstimulated cells (Fig. 1a–f) to accumulation at BCR clusters in stimulated cells (Fig. 1g–l and Video S1).
Fig. 1.
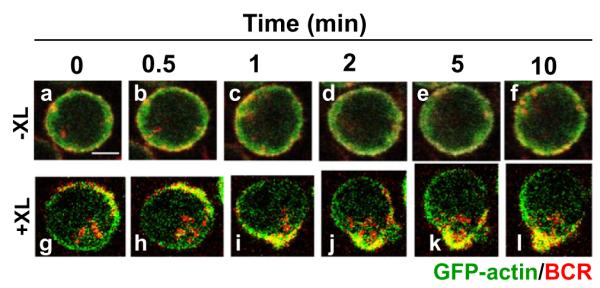
Multivalent soluble antigens induce actin reorganization. A20 B cells that expressed GFP–actin were incubated with AF546-mB-Fab’-anti-Ig for 10 min at 37 °C to label the BCR. Then the cells were either incubated with the medium along (−XL, a–f) or with streptavidin (+XL, g–l) at 37 °C. Time lapse images were acquired using a confocal fluorescence microscope. Shown are selected images. Bar, 2.5 μm.
3.2. Membrane associated antigens induce the redistribution of surface BCRs and the actin cytoskeleton
To closely examine the reorganization of the actin cytoskeleton and surface BCRs at the B cell surface, we analyzed GFP–actin expressing A20 B cells stimulated by AF546-mB-Fab’-anti-Ig tethered onto lipid bilayers by streptavidin as the membrane associated antigen using total internal reflection fluorescence microscopy (TIRFm). Additionally, we used interference reflection microscopy (IRM) to quantitatively determine the surface area of B cells contacting the antigen tethered lipid bilayer, the B cell contact area. When incubated on lipid bilayers that were tethered with Fab fragment of a negative control antibody as a non-specific antigen, A20 B cells were able to establish contact with the lipid bilayer, and actin was detected in the contact zone (Fig. 2Aa–d). However, there was neither expansion of the B cell contact area nor increase in the fluorescence intensities of actin and the antigen over time (Fig. 2D). Upon activation by membrane-associated antigens, B cells spread over antigen-tethered lipid bilayers (Fig. 2Ae and D). Since a non-specific antigen was unable to induce aggregation and clustering, the clustering of BCR-specific antigen shown here reflects aggregation of surface BCRs. In B cells stimulated with membrane associated antigens, GFP–actin was rapidly accumulated at the B cell contact zone as BCR microclusters formed (Fig. 2D) and particularly concentrated at the outer edge of BCR clusters (Fig. 2Ae–i, C and Video S2). Time lapse images showed an outward flow of actin as B cells contacted and spread over antigen-tethered lipid bilayers, followed by a centripetal flow as antigen microclusters merged into central cluster (Video. S2). Quantitative analysis of TIRFm time lapse images showed that the fluorescence intensities of both GFP–actin and the antigen in the B cell contact zone as well as the B cell contact area increased over time, but with different kinetics (Fig. 2D). The increases in the fluorescence intensities of the antigen and actin in the B cell contact zone appeared to have three phases: a fast rate of increase at the earlier time followed by a period of slow increases before reaching a plateau (Fig. 2D). The contact area of A20 B cells did not reduce after reaching its peak, suggesting that A20 B cells do not contract after spreading (Fig. 2D and Video. S2). In fully spread cells, actin underwent retrograde flow at the leading edge. A correlation-based algorithm was used to track the (Fig. 2B) the direction and speed of this retrograde flow. The length of the arrows correspond to velocity vectors and the peak values of velocity were approximately 5 μm/min, which is somewhat faster than adherent cells such as fibroblasts, but slower than in T lymphocytes [22], and this quantification achieves more detailed information on the actin dynamics.
Fig. 2.

Membrane associated antigens induce actin reorganization GFP–G-actin expressing. A20 B cells were incubated with lipid bilayers tethered with AF546-mB-Fab’-anti-Ig (+Ag) or a non-specific antibody (Control) at 37 °C. Time lapse images were acquired using TIRFm. Shown are representative images at 5 min (A), actin flow speed indicated by the arrows (B), relative fluorescence and IRM intensity across the cell (blue line) (C), and the average values (±SD) of the contact area, and the total fluorescence intensities (±SD) of antigens and GFP–F-actin in the contact zone (D) from >20 cells of three independent experiments. Bar, 2.5 μm.
3.3. Engagement of the BCR with membrane-associated antigens induces actin polymerization at BCR clusters
The actin cytoskeleton is highly dynamic and rearranges its organization by polymerization and depolymerization. To examine how actin reorganization coordinates with surface BCR clustering, we analyzed the cellular distribution and levels of actin polymerization in relation to surface BCRs. Actin polymerization sites were detected by the incorporation of AF488-G-actin into polymerizing ends of actin filaments. AF488-G-actin was introduced to cells in the presence of a low concentration of non-ionic detergent after splenic B cells were stimulated by membrane antigens. In splenic B cells activated by membrane associated antigen, actin polymerization activity was detected at the B cell contact zone (Fig. 3A andVideo S3). Actin polymerization sites was preferentially located at the cell periphery of the spreading membrane and the outer edge of BCR microclusters as they were moving to the center of B cell contact zones (Fig. 3A and Video S3). Quantitative analysis of TIRFm time lapse images showed that the fluorescence intensities of AF488-G-actin in the B cell contact zone increased over time. The increases in the fluorescence intensities of actin in the B cell contact zone appeared to have two phases: a steady state of undetectable polymerized actin period around 4 min followed by a period of quick increases (Fig. 3B). Non-stimulated antigens did not elicit detectable accumulation of actin polymerization in the contact area (data not shown). These results indicate that membrane-associated antigens induce actin polymerization at BCR microclusters and the outer edges of BCR central clusters, and that it takes a short time for the initiation of actin polymerization after BCR activation.
Fig. 3.
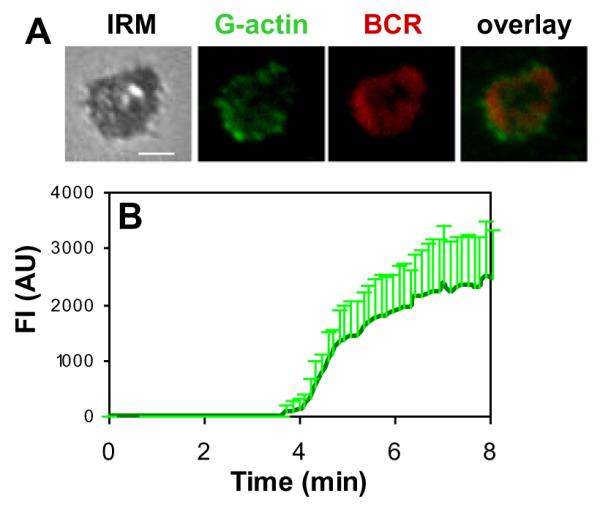
Membrane associated antigens induce actin polymerization at BCR clusters. Splenic B cells were incubated with AF546-mB-Fab’-anti-Ig tethered on lipid bilayers at 37 °C in the presence of AF488-G-actin and 0.025% saponin. Time lapse images were acquired using TIRFm. Shown are representative images at 5 min (A) and the average fluorescence intensities of AF488-G-actin in the contact area from three independent experiments (B). Bar, 2.5 μm.
3.4. Analyzing actin dynamics using photo-convertible mEos2–actin
In order to investigate the actin polymerization and depolymerization in a live cell, a region of an A20 B cell expressing mEos2–actin stimulated by F (ab’)2 -anti-Ig was photoactivated using a UV laser, mEos2 fluoresces in green before photoactivation and fluoresces in red after photoactivation. For the photoactivated area, the red fluorescence signal of photo-converted actin increased rapidly and peaked at approximately 5 s following photoactivation, indicating that actin polymerization occurred from the photo-activated G-actin pool (Fig. 4A and B and Video S4). The actin polymerization rate was calculated from the slope of initial increase in fluorescent signal to be 0.140 AU (arbitrary unit)/second. These areas were tracked for 45 additional seconds during which time the signal of photo-converted actin decreased slowly (Fig. 4A and B and Video S4). This is the expected pattern for actin depolymerization in this system, since the photoconverted G-actin monomer will separate from the F-actin. Similarly, we calculated the actin depolymerization rate from the slope of the second phase to be 0.005 AU/s. In contrast, the signal of photo-converted actin in an area without photo-activation increased slowly over the entire period of observation (Fig. 4A and B and Video S4), from which we calculated the actin polymerization rate from the photo-converted actin monomer in absence of photo-activation to be 0.040 AU/s. These results show that this technique can track the actin polymerization and depolymerization dynamics at the same time, and also the actin polymerization rate at different regions of the cell.
Fig. 4.

Analyzing actin dynamics using photo-convertible mEos2–actin. (A) A region of an A20 B cell that expresses mEos2-G-actin stimulated by Fab2-anti-Ig at 37 °C was photoactivated using a UV laser. (B) The fluorescence intensity of photoconverted G-actin in the photoactivated area (white region) and in the unphotoactivated area (red region) was measured over time. Bar, 2.5 μm.
4. Discussion
The reorganization of actin upon BCR activation in response to soluble antigens was studied, and we showed in vivo cell actin reorganization after BCR stimulation in response to soluble antigens. We could clearly see the polarization process of actin in accordance with the BCR polarization. This shows that the live cell imaging technique described here could provide further insight on the dynamics of actin polarization. Next we tracked the actin reorganization and dynamics lively through the high resolution images upon BCR activation in response to membrane antigens through TIRF. While actin had been studied by TIRF without antigen stimulation in the coverslip before [9], no quantification was reported. Using the techniques described above, we not only observed the retrograde actin flow, but also could quantify overall actin dynamics. Quantification of the speed of actin flow and detailed analysis of the actin dynamics in the specific regions are key to a more complete understanding of cytoskeletal dynamics in the cell.
The de novo actin polymerization sites and dynamics were tracked live in primary B cells, which are more closely related to the physiological situation compared to immortal cell lines used extensively in previous research. Actin polymerization sites were mainly outside the central BCR cluster and showed up later than the BCR cluster. We could track the total actin dynamics in live cells with a clear and contrast background. Our technique allows further research on the dynamics of a single actin protein on the de novo actin polymerization sites. In this way, we could also get more detail information as the way to track the speed of actin flow. Compared to the previous GFP-tagged actin assay, the de novo actin polymerization assay could eliminate the effect of GFP on the actin. Although FRAP could be also used to track the actin polymerization kinetics in lymphocytes with good spatial and temporal resolution, it requires the use of high laser power that can damage the cell membrane as we as a relative large bleached region in the cell. Through the photo-convertible actin expression, we could successfully track the actin dynamics, especially the actin depolymerization with lower laser power, and we could also track the actin polymerization and depolymerization at the same time, which is not easy to be done in the combination of FRAP and fluorescence loss in photobleaching (FLIP). Further research could be done on the actin particles by fluorescence speckle microscopy. Low power illumination with UV light photoactivates small numbers of photoconvertible G-actin molecules whose movement can be tracked using particle tracking methods. However, we still can’t exclude the effect of photo-convertible protein on the actin. Although Lifeact can eliminate the effect of tag protein on actin, but it has lower affinity to bind to the actin. It would be a potential direction to develop a photo-convertible Lifeact with high affinity for actin in the future.
In summary, we have shown that using GFP or photo-convertible actin and de novo actin polymerization assay by confocal and TIRF microscopy can offer a more complete and accurate dynamic picture of actin dynamics during BCR-antigen recognition and BCR cluster formation in both lymphoma immortal cells and in primary B cells.
Supplementary Material
Acknowledgments
We thank Dr. Katharina Richard and Dr. Eduardo Zattara for critically reading the manuscript and this work was supported by a Grant from National Institutes of Health (AI059617 to W.S.).
Footnotes
Appendix A. Supplementary data Supplementary data associated with this article can be found, in the online version, at http://dx.doi.org/10.1016/j.bbrc.2012.09.046.
References
- [1].Fooksman DR, Vardhana S, Vasiliver-Shamis G, Liese J, Blair DA, Waite J, Sacristán C, Victora GD, Zanin-Zhorov A, Dustin ML. Functional anatomy of T Cell activation and synapse formation. Annu. Rev. Immunol. 2010;28:79–105. doi: 10.1146/annurev-immunol-030409-101308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Billadeau DD, Nolz JC, Gomez TS. Regulation of T-cell activation by the cytoskeleton. Nat. Rev. Immunol. 2007;7:131–143. doi: 10.1038/nri2021. [DOI] [PubMed] [Google Scholar]
- [3].Burkhardt JK, Carrizosa E, Shaffer MH. The actin cytoskeleton in T cell activation. Annu. Rev. Immunol. 2008;26:233–259. doi: 10.1146/annurev.immunol.26.021607.090347. [DOI] [PubMed] [Google Scholar]
- [4].Suzuki J, Yamasaki S, Wu J, Koretzky GA, Saito T. The actin cloud induced by LFA-1-mediated outside-in signals lowers the threshold for T-cell activation. Blood. 2007;109:168–175. doi: 10.1182/blood-2005-12-020164. [DOI] [PubMed] [Google Scholar]
- [5].Kaizuka Y, Douglass AD, Varma R, Dustin ML, Vale RD. Mechanisms for segregating T cell receptor and adhesion molecules during immunological synapse formation in Jurkat T cells. Proc. Natl. Acad. Sci. USA. 2007;104:20296–20301. doi: 10.1073/pnas.0710258105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Hao S, August A. Actin depolymerization transduces the strength of B-cell receptor stimulation. Mol. Biol. Cell. 2005;16:2275–2284. doi: 10.1091/mbc.E04-10-0881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Fleire SJ, Goldman JP, Carrasco YR, Weber M, Bray D, Batista FD. B cell ligand discrimination through a spreading and contraction response. Science. 2006;312:738–741. doi: 10.1126/science.1123940. [DOI] [PubMed] [Google Scholar]
- [8].Treanor B, Batista FD. Organisation and dynamics of antigen receptors: implications for lymphocyte signalling. Curr. Opin. Immunol. 2010;22:299–307. doi: 10.1016/j.coi.2010.03.009. [DOI] [PubMed] [Google Scholar]
- [9].Treanor B, Depoil D, Gonzalez-Granja A, Barral P, Weber M, Dushek O, Bruckbauer A, Batista FD. The membrane skeleton controls diffusion dynamics and signaling through the B cell receptor. Immunity. 2010;32:187–199. doi: 10.1016/j.immuni.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Liu C, Miller H, Hui KL, Grooman B, Bolland S, Upadhyaya A, Song W. A balance of Bruton’s tyrosine kinase and SHIP activation regulates B cell receptor cluster formation by controlling actin remodeling. J. Immunol. 2011;187:230–239. doi: 10.4049/jimmunol.1100157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Barois N, Forquet F, Davoust J. Actin microfilaments control the MHC class II antigen presentation pathway in B cells. J. Cell Sci. 1998;13:1791–1800. doi: 10.1242/jcs.111.13.1791. [DOI] [PubMed] [Google Scholar]
- [12].Brown BK, Song W. The actin cytoskeleton is required for the trafficking of the B cell antigen receptor to the late endosomes. Traffic. 2001;2:414–427. doi: 10.1034/j.1600-0854.2001.002006414.x. [DOI] [PubMed] [Google Scholar]
- [13].Freeman SA, Lei V, Dang-Lawson M, Mizuno K, Roskelley CD, Gold MR. Cofilin-mediated F-actin severing is regulated by the Rap GTPase and controls the cytoskeletal dynamics that drive lymphocyte spreading and BCR microcluster formation. J. Immunol. 2011;187:5887–5900. doi: 10.4049/jimmunol.1102233. [DOI] [PubMed] [Google Scholar]
- [14].Riedl J, Flynn KC, Raducanu A, Gärtner F, Beck G, Bösl M, Bradke F, Massberg S, Aszodi A, Sixt M, Wedlich-Söldner R. Lifeact mice for studying F-actin dynamics. Nat. Methods. 2010;3:168–169. doi: 10.1038/nmeth0310-168. [DOI] [PubMed] [Google Scholar]
- [15].Harwood NE, Batista FD. Early events in B cell activation. Annu. Rev. Immunol. 2010;28:185–210. doi: 10.1146/annurev-immunol-030409-101216. [DOI] [PubMed] [Google Scholar]
- [16].Patterson GH, Lippincott-Schwartz J. A photoactivatable GFP for selective photolabeling of proteins and cells. Science. 2002;297:1873–1877. doi: 10.1126/science.1074952. [DOI] [PubMed] [Google Scholar]
- [17].Peluso P, Wilson DS, Do D, Tran H, Venkatasubbaiah M, Quincy D, Heidecker B, Poindexter K, Tolani N, Phelan M, Witte K, Jung LS, Wagner P, Nock S. Optimizing antibody immobilization strategies for the construction of protein microarrays. Anal. Biochem. 2003;312:113–124. doi: 10.1016/s0003-2697(02)00442-6. [DOI] [PubMed] [Google Scholar]
- [18].Grakoui A, Bromley SK, Sumen C, Davis MM, Shaw AS, Allen PM, Dustin ML. The immunological synapse: a molecular machine controlling T cell activation. Science. 1999;285:221–227. doi: 10.1126/science.285.5425.221. [DOI] [PubMed] [Google Scholar]
- [19].Sohn HW, Pierce SK, Tzeng SJ. Live cell imaging reveals that the inhibitory FcγRIIB destabilizes B cell receptor membrane-lipid interactions and blocks immune synapse formation. J. Immunol. 2008;180:793–799. doi: 10.4049/jimmunol.180.2.793. (2008) [DOI] [PubMed] [Google Scholar]
- [20].Hebert B, Costantino S, Wiseman PW. Spatiotemporal image correlation spectroscopy (STICS) theory, verification, and application to protein velocity mapping in living CHO cells. Biophys. J. 2005;88:3601–3614. doi: 10.1529/biophysj.104.054874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Chan AY, Raft S, Bailly M, Wyckoff JB, Segall JE, Condeelis JS. EGF stimulates an increase in actin nucleation and filament number at the leading edge of the lamellipod in mammary adenocarcinoma cells. J. Cell Sci. 1998;111:199–211. doi: 10.1242/jcs.111.2.199. [DOI] [PubMed] [Google Scholar]
- [22].Yu CH, Wu HJ, Kaizuka Y, Vale RD, Groves JT. Altered actin centripetal retrograde flow in physically restricted immunological synapses. PLoS ONE. 2010;5:e11878. doi: 10.1371/journal.pone.0011878. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.