

Abstract
Peptibodies or peptide-Fc fusions are an attractive alternative therapeutic format to monoclonal antibodies. They consist of biologically active peptides grafted onto an Fc domain. This approach retains certain desirable features of antibodies, notably an increased apparent affinity through the avidity conferred by the dimerization of two Fcs and a long plasma residency time. Peptibodies can be made in E. coli using recombinant technology. The manufacturing process involves fermentation and downstream processing, including refolding and multiple column chromatographic steps, that result in overall yields and quality suitable for commercial development. Romiplostim, marketed under the brand name Nplate®, is the first peptibody to be approved by the United States Food and Drug Administration and the European Medicines Agency and is indicated for the treatment of immune thrombocytopenic purpura. AMG 386, a peptibody antagonist to angiopoietins 1 and 2, is being evaluated in Phase 3 clinical testing in combination with chemotherapy in women with ovarian cancer. AMG 819, a peptibody targeting nerve growth factor for pain has also progressed to clinical trials. These peptibodies illustrate the versatility of the modality.
Keywords: AMG 386, AMG 531, AMG 819, Nplate®, peptibody, romiplostim
Introduction
The use of peptides as pharmacological agents is an attractive therapeutic approach. Peptides can be designed to modulate various biological processes with high specificity and can be synthetically made with high purity. Because of their low cellular permeability, peptidic drugs mostly target extracellular receptors, the best known being an agonist to the glucagon-like peptide-1 receptor (Byetta®, Amylin Pharmaceuticals, Inc.) for type 2 diabetes mellitus and agonists to the gonadotropin-releasing hormone receptor (Zoladex®, AstraZeneca; Lupron®, Abbott) for the treatment of prostate cancer and endometriosis. Other peptide-based drugs are progressing in clinical development in oncology. The most advanced, cilengitide (Merk Serono), an αvβ3 antagonist, and ABT510 (Abbott), a thrombospondin-1 mimetic, have advanced to Phase 2 and target the process of angiogenesis. Despite these successes and early promise, the use of peptides as drugs is limited. Peptides usually suffer from a rapid clearance, the consequence of a combination of poor metabolic stability and a hydrodynamic radius that falls below the limit of the glomerular filtration of the kidney. Strategies to extend stability and improve pharmacokinetic (PK) properties have been described.1 One of these strategies has been to graft peptides to the Fc region of an immunoglobulin G (IgG), thereby hijacking the FcRn recycling process that provides a long circulating half life to human IgG.2 These peptides fused in frame to IgG Fc, called peptibodies by Amgen, represent a novel class of therapeutics. The prototype peptibody, romiplostim (Nplate®, AMG 531) was approved by the United States (US) Food and Drug Administration in 2008 and European Medicines Agency in 2009 for the treatment of immune thrombocytopenic purpura.3 The objective of this review is to describe the generation, properties and production of peptibodies, and to provide an overview of their status in clinical development. We describe the development of Nplate® and two peptibody drugs that entered human clinical trials: AMG 386, targeting angiopoietins4 and AMG 819, an antagonist of nerve growth factor (NGF).
Process for the generation of a peptibody
A peptibody is composed of two moieties, a biologically active peptide and an Fc region (Fig. 1). Peptides with diverse biological activities have successfully been grafted onto Fc, e.g., an agonist to a growth factor receptor (romiplostim), and antagonists to soluble proteins (AMG 819, AMG 386). Peptibodies developed by Amgen that have progressed to human testing are listed in Table 1.
Figure 1.
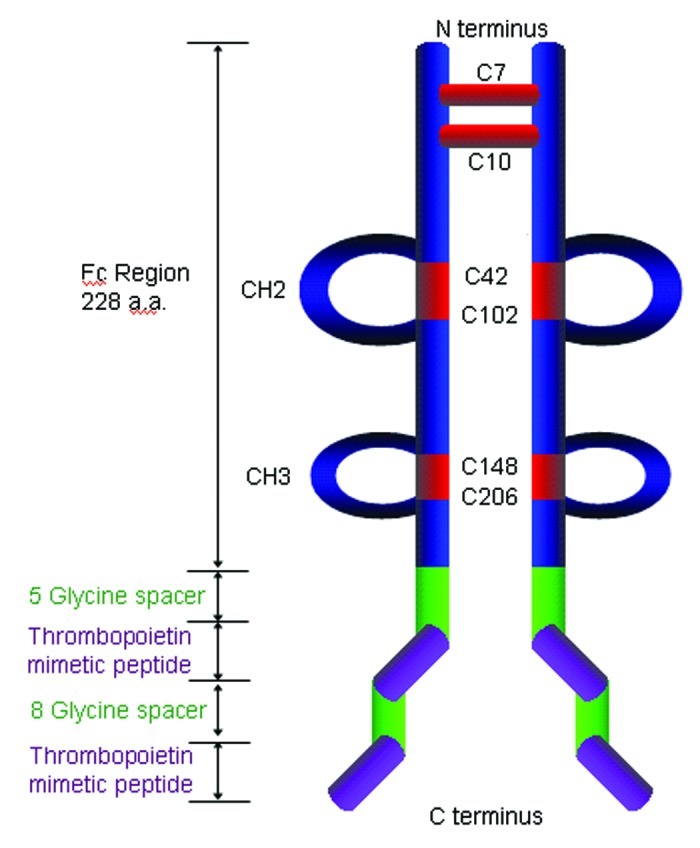
Structure of the prototypical peptibody AMG531/romiplostim/Nplate©. Two thrombopoietin mimetic peptides separated by an eight-glycine spacer are fused to the carboxy-terminal end of the IgG1 heavy chain. A five glycine linker separates the peptide and IgH.
Table 1. Peptibody Programs in Clinical Trials Sponsored by Amgen.
| MOA | Disease indication | Highest stage of development | Developer | |
|---|---|---|---|---|
|
AMG 531 (romiplostim) |
TPO mimetic, TPOR agonist |
Immune thrombocytopenia Purpura |
Marketed |
Amgen |
|
AMG 386 |
Angiopoietins antagonist |
Oncology – anti-angiogenic |
Phase III |
Amgen |
| AMG 819 | NGF antagonist | Pain | Terminated – Phase I | Amgen |
Source: clinicaltrials.gov and Thomson Pharma® Partnerin
The generation of a peptibody starts with the identification of a peptide with the desired binding and biological properties for the target of interest. The peptide component of romiplostim was identified by Cwirla et al. from screening libraries of random peptides displayed as fusions to the major coat protein of filamentous phage or to the E. coli lac repressor protein.5 The peptides’ affinity was optimized by mutagenesis and addition of flanking residues.5 The peptides used for the generation of AMG 386 and AMG 819 were also identified by screening phage libraries.6 Typically the respective peptides are affinity matured and optimized for the desired biological activity.7 In the case of romiplostim, the optimized peptide IEGPTLRQWLAARA was shown to displace thrombopoietin (TPO) from its receptor hTPOR and to stimulate the proliferation of Ba/F3 cells engineered to express hTPOR.5 After bolus intravenous (iv) administration in mice, the peptide was able to stimulate, albeit transiently, the formation of platelets.8 Continuous infusion in mice resulted in a prolonged increase in platelet counts, indicating that the activity of the peptide was limited by its rapid clearance. Extension of the plasma half-life of the biologically active peptides and the concomitant prolongation of the pharmacodynamic (PD) effect was achieved by the grafting of hTPOR binding peptides onto a human Fc, which led to the generation of romiplostim. The half-life improvement can be attributed to at least two factors: the increased molecular weight (about 60kD), which increased the hydrodynamic volume above the threshold of glomerular filtration by the kidney, and the ability to use the neonatal Fc receptor-mediated recycling process.2 The average plasma half-life of peptibodies in humans ranges from 3 to 8 d.9,10 While this is shorter than that of some monoclonal antibodies (mAbs) in the clinic, it is a substantial improvement over that observed for iv administered naked peptides. Fc fusion can also be more attractive than chemical modification, as the resulting polypeptide can be completely manufactured in a recombinant manner. In addition, the homodimerization of the two Fc moieties provides at least two peptides per peptibody, resulting in an increased avidity to the target. Further concatemerization of the peptide spaced by a linker, as is the case for romiplostim and AMG 386, may further increase the avidity. For example, the potency of AMG 386, which contains two peptides per Fc strand, is ten times greater in an Ang2 neutralization assay than the equivalent molecule with a single peptide per Fc strand (IC50 = 0.02 nM vs. IC50 = 0.2 nM; unpublished data). Subsequent improvement in peptibody activity is generally achieved with variation in the linker and the context of the peptides. Interestingly, and for reasons still unknown, certain peptides seem to be more active when fused to the carboxy terminus of the Fc. In a NGF neutralizing assay, the carboxy terminus fused peptibody is 1500 times more potent than the N-terminus (data not shown). It should however be noted that this position is not always preferred (data not shown).
An extension of these peptibody design strategies involves engineering the therapeutic warhead peptides as insertion sequences within the Fc sequence (Fig. 2). As non-terminal fusions, these novel peptibodies are influenced by both the context of the selected insertion site and the composition of the inserted peptide warhead. For that reason several exposed loop sequences in the Fc CH2 and CH3 domains were explored for peptide insertion using phage derived peptides from the disulfide constrained libraries.11 These internally fused Fc:peptide molecules are called FcLoop peptibodies. When insertion site and peptide composition are correctly paired, significant improvements in refolding efficiency, purification, yield, and in vitro stability can be observed relative to a conventional terminal Fc fusion while retaining equivalent bioactivity. Further, in vivo studies indicate significant improvements in PK exposure (unpublished data). The improved PK of FcLoop peptibodies relative to terminal Fc fusions has been attributed to decreased rates of warhead proteolysis in the FcLoop context,12 indicating a protective influence conferred by the Fc domain.
Figure 2.
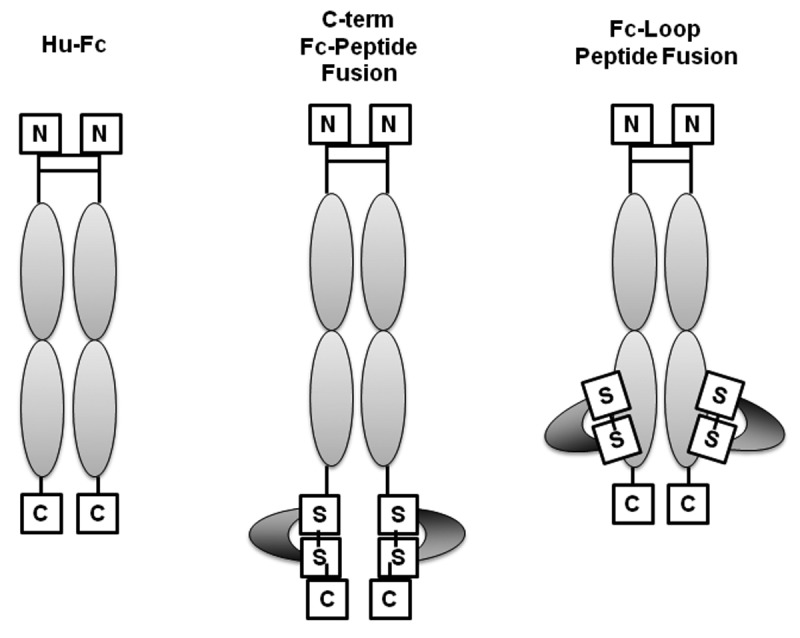
Schematic representation of various peptibody formats. From left to right, Fc, Fc with terminal fusion of the biologically active peptide and Fc with peptide insertion (FcLoop peptibody).
Peptibody Manufacturing
The manufacturing of peptibodies in E. coli consists of the basic unit operations found in processes designed to recover insolubly expressed product from E. coli fermentation cell mass13 and can be divided into three main stages: 1) the cellular expression and isolation of raw starting material, followed by 2) the refolding and purification of the recombinant product, and finally 3) the formulation of the bulk product and the production of the final dosage form. The three primary advantages of E. coli-based expression compared with mammalian cell culture systems are the short time of culture, a high expression level, and the lack of post-translational modifications. For these reasons, peptibodies to date have been expressed for manufacturing purposes only in E. coli.
The first stage of manufacturing E. coli-expressed peptibodies begins with the fermentation unit operations that include the cell culturing, cell harvesting, homogenization of cell paste, followed by the centrifugal isolation and washing of the insoluble inclusion bodies (IB) that contain the denatured recombinant product. Kilogram quantities of cells can be produced from single 10-L fermentations during this operation. The purification stage begins with the solubilization and refolding unit operations to generate the homodimeric peptibodies. The IB slurry is dissolved using a solubilization buffer consisting of a chaotrope with a reducing agent at an alkaline pH. The solubilized inclusion bodies are then diluted with a folding solution of alkaline-buffered urea with stabilizing agents and a defined redox couple. The reaction mixture is then stirred at 5°C to allow for folding and cysteine disulfide formation by air oxidation.
The completed refold is depth-filtered and processed immediately in the next unit operation which, for most peptibodies, is tangential flow filtration where the refold is concentrated to allow for the subsequent differential precipitation and column chromatography operations.
The chromatography unit operations are optimized for each peptibody to provide the best capture of product while optimally clearing proteases, product-related contaminants such as mis-folded product, aggregates and host cell contaminants such as endotoxins, nucleic acids, lipids and proteins. The best liquid formulation is used for the final column pool processing using a tangential flow filtration unit operation to achieve both a concentration to the target value and formulation buffer exchange.
The development of the formulation buffer is a critical component that not only affects the stability of the peptibody but also substantially affects the preclinical development timeline because the production of test material cannot be initiated without knowing the formulation in which to store the final purified bulk. Therefore, formulation development begins at the research stage of the peptibody program. To expedite the development, the initial formulation strategy typically involves determination of a frozen liquid form of the product with additional investigation into a lyophilized form. The overall yield range has been acceptable for manufacturing based on dosage, manufacturing scale limitations and potential market sizes.
Development of Romiplostim, the First Marketed Peptibody
Romiplostim is a peptibody that mimic the activity of thrombopoietin (TPO), a hormone that stimulates the production of platelets.3 Romiplostim potently displaces TPO from its receptor, TPOR (also known as c-MPL) with EC50 value between 5 and 170 pM and stimulates the generation and the maturation of megakaryocytes, the cells giving rise to platelets.14 Romiplostim is composed of two tandem repeats of a TPOR-binding agonist peptide5 separated by eight glycine residues and fused to the carboxy-terminus of a human IgG1 Fc by another five-glycine linker3 (Fig. 1). Preclinical studies in mouse, rat and cynomolgus monkey demonstrated that romiplostim, iv or subcutaneously (sc) delivered, is a potent stimulator of platelet formation. Stimulation of TPOR by recombinant TPO protein had previously validated the pathway in platelet donors and in patients with immune thrombocytopenic purpura (ITP) and chemotherapy-induced thrombocytopenia. While the administration of recombinant TPO led to the expected rise in platelet counts, it was soon recognized that a few patients could develop a neutralizing antibody to recombinant TPO that cross-reacted to endogenous TPO. These patients became thrombocytopenic and dependent on platelet transfusion, thus leading to the abandonment of the clinical development of this first generation of thrombopoietic stimulating agents.15 Since the bioactive peptides comprising romiplostim bear no homology to native TPO, romiplostim was expected to avoid this important problem. The initial single dose iv (0.3–10 μg/kg) or sc (0.1–2 μg/kg) clinical phase I trials in healthy volunteers established romiplostim’s favorable safety profile as well as its ability to elevate platelet counts in a dose dependent manner.10 In patients with chronic ITP, a single sc dose of 1 to 10 μg/kg romiplostim increased the peak platelet count by 1.3 to 14.9 times over the baseline count for a 2 to 3 week period.16 Romiplostim was approved in the US and in the European Union for the treatment of adult ITP patients on the basis of the results of two double blind placebo controlled Phase 3 studies.17 In the US trial, most patients had undergone splenectomy, a standard procedure to limit platelet destruction. The European trial enrolled non-splenectomized patients. 83% of the treated patients, but only 7% of the placebo control patients, were able to increase their platelet level above 50 x 109/L. These responses were durable in 38% of splenectomized patients and 61% of non-splenectomized patients. Romiplostim was generally well-tolerated; the most serious adverse events (observed in 2% of patients) were reticulin deposition in the bone marrow and arterial embolism. Conversely, romiplostim reduced almost by half the risk of serious bleeding events. In a comparison with standards of care, romiplostim provided more than double the rate of platelet response, lowered the incidence of treatment failure and splenectomy, bleeding, and requirement for blood transfusion.18
Romiplostim efficacy and acceptable safety profile in ITP patients have recently been confirmed in an open-label extension study in patients with ITP.19 Romiplostim has a starting dose of 1 μg/kg sc, which is then adjusted weekly by increments of 1 μg/kg until the patient achieves a platelet count above 50 x 109/L. In this study, the serum half-life of romiplostim ranged from 1 to 34 d with a median of 3.5 d. It should be noted that, like TPO, the degradation of romiplostim is partly receptor-mediated; the serum half-life is thus inversely correlated with the platelet count, i.e., patients with high platelet counts have lower serum concentrations. The immunogenicity of romiplostim is evaluated with a sensitive Biacore assay. Any positive sample is then tested in a biological assay for the presence of neutralizing activity. Despite the presence of tandem repeats of non-naturally occurring peptides, the presence of anti-drug antibodies is on par with what is generally observed for therapeutic mAbs. In the 143-patient, 256-week extension study, neutralizing antibodies to romiplostim were transiently detected in one patient. As expected, these antibodies did not cross react with TPO.19 Romiplostim is currently not indicated for use for thrombocytopenia in other disease conditions.
AMG 386, an Anti-Angiogenic Peptibody that Neutralizes Angiopoietins 1 and 2
Angiogenesis, the formation of new blood vessels, is a well-orchestrated process during embryonic development and adult life. This process is co-opted in a number of diseases such as cancer, diabetic retinopathy and age-related macular degeneration. Intervention using large molecules (e.g., bevacizumab, ranibizumab) or small molecule kinase inhibitors (e.g., sorafinib, sunitinib, pazopanib) at the level of the VEGF-VEGFR axis has been validated in the clinic, but is not free of side effects or development of tumor resistance.20 This is particularly the case in solid tumors where angiogenesis is required to provide nutrients and oxygen to the growing tumor and is driven by multiple pathways. The angiopoietins Ang1 and Ang2 and their receptor, the tyrosine kinase, Tie2, contribute to angiogenesis through the regulation of the coordinated processes of vessel destabilization, sprouting, and maturation.21 Ang1 binding to Tie2 induces the autophosphorylation and activation of the receptor. Ang1 is generally viewed as a stabilizing force acting in concert with other signaling molecules to induce smooth muscle cell migration, and to recruit pericytes to the endothelium. By contrast Ang2 expression is markedly increased during vascular remodeling, such as during tumor growth. Ang2 is thought to destabilize vessels and to promote sprouting of new vessels. Ang2 generally acts as an antagonist or partial agonist of Tie2. Both signaling molecules work in concert to build new blood vessels. Peptibodies with dual specificity to Ang1 and Ang2 such as AMG 386, or with single specificity to Ang122 or Ang2,6 were generated by screening phage libraries. These agents potently and selectively inhibited the binding of human Ang1 and Ang2 to Tie2 and displayed similar potency toward the murine orthologs, thus enabling fine dissection of the relative contribution of Ang1 and Ang2 in physiological and tumor angiogenesis in mouse models.6,22,23 Selective inhibition of Ang2 reduced endothelial cell proliferation, thus decreasing the abundance of intratumoral blood vessels. Because of the unopposed activity of Ang1 on Tie2, inhibition of Ang2 alone led to blood vessel normalization, as evidence by increased endothelial tight junctions and pericyte recruitment. While inhibition of Ang1 had little consequence on tumor angiogenesis, the dual inhibition of Ang1 and Ang2 achieved by AMG 386 or by the combination of the peptibodies selective for Ang1 and Ang2 resulted in both reduced number and increased abnormality of blood vessels and was required to achieve the maximal antitumor activity.22,23 The safety, PK, PD and efficacy in solid tumors was first evaluated in a Phase 1 clinical study.9 AMG 386 administered weekly by iv infusion up to 30 mg/kg appeared well-tolerated, with no maximum tolerated dose reached. Interestingly, the side effects typically associated with the blockage of the VEGF pathway24 were not observed for AMG 386. AMG 386 was also well-tolerated in combination with chemotherapy regimens (FOLFOX-4, carboplatin-paclitaxel and docetaxel) and with inhibitors of the VEGF pathway (bevacizumab, motesanib and sorafinib).4,25-27 Serum exposure to AMG 386 increased proportionally with dose and the half-life ranged from 3.1 to 6.3 d. In the first human trial, as well as in a following trial in combination with chemotherapy, no neutralizing anti-drug antibodies were detected.9,27 Significant reduction of tumor blood flow was observed after AMG 386 administration by dynamic contrast-enhanced magnetic resonance imaging. Promising activity as a single agent and in combination led to the initiation of a randomized, placebo-controlled Phase 2 study of AMG 386 in combination with paclitaxel in patients with recurrent ovarian cancer. Two weekly dose levels of AMG 386 at 3 and 10 mg/kg were evaluated. The trial met its primary endpoint and demonstrated a dose-dependent increase in progression-free survival and response for CA-125, an ovarian cancer biomarker.26 Peripheral edema was the most common treatment-emergent adverse event in the 10 mg/kg AMG 386 paclitaxel combination group, and 96% were grade 1 or 2.27 No thromboembolic events and no bowel perforation occurred in AMG 386 treatment groups. AMG 386 is currently being evaluated in three randomized, double-blind Phase 3 trials in women with recurrent ovarian cancer, primary peritoneal cancer or, fallopian tube cancer. In addition to the Phase 3 program, a broad Phase 1 and 2 program is underway in multiple other types of cancer.
AMG 819, Rise and Fall for the First NGF-Neutralizing Peptibody
A considerable body of evidence in preclinical models and in human subjects supports the notion that nerve growth factor (NGF) is an important mediator of pain associated with various inflammatory diseases.28 These data have fueled the drive of multiple biopharmaceutical companies to test NGF-neutralizing agents to investigate benefit in pain associated with conditions such as osteoarthritis, low back pain, diabetes and cancer. In 2003, AMG 819 became the first biological anti-NGF agent to enter a clinical study. AMG 819 was generated by screening a peptide phage-display library for the ability to bind to recombinant human NGF. An in vitro functional assay was then employed to determine the NGF neutralizing ability of these peptides when expressed as fusions with Fc; these peptides were further affinity matured, leading to the identification of several sub-nM NGF-neutralizing peptibodies. AMG 819, consisting of a single 20 amino-acid peptide fused to the carboxy-terminus of IgG1 Fc, binds a conserved domain on NGF mediating the interaction with the NGF receptor TrkA. Not surprisingly, AMG 819 neutralizes human and rat NGF with equivalent potency, thus enabling the preclinical evaluation in rat models of hypersensitivity and allodynia (unpublished data). The efficacy observed in these models prompted the progression of AMG 819 to a Phase 1 clinical study. The median terminal half-life of AMG 819 dosed iv or sc in healthy volunteered was 3.5 d, which was similar to other peptibodies in humans (unpublished data). The immunogenicity profile of AMG 819 proved to be a major impediment to further development. Thirty seven percent of all subjects developed detectable antibodies after a single injection of AMG 819. These antibodies were of the IgG subtype, indicating a T cell dependent response. In silico T-helper epitope prediction highlighted a 14 amino-acid stretch in the carboxy-terminus of the bioactive peptide as a potential promiscuous immunogenic epitope for a broad range of HLA alleles.29 Confirming the prediction, a memory T-cell response against the peptide was detected in antibody-positive subjects, but not in antibody-negative subjects, thus providing clinical validation for the in silico epitope prediction tool and hope that in the future such immunogenicity issues can be eliminated at an early stage of lead selection.
Conclusion
Peptibodies, or peptides fused to an IgG1 Fc moiety were pioneered at Amgen and have been successful in providing biological agents endowed with potency, PK characteristics, and manufacturability profiles warranting clinical evaluation. The peptibody format was found to be flexible, addressing a wide range of pharmacological design goals. The first peptibody, romiplostim (Nplate®), successfully gained marketing approval for the treatment of ITP. It is an agonist of the TPO receptor and stimulates platelet production. AMG 386, currently being evaluated in Phase 3 trials, neutralizes the binding of two soluble ligands, Ang1 and Ang2, to the Tie2 receptor tyrosine kinase and inhibits tumor angiogenesis. Neutralization of a secreted ligand is also a property of AMG 819, an additional peptibody that progressed to human testing.
The benefit of the peptibody format over peptides as a pharmacological agent is 2-fold. First, a peptibody is composed of two to four peptides per molecule, thus providing an increased avidity for the target. Second, peptibodies benefit from an improvement of plasma residency time because of an increase in molecular weight compared with simpler peptides and recycling through the neonatal Fc receptor. In our experience, the peptibody format has also facilitated preclinical evaluation of efficacy and safety, as every lead peptibody was able to cross-react with the rodent ortholog. The completely artificial nature of the bioactive peptide within a peptibody also leads to a reduced risk of generating cross-neutralizing antibodies to endogenous proteins. It should however be noted that, like all biological drugs, peptibodies carry the risk of developing immunogenicity. AMG 819 is an extreme example of a highly immunogenic epitope that has led to termination of its clinical development and to the emergence of screening strategies to prevent such issues in the future. The immunogenicity profile of Nplate® and AMG 386, for which clinical data on a large number of patients are available, is not out of the range of what is observed for therapeutic mAbs. Peptibodies are made from a single chain polypeptide in E. coli. The manufacturing of peptibodies in E. coli is achieved as insoluble products that are expressed in inclusion bodies and subsequently refolded, purified by multiple rounds of differential precipitation and chromatography and finally formulated to the final dosage form. In addition to the high expression level and the faster manufacturing process compared with mammalian cell culture systems, the homogeneity of the final product and the absence of glycosylated forms are important advantages of the production in E. coli.
In conclusion, the peptibody, although rarely explored by the biopharmaceutical industry when compared with the classical mAb, is a versatile format that has now been clinically validated, particularly with Nplate® gaining marketing approval. Additional opportunities of this format include the potential to insert biologically active peptides within Fc-loops or to generate agents with multiple specificities to engage multiple receptors or ligands that may synergistically contribute to a disease process.
Acknowledgments
We are indebted to the following people at Amgen for providing data from their respective project teams and for critically reviewing the manuscript Kenneth Wild, Ping Wei, Jonathan Oliner, Vibha Jawa, Margaret Karow and Lanny Kirsch.
Glossary
Abbreviations:
- Ang 1 and Ang2
angiopoietin 1 and angiopoietin 2
- EMA
European Medicine Agency
- FDA
US Food and Drug Administration
- IB
inclusion bodies
- ITP
immune thrombocytopenic purpura
- NGF
nerve growth factor
- TPO
thrombopoietin
- TPOR
thrombopoietin receptor
- VEGF
Vascular endothelial growth factor
Disclosure of Potential Conflicts of Interest
All the authors are or were Amgen’s employees and/or stockholders.
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/21024
References
- 1.Kontermann RE. Strategies to extend plasma half-lives of recombinant antibodies. BioDrugs. 2009;23:93–109. doi: 10.2165/00063030-200923020-00003. [DOI] [PubMed] [Google Scholar]
- 2.Roopenian DC, Akilesh S. FcRn: the neonatal Fc receptor comes of age. Nat Rev Immunol. 2007;7:715–25. doi: 10.1038/nri2155. [DOI] [PubMed] [Google Scholar]
- 3.Molineux G, Newland A. Development of romiplostim for the treatment of patients with chronic immune thrombocytopenia: from bench to bedside. Br J Haematol. 2010;150:9–20. doi: 10.1111/j.1365-2141.2010.08140.x. [DOI] [PubMed] [Google Scholar]
- 4.Neal J, Wakelee H. AMG-386, a selective angiopoietin-1/-2-neutralizing peptibody for the potential treatment of cancer. Curr Opin Mole Ther; 12:487-95. [PubMed]
- 5.Cwirla SE, Balasubramanian P, Duffin DJ, Wagstrom CR, Gates CM, Singer SC, et al. Peptide agonist of the thrombopoietin receptor as potent as the natural cytokine. Science. 1997;276:1696–9. doi: 10.1126/science.276.5319.1696. [DOI] [PubMed] [Google Scholar]
- 6.Oliner J, Min H, Leal J, Yu D, Rao S, You E, et al. Suppression of angiogenesis and tumor growth by selective inhibition of angiopoietin-2. Cancer Cell. 2004;6:507–16. doi: 10.1016/j.ccr.2004.09.030. [DOI] [PubMed] [Google Scholar]
- 7.Lowman HB. Bacteriophage display and discovery of peptide leads for drug development. Annu Rev Biophys Biomol Struct. 1997;26:401–24. doi: 10.1146/annurev.biophys.26.1.401. [DOI] [PubMed] [Google Scholar]
- 8.Molineux G. The development of romiplostim for patients with immune thrombocytopenia. Ann N Y Acad Sci. 2011;1222:55–63. doi: 10.1111/j.1749-6632.2011.05975.x. [DOI] [PubMed] [Google Scholar]
- 9.Herbst RS, Hong D, Chap L, Kurzrock R, Jackson E, Silverman JM, et al. Safety, pharmacokinetics, and antitumor activity of AMG 386, a selective angiopoietin inhibitor, in adult patients with advanced solid tumors. J Clin Oncol. 2009;27:3557–65. doi: 10.1200/JCO.2008.19.6683. [DOI] [PubMed] [Google Scholar]
- 10.Wang B, Nichol JL, Sullivan JT. Pharmacodynamics and pharmacokinetics of AMG 531, a novel thrombopoietin receptor ligand. Clin Pharmacol Ther. 2004;76:628–38. doi: 10.1016/j.clpt.2004.08.010. [ast] [DOI] [PubMed] [Google Scholar]
- 11.Gegg C, Xiong F, Sitney KC. US7750128: Modified Fc molecules. United States Patent. US United States of America: Amgen Inc., Thousand Oaks, CA, United States of America, 2010.
- 12.Hall MP, Gegg C, Walker K, Spahr C, Ortiz R, Patel V, et al. Ligand-binding mass spectrometry to study biotransformation of fusion protein drugs and guide immunoassay development: strategic approach and application to peptibodies targeting the thrombopoietin receptor. AAPS J. 2010;12:576–85. doi: 10.1208/s12248-010-9218-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Graumann K, Premstaller A. Manufacturing of recombinant therapeutic proteins in microbial systems. Biotechnol J. 2006;1:164–86. doi: 10.1002/biot.200500051. [DOI] [PubMed] [Google Scholar]
- 14.Broudy VC, Lin NL. AMG531 stimulates megakaryopoiesis in vitro by binding to Mpl. Cytokine. 2004;25:52–60. doi: 10.1016/j.cyto.2003.05.001. [DOI] [PubMed] [Google Scholar]
- 15.Basser RL, O’Flaherty E, Green M, Edmonds M, Nichol J, Menchaca DM, et al. Development of pancytopenia with neutralizing antibodies to thrombopoietin after multicycle chemotherapy supported by megakaryocyte growth and development factor. Blood. 2002;99:2599–602. doi: 10.1182/blood.V99.7.2599. [DOI] [PubMed] [Google Scholar]
- 16.Bussel JB, Kuter DJ, George JN, McMillan R, Aledort LM, Conklin GT, et al. AMG 531, a thrombopoiesis-stimulating protein, for chronic ITP. N Engl J Med. 2006;355:1672–81. doi: 10.1056/NEJMoa054626. [DOI] [PubMed] [Google Scholar]
- 17.Kuter DJ, Bussel JB, Lyons RM, Pullarkat V, Gernsheimer TB, Senecal FM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet. 2008;371:395–403. doi: 10.1016/S0140-6736(08)60203-2. [DOI] [PubMed] [Google Scholar]
- 18.Kuter DJ, Rummel M, Boccia R, Macik BG, Pabinger I, Selleslag D, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med. 2010;363:1889–99. doi: 10.1056/NEJMoa1002625. [DOI] [PubMed] [Google Scholar]
- 19.Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood. 2009;113:2161–71. doi: 10.1182/blood-2008-04-150078. [DOI] [PubMed] [Google Scholar]
- 20.Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–91. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- 21.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol. 2009;10:165–77. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 22.Coxon A, Bready J, Min H, Kaufman S, Leal J, Yu D, et al. Context-dependent role of angiopoietin-1 inhibition in the suppression of angiogenesis and tumor growth: implications for AMG 386, an angiopoietin-1/2-neutralizing peptibody. Mol Cancer Ther. 2010;9:2641–51. doi: 10.1158/1535-7163.MCT-10-0213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Falcón BL, Hashizume H, Koumoutsakos P, Chou J, Bready JV, Coxon A, et al. Contrasting actions of selective inhibitors of angiopoietin-1 and angiopoietin-2 on the normalization of tumor blood vessels. Am J Pathol. 2009;175:2159–70. doi: 10.2353/ajpath.2009.090391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Eskens FALM, Verweij J. The clinical toxicity profile of vascular endothelial growth factor (VEGF) and vascular endothelial growth factor receptor (VEGFR) targeting angiogenesis inhibitors; a review. Eur J Cancer. 2006;42:3127–39. doi: 10.1016/j.ejca.2006.09.015. [DOI] [PubMed] [Google Scholar]
- 25.Hong D, Gordon M, Appleman L, Kurzrock R, Sun YN, Rasmussen E, et al. Interim results from a phase 1b study of safety, pharmacokinetics (PK) and tumor response of the angiopoietin1/2-neutralizing peptibody AMG 386 in combination with AMG 706, bevacizumab (B) or sorafenib (S) in advanced solid tumors. Ann Oncol 2008; 19 (Supplement 8): viii154, abstract 462PD. http://annonc.oxfordjournals.org/content/19/suppl_8/viii153.full.pdf
- 26.Karlan BY, Oza AM, Hansen VL, Richardson GE, Provencher D, Ghatage P, et al. Randomized, double-blind, placebo-controlled phase 2 study of AMG 386 combined with weekly paclitaxel in patients with recurrent ovarian carcinoma. J Clin Oncol 2010; 28:15s, (suppl; abstr 5000). www.asco.org/ascov2/Meetings/Abstracts?&vmview=abst_detail_view&confID=74&abstractID=50835
- 27.Vergote IB, Oza AM, Hansen VL, Richardson GE, Provencher D, Ghatage P, et al. A randomized, double-blind, placebo-controlled phase 2 study of AMG 386 plus weekly paclitaxel in patients with advanced ovarian cancer. 35th European Society for Medical Oncology (ESMO) Congress, 2010. [Google Scholar]
- 28.Seidel MF, Herguijuela M, Forkert R, Otten U. Nerve growth factor in rheumatic diseases. Semin Arthritis Rheum. 2010;40:109–26. doi: 10.1016/j.semarthrit.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Koren E, De Groot AS, Jawa V, Beck KD, Boone T, Rivera D, et al. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol. 2007;124:26–32. doi: 10.1016/j.clim.2007.03.544. [DOI] [PubMed] [Google Scholar]