

Abstract
Epigenetic modifications to peripheral white blood cell DNA occur in response to a wide variety of exposures. In prior work, we and others have shown that broad changes in DNA methylation, particularly at the aryl hydrocarbon receptor repressor (AHRR) locus, occur in samples from subjects with long histories of smoking. However, given the large number of epigenetic changes that occur in response to prolonged smoking, the primacy of the response at AHRR and the sensitivity of these changes to low levels of smoking are not known. Therefore, we examined the association of smoking to genome lymphocyte DNA methylation status in a representative sample of 399 African American youths living in the rural South that includes 72 subjects with less than one half-pack year of exposure. Consistent with our prior findings, we found a stepwise effect of smoking on DNA methylation among youth with relatively brief exposure histories at a CpG residue in AHRR (cg05575921) (FDR corrected p values; 3 × 10−7 and 0.09 in the male and female samples, respectively) that was identified in previous studies and at which the effects of smoking were significant, even in those subjects with less than one half pack year exposure. We conclude that AHRR demethylation at cg05575921 in peripheral cells may serve as an early, sensitive biomarker for even low levels of exposure to tobacco smoke, providing a non-self-report alternative for nascent exposure to tobacco smoke. We also suggest that the AHRR/AHR pathway may be functional in the response of peripheral white blood cells to tobacco smoke exposure.
Keywords: aryl hydrocarbon receptor repressor, smoking, biomarker, lymphocytes, DNA methylation, epigenetics
Introduction
Smoking is the single largest potentially preventable cause of morbidity and mortality in the United States. Despite a plethora of public policy prevention measures, approximately 21% of US adults are daily cigarette smokers.1 Unfortunately, once smoking has become behaviorally ingrained, it is extremely difficult to stop. In particular, those individuals who start smoking early are more likely to smoke as adults and twice as less likely to stop.2 Hence, methods to identify nascent smokers could have substantial utility in prevention efforts. Similarly, a biological method to quantify cumulative exposure would provide a useful tool for researchers studying health outcomes linked to smoking.
Smoking has a gradual onset.3,4 The majority of eventual users begin with an irregular pattern of use over extended periods of time that can last several years. The frequency during this initial “experimentation” phase is low, ranging as low as 1 to 2 cigarettes per month.4,5 This periodic puffing can either gradually or rapidly increase until regular usage is achieved. Unfortunately, once a pattern of regular smoking has been achieved, it typically signals addiction and, therefore, increased difficulty in treatment, suggesting that intervening during the experimentation phase is desirable.4
Because the ideal time for intervention may be shortly after experimentation has ensued, a large number of investigators have examined genetic and environmental risk factors for the onset of smoking. Several partially overlapping psychosocial factors have been defined. In particular, studies have shown that family socioeconomic status, parental and peer smoking, psychological distress and family attachment factors play an important role in predicting early-onset of smoking.6 The genetic risk factors have also been extensively examined (for a review see ref. 7). The chromosome 15q11 locus containing rs1051730 is perhaps the best characterized and has unequivocally been identified as a locus containing variability for early onset smoking.8 But the relative risk conveyed by the genetic variation at this site is still relatively low (odds ratio of ~1.2) with the remainder of the genetic variation for early onset smoking identified to date conveying even less relative risk. Although investigation of risk factors has contributed to more effective smoking prevention measures, they do not identify all of the individuals at risk. Nor do any of these factors identify those who have initiated the experimental phase of smoking, i.e., the group that is arguably at highest risk for eventual addition. Therefore, a sensitive index of smoking initiation would have considerable utility for both practical and research applications.
The easiest mechanism through which to identify those who have actually initiated smoking is self-report. Self-report measures with respect to smoking status in situations where duress is not present are both cheap and generally accurate.9,10 However, in certain clinical situations where smoking is viewed as less desirable or in prevention interventions, the reliability of self-report data are often decreased and the use of more sensitive measures is indicated.11,12 Likewise, if intervention were tied to assessment results, under reporting could quickly emerge as a vulnerability of self-reports.
Potentially, the use of biomarkers for smoking status could increase the sensitivity of smoking assessments by identifying individuals who may deny smoking despite nascent smoking exposure that renders them high-risk for subsequent addiction. Smoking may be denied for a variety of reasons, giving rise to investigation of several biological assessment approaches. One of the most commonly used biological assessment measures is level of cotinine in serum or urine. Cotinine is an alkaloid with a serum half-life of 20 h which is both found in tobacco and is a metabolite of nicotine.13 Unfortunately, cotinine assessments are not particularly sensitive to low levels of tobacco use.14 In a recent meta-analysis by Wells and colleagues of 14,554 subjects from the US, 1% and 6% of regular and occasional smokers of European ancestry were misclassified as non-smokers, while 3% and 15% of regular and occasional smokers of non-European ancestry were similarly misclassified using clinical algorithms based on cotinine levels.15 The specificity of cotinine levels may also be a problem with 1 to 3% of self-reported non-smokers being classified as smokers using serum cotinine measures. These considerations suggest that cotinine may not be ideal for the assessment of tobacco smoke exposure during the early experimentation phase that is of great potential interest to prevention researchers. Finally, the assessment of cotinine in biological assessments can be time-consuming and expensive.14 Hence, there is a need to identify additional biomarkers for smoking status.
The assessment of exhaled carbon monoxide is another good biological measure of smoking. Unlike serum cotinine levels, it is both relatively cheap to assay and easy to measure in the field.16 Unfortunately, given the relatively short life of carbon monoxide in the blood, this assessment is also best suited for regular smokers, i.e., individuals who are likely to have smoked in the past 8 to 12 h.17 Exhaled carbon monoxide is much less sensitive in detecting those individuals who smoke only periodically, i.e., nascent smokers. Therefore, the assessment of exhaled carbon monoxide does not fully meet the need for additional biomarkers for smoking when the targets of interest are not yet regular smokers.
In previous communications, we and others have shown that alterations in DNA methylation are associated with smoking status.18-22 In particular, we have demonstrated in two independent samples that differential methylation at the aryl hydrocarbon receptor repressor (AHRR), a known cancer susceptibility gene and a key regulator of the catabolism of xenobiotics,23,24 is associated with smoking status.21 Recently, this line of research was extended in an examination of cord blood, finding lower methylation at cg05575921 associated with higher levels of self-reported or cotinine-based assessment of maternal smoking.25 In this communication, in the hopes of demonstrating the sensitivity of methylation assessments, we again examine the relationship of genome wide DNA methylation to current smoking status—this time using a population of subjects with a more recent onset of smoking.
Results
The clinical characteristics of the 399 subjects included in the study are shown in Table 1. In brief, the subjects were all between the ages of 18 to 20 y old. The vast majority of both male and female subjects were non-smokers. However, even among the smoking subjects, the consumption of cigarettes was relatively light and occasionally intermittent. Only 27 of the female subjects of the 37 subjects with a history of smoking reported smoking at the time of the last interview. Furthermore, of those 27 females who reported current smoking at the last interview, only 4 of those subjects smoked more than 3 cigarettes per day with no female subject having greater than two pack years of total smoking exposure. In contrast, the males tended to be more consistent consumers with 62 of the 70 male subjects with a substantive history of smoking reporting current consumption of cigarettes. In addition, there were 15 male subjects with at least five pack years of smoking history. However, their daily cigarette consumption was also relatively low with a modal consumption of 3 cigarettes per day.
Table 1. Clinical and demographic characteristics of African American subjects.
| Male | Female | |
|---|---|---|
|
N |
181 |
218 |
|
Age |
19.2 ± 0.7 |
19.3 ± 0.7 |
|
Smoking Status |
|
|
| Non-Smoker |
111 |
181 |
| Less than ½ Pack Years |
42 |
30 |
| ≥ 1/2 and < 2 Pack Years |
12 |
7 |
| ≥ 2 and < 5 Pack Years |
1 |
0 |
| ≥ 5 Pack Years | 15 | 0 |
The lymphocyte DNA methylation status of each of the samples was determined using the HumanMethylation 450 Beadchip. Not unexpectedly, the average genome wide methylation (methylated CpG/total CpG) for females was greater than that for males (males 0.510 ± 0.011 vs. females 0.514 ± 0.011, p < 0.003). The correlation of the eight control replicate DNA samples was excellent with an average correlation of 0.995 with no evidence of bisulfite conversion batch effects.
In this population of young adults, there was no effect of smoking on average methylation. The average methylation of the male non-smokers was 50.97% while that of the male smokers was 50.94%. The average methylation of the female non-smokers was 51.42% while that of the female smokers was 51.22%.
After inspection of the distribution of smoking consumption variable, we divided the male subjects into three distinct into three classes: (1) non-smokers, (2) those with < 1/2 pack year exposure (n = 42) and (3) those with > 1/2 pack year of exposure (n = 28). We then conducted genome wide regression analysis with respect to smoking status. The list of the 30 most highly associated probes is given in Table 2 while a Q-Q plot of the analyses is given in Figure S1. Using a three-class regression paradigm, only methylation at cg05575921, a probe localizing to intron 3 of the AHRR was significantly associated with smoking status (see Fig. 1; FDR corrected p value, 2.7 × 10−7). Although no other probe was significantly associated after correction for genome wide comparison, it is interesting to note that the probe immediately adjacent to cg05575921, cg23576855, which is a CpG residue only 79 base pairs more 3′ in the intron, was the 3rd ranked probe with a nominal p value of 4.9 × 10−5 (FDR corrected p < 0.80).
Table 2. The 30 most significantly associated probes in DNA from male subjects.
| |
|
|
|
Average β values for each usage class |
||||
|---|---|---|---|---|---|---|---|---|
| Probe ID | GENE | Placement | Island Status | None | Light | Heavy | T-test | Corrected p value |
| cg05575921 |
AHRR |
Body |
N Shore |
0.878 |
0.829 |
0.772 |
5.65 E-13 |
2.74E-07 |
| cg17268033 |
TUBB8 |
TSS200 |
S Shore |
0.529 |
0.557 |
0.581 |
5.14 E-07 |
0.12 |
| cg23576855 |
AHRR |
Body |
N Shore |
0.719 |
0.675 |
0.652 |
4.86 E-06 |
0.55 |
| cg11620896 |
MUS81 |
TSS1500 |
Island |
0.057 |
0.053 |
0.051 |
6.94 E-06 |
0.55 |
| cg01060358 |
RNASEH2B |
TSS200 |
Island |
0.155 |
0.148 |
0.145 |
7.05 E-06 |
0.55 |
| cg17816357 |
|
|
|
0.427 |
0.452 |
0.476 |
8.86 E-06 |
0.55 |
| cg03716937 |
ADAM2 |
TSS200 |
|
0.851 |
0.863 |
0.867 |
9.02 E-06 |
0.55 |
| cg06422309 |
ADARB2 |
Body |
|
0.636 |
0.659 |
0.687 |
1.03 E-05 |
0.55 |
| cg13880034 |
GNAQ |
Body |
|
0.730 |
0.736 |
0.741 |
1.23 E-05 |
0.55 |
| cg19162075 |
DPP6 |
Body |
|
0.718 |
0.725 |
0.747 |
1.60 E-05 |
0.55 |
| cg05105069 |
TCEAL7 |
5′UTR |
|
0.558 |
0.593 |
0.590 |
1.81 E-05 |
0.55 |
| cg09754550 |
|
|
S Shore |
0.806 |
0.813 |
0.832 |
2.07 E-05 |
0.55 |
| cg01588546 |
GPX4 |
Body |
Island |
0.067 |
0.064 |
0.062 |
2.19 E-05 |
0.55 |
| cg14839809 |
|
|
|
0.934 |
0.935 |
0.938 |
2.21 E-05 |
0.55 |
| cg07777270 |
NR2F1 |
Body |
N Shore |
0.554 |
0.570 |
0.583 |
2.27 E-05 |
0.55 |
| cg13601565 |
|
|
|
0.779 |
0.788 |
0.809 |
2.30 E-05 |
0.55 |
| cg24996985 |
|
|
|
0.588 |
0.604 |
0.636 |
3.23 E-05 |
0.55 |
| cg07333872 |
|
|
|
0.856 |
0.841 |
0.789 |
3.43 E-05 |
0.55 |
| cg04425458 |
KCNQ3 |
1stExon |
Island |
0.119 |
0.113 |
0.115 |
3.45 E-05 |
0.55 |
| cg05131102 |
|
|
|
0.720 |
0.728 |
0.744 |
3.49 E-05 |
0.55 |
| rs10846239 |
|
|
|
0.769 |
0.826 |
0.921 |
3.60 E-05 |
0.55 |
| cg07590705 |
CHCHD3 |
Body |
Island |
0.069 |
0.072 |
0.079 |
3.67 E-05 |
0.55 |
| cg00462971 |
|
|
N Shore |
0.471 |
0.498 |
0.569 |
3.68 E-05 |
0.55 |
| cg02662576 |
MIR495 |
TSS200 |
|
0.866 |
0.874 |
0.878 |
3.87 E-05 |
0.55 |
| cg09028935 |
TGM6 |
Body |
S Shore |
0.957 |
0.958 |
0.962 |
4.07 E-05 |
0.55 |
| cg06574139 |
|
|
|
0.721 |
0.741 |
0.740 |
4.14 E-05 |
0.55 |
| cg17563789 |
|
|
|
0.825 |
0.831 |
0.838 |
4.26 E-05 |
0.55 |
| cg08525989 |
NECAB3 |
TSS200 |
Island |
0.060 |
0.054 |
0.055 |
4.37 E-05 |
0.55 |
| cg19318450 |
TMEM50B |
TSS200 |
Island |
0.092 |
0.085 |
0.086 |
4.43 E-05 |
0.55 |
| cg04318534 | PLXDC2 | Body | Island | 0.098 | 0.091 | 0.089 | 4.73 E-05 | 0.55 |
All average methylation values are non-log transformed β-values. Island status refers to the position of the probe relative to the island. Classes include: (1) Island, (2) N (north) shore, (3) S (south) shore, (4) N (north shelf), (5) S (south) shelf and (6) blank denoting that the probe does not map to an island.
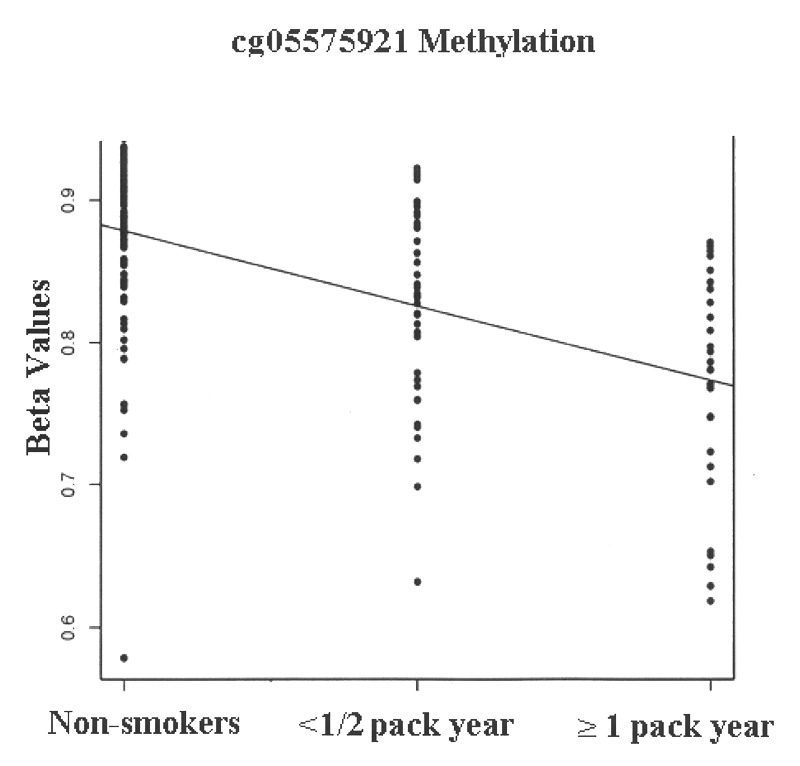
Figure 1. The methylation at the AHRR CpG residue interrogated by cg05575921 as a function of smoking history in male subjects. Group sizes: non-smokers (n = 111), smokers with less than 1 pack year (n = 42) and smokers with at least one pack year (n = 28).
Because only one female had greater than 1 pack year of smoking consumption, we analyzed the methylation data from the female subjects using a two class paradigm. The results of those analyses are shown in Table 3 and Figure 2. This time, cg05575921 was the 2nd ranked probe (p < 4.5 × 10−7, FDR corrected value p < 0.09). But in this instance, after genome wide correction, neither it nor the slightly more highly associated probe cg11685249 was significant at a p of 0.05 after FDR correction. Interestingly, the AHRR probe cg23576855, also identified above, was once again one of the more highly ranked probes (371st) with a nominal p value of < 0.002.
Table 3. The 30 most significantly associated probes in DNA from female subjects.
| |
|
|
|
Average β values for each usage class |
|||
|---|---|---|---|---|---|---|---|
| Probe ID | GENE | Placement | Island status | None | Light | T-test | Corrected p value |
| cg11685249 |
TSSK1B |
TSS1500 |
|
0.817 |
0.740 |
2.85E-07 |
0.09 |
| cg05575921 |
AHRR |
Body |
N Shore |
0.904 |
0.864 |
4.51E-07 |
0.09 |
| cg06961873 |
TMEM57 |
3′UTR |
|
0.716 |
0.510 |
2.35E-06 |
0.32 |
| cg12500300 |
|
|
|
0.719 |
0.560 |
3.18E-06 |
0.33 |
| cg01038172 |
|
|
Island |
0.709 |
0.667 |
4.32E-05 |
0.81 |
| cg21085686 |
|
|
N Shelf |
0.761 |
0.721 |
4.74E-05 |
0.81 |
| cg06875704 |
MIR510 |
TSS1500 |
|
0.868 |
0.801 |
4.89E-05 |
0.81 |
| cg22159835 |
NRXN2 |
Body |
|
0.766 |
0.722 |
4.95E-05 |
0.81 |
| cg06768670 |
|
|
|
0.843 |
0.824 |
5.12E-05 |
0.81 |
| cg14611816 |
BAIAP2 |
Body |
|
0.511 |
0.460 |
6.73E-05 |
0.81 |
| cg16879197 |
C4orf50 |
Body |
|
0.808 |
0.734 |
6.99E-05 |
0.81 |
| cg10328548 |
SS18L1 |
Body |
S Shore |
0.897 |
0.825 |
7.04E-05 |
0.81 |
| cg06578276 |
BAIAP2 |
Body |
|
0.514 |
0.463 |
7.96E-05 |
0.81 |
| cg14696820 |
LCE1A |
1stExon |
|
0.651 |
0.614 |
8.54E-05 |
0.81 |
| cg06619428 |
LCE2B |
3′UTR |
|
0.671 |
0.629 |
9.38E-05 |
0.81 |
| cg03640465 |
SLC2A9 |
TSS1500 |
|
0.866 |
0.823 |
0.0002 |
0.81 |
| cg01177709 |
COMMD8 |
Body |
N Shore |
0.514 |
0.471 |
0.0002 |
0.81 |
| cg08276328 |
MAGEC1 |
TSS1500 |
|
0.698 |
0.675 |
0.0002 |
0.81 |
| cg00848461 |
PRELID1 |
Body |
S Shore |
0.690 |
0.632 |
0.0002 |
0.81 |
| cg23268879 |
TNRC18 |
Body |
N Shore |
0.885 |
0.866 |
0.0002 |
0.81 |
| cg06587435 |
|
|
N Shore |
0.761 |
0.696 |
0.0002 |
0.81 |
| cg02716826 |
SUGT1P1 |
Body |
N Shore |
0.399 |
0.363 |
0.0002 |
0.81 |
| cg11124142 |
LOC100129055 |
TSS1500 |
|
0.727 |
0.699 |
0.0002 |
0.81 |
| cg13632983 |
|
|
S Shelf |
0.689 |
0.658 |
0.0002 |
0.81 |
| cg27348515 |
ARMCX2 |
TSS200 |
|
0.331 |
0.359 |
0.0002 |
0.81 |
| cg09083945 |
BAIAP2 |
Body |
|
0.401 |
0.363 |
0.0002 |
0.81 |
| cg12873476 |
|
|
S Shore |
0.760 |
0.730 |
0.0002 |
0.81 |
| cg10978503 |
CNR2 |
3′UTR |
|
0.804 |
0.767 |
0.0002 |
0.81 |
| cg00294152 |
|
|
|
0.686 |
0.637 |
0.0002 |
0.81 |
| cg21854924 | MIR510 | TSS200 | 0.548 | 0.464 | 0.0002 | 0.81 | |
All average methylation values are non-log transformed β-values. Island status refers to the position of the probe relative to the island. Classes include: (1) Island, (2) N (north) shore, (3) S (south) shore, (4) N (north shelf), (5) S (south) shelf and (6) blank denoting that the probe does not map to an island.

Figure 2. The relationship between methylation at Cg05575921 and lifetime consumption for the female subjects. Group size: non-smoker, n = 181; smokers, n = 37. Although the methylation is expressed here as untransformed B-value, all calculations were performed using log transformed values.
In prior communications, we have shown that genetic variation near a given residue may have effects on the degree of methylation.26 To determine whether this was true for this locus as well, we examined the relationship of two single nucleotide polymorphisms, rs6869832 and rs6894195, flanking the CpG residue recognized by cg05575921 to average methylation. Consistent with the data provided on the UCSC Genome Browser,27 the two polymorphisms were relatively uninformative (MAF 0.0196), with the average methylation of the 15 heterozygous individuals (AG, 15 of 383 successfully genotyped) at rs6869832 not differing from the average methylation of the 368 GG subjects (0.887 ± 0.051 vs. 0.876 ± 0.063, p < 0.53)
Discussion
In this group of 399 young adults, we replicate prior findings by ourselves and others that methylation at AHRR is associated with smoking status21,25 and demonstrate that these changes in methylation are significant even among subjects reporting a relatively restrained history of smoking. The current findings also confirm prior reports that induction of the xenobiotic pathway in lymphocytes may be an important portion of the extra hepatic response to smoking.28 The induction of this nuclear receptor mediated pathway is initiated by the sequential binding of dioxin-like compounds to AHR, the complexing of AHR to its cognate partner, the AhR nuclear translocator (ARNT), and finally the binding of the AHR/ARNT dimer to a six base pair xenobiotic responsive elements (XRE) in the promoter regions of xenobiotic metabolism related genes.29 AHRR functions as a competitive antagonist of AHR stimulation of this pathway by competing with AHR for binding to the AhNT and also serves as a tumor suppressor by suppressing the pro-tumorigenic activities of unrestrained AHR activity.23 Exposure of bovine lymphocytes to low levels dioxin-like compounds has been demonstrated to affect transcription of AHR, AHRR and downstream members of the xenobiotic pathway.28 Since cigarette smoke contains low levels of dioxin like compounds,29 the current findings are highly plausible and suggest that the intron 3 region corresponding to cg0557921 may be very important in regulating the strength of that metabolic response.
The current studies also indicate that the extent of AHRR methylation may be a potential biomarker for the initiation of smoking. AHRR has five advantages as a potential biomarker for investigations of smoking. First, as demonstrated in the current investigation, it appears to be sensitive to even relatively low levels of smoking. This may make it useful in studies of nascent smokers, as in the current study, and also in studies of smoke exposure where smoking exposure may be variable and more difficult to assess—such as those of second hand smoke. Second, AHRR methylation may have a dose-response curve that provides a window on cumulative exposure. Although additional work is necessary to establish the precise shape of the dose-response curve and its independence from recent smoking, the development of a sensitive index of cumulative exposure to smoking would be a significant step forward both because cumulative exposure may be less reliably indexed by self-report recent consumption and because, as noted above, alternative biomarkers provide an index only of relatively recent smoking behavior. Accordingly, to the extent that AHRR provides a useful metric for cumulative exposure effects this should help increase precision of current measurement of computing pack years and make it less dependent on retrospective recall. Third, prior work suggests good correspondence between peripheral assessment of AHRR methylation and assessments of alveolar macrophages. Correspondence across tissues suggests the potential for a relatively accessible peripheral measure (methylation of AHRR in white blood cells) to provide a window on methylation changes occurring in other tissues of interest (e.g., changes in alveolar tissue), but at much reduced cost. Fourth, because AHRR has a role in moderating the detoxification of smoking related carcinogens,23 assessment of AHRR may be useful for future studies of addiction mechanisms as well as susceptibility to a range of adverse health outcomes secondary to smoking. In this regard, identification of predictors of variability in AHRR response to smoking will be useful and will also serve to enhance the utility of AHRR methylation as a biomarker of smoking exposure.
Despite the several potential advantages of AHRR as a potential biomarker for nascent smoking behavior, it is important to realize that methodological shortcomings in the current study may have underestimated the relationship between changes in DNA methylation and smoking status. First, our method of smoking assessment used data from the most recent month. Since the use of cigarettes by smokers tend to escalate over time, the use of the past month measure as the average use over the past year probably overestimates the number of cigarettes smoked. Second, our primary measure was based on total cigarette consumption. Since the pathway to daily smoking typically pursues an indolent course, many of individuals with smoking histories may have not smoked in the past month. Because we have previously demonstrated that smoking induced methylation signatures may change after cessation of smoking,26 it may well be that by the time phlebotomy was performed, their methylation at this locus may have reverted to the mean. Therefore, in future studies it will be important to more exactly understand the temporal relationship between cigarette consumption and changes in DNA methylation. When considering these results, it is also important to note that all the subjects in the study were of African American ancestry and that smoking histories were not verified by independent measures. So, results should be replicated with other ethnic groups and using other biological measure of exposure to further validate the current findings. Finally, our analyses did not take into account the possibility of exposure of the subjects to other forms of toxin exposures such as second hand smoke. If the observed changes in methylation are due to induction of the AHR pathway by the dioxins contained within tobacco smoke, in order to produce the most sensitive results, it will be important to more completely assess exposure to other sources of toxins and include these measures in any future epigenetic analyses.
In contrast for prior studies at monoamine oxidase A,26 we did not find any relationship between genotype and methylation. However, the two SNPs assayed in this study, rs6869832 and rs6894195, are relatively uninformative in this population. But according to the dbSNP database their minor allele frequency in other populations is substantially higher (0.09).30 Hence, examination in other populations will be important. It is also important to note that there are a number of CpG residues in the area interrogated by cg0557921 and that methylation status at these other residues may be even more predictive of smoking status and more associated with any biological response at this locus. This is particularly relevant because, inspection of the area near the probe using the UCSC Genome Browser27 and data from the ENCODE database31 shows that the region identified by cg0557921 is enriched for motifs associated with gene transcription including H3K27 acetylation, DNAase I hypersensitivity, and transcription factor binding. Furthermore, review of the AceView database reveals that the gene has five probable alternative promoters that regulate the production of seven alternatively spliced mRNAs, at least five partially confirmed protein isoforms.32 Taken together with the current findings, these additional data suggest that differential methylation in the region identified by cg0557921 may be integral to the initial response to cigarette smoke exposure and demonstrates the need for further studies to examine the relationship of gene methylation to AHRR transcription and translation.
In summary, we replicate prior findings that methylation at AHRR is associated with smoking status. We identify methylation changes occurring early in the course of smoking exposure, and show evidence of a stepwise response to level of cumulative exposure. Accordingly, we suggest that further studies to examine the importance of this pathway in the detoxification of smoking carcinogens and its utility as a biomarker for low levels of tobacco smoke exposure are needed.
Materials and Methods
The 399 subjects in this study are drawn from the Strong African-American Healthy Adults project, which is a continuation of a longitudinal study of young African-Americans and their primary care caregivers.33 The young adults in this study were originally recruited in 2000 when they were approximately 11 to 12 y old. Each of the subjects and their primary caregivers gave consent for participation the study. All protocols and procedures were approved by the University of Georgia institutional review board.
The clinical data in the study is derived from 8 consecutive yearly computerized assessments of health behaviors including cigarette consumption under the supervision of a trained research assistant. At each wave of data collection, subjects were asked “In the past month, how often did you smoke cigarettes?” The number of cigarettes given in reply was used as that year’s estimated average daily consumption with that number being divided by 20 to give the number of pack years. A positive response at any time point from a subject resulted in the categorization of that subject as a smoker.
During the last wave of data collection, individuals were also phlebotomized to provide blood for the current study. Mononuclear (lymphocyte) cell pellets were prepared from the samples by standard Ficoll gradient centrifugation. DNA was prepared from the cell pellets using a Qiagen QIAamp DNA Minikit according to manufacturer's instructions and stored at -20°C until use.
Sequence variation at rs6869832 and rs6894195 was determined by first amplifying the loci using reagents and directions provided by KB Biosciences. Genotypes were then called using an ABI 7900 HT Genetic Analysis System (Applied Biosystems) and the proprietary allele discrimination software.
Genome wide DNA methylation was assessed using the Illumina HumanMethylation450 Beadchip by the University of Minnesota Genome Center using the protocol specified by the manufacturer as previously described.21 This chip contains 485,577 probes recognizing at least 20,216 transcripts, potential transcripts or CpG islands. Male and female subjects were randomly assigned to 12 sample “slides” with groups of 8 slides being bisulfite converted in a single batch. Eight replicates of the same DNA were also included to monitor for slide to slide and batch bisulfite conversion variability. The resulting data were inspected for complete bisulfite conversion and average β values for each targeted CpG residue determined using the Illumina Genome Studio Methylation Module, Version 3.2. The resulting data was then cleaned to remove unreliable beta values, which were identified by the Genome Studio software as having as not having a less than 0.05 probability of representing real signal were removed from the data set.
These methylation values were log transformed, then analyzed using MethLAB version 1.21.34 Because a portion of the methylation signature is gender dependent and the clinical phenomenology of substance use may differ between the two sexes, the data from male and female subjects were analyzed separately with chip assignment being used as a covariate. Because in previous work we have demonstrated that alcohol use does not significantly affect methylation at the AHRR or other associated loci,21,35 we did not use alcohol use as a covariate. Where indicated, false discovery rate (FDR) correction for genome wide comparisons was applied via the method of Benjamini and Hochberg36 at a rate of 0.05. The comparison of methylation values at rs6869832 was conducted using Student’s t-test.37
Supplementary Material
Acknowledgments
The work in this study was supported by 5R01HD030588-16A1 to G.H.B. and R21DA034457 to R.A.P. Additional support for these studies was derived from the Center for Contextual Genetics and Prevention Science (Grant Number P30 DA027827, GB) funded by the National Institute on Drug Abuse. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. The University of Iowa has filed intellectual property right claims on some of the material related to this manuscript on behalf of
Glossary
Abbreviations:
- AHRR
aryl hydrocarbon receptor repressor
- AHR
aryl hydrocarbon receptor
- FDR
false discovery rate
- MAF
minor allele frequency
Disclosure of Potential Conflicts of Interest
The University of Iowa has filed intellectual property right claims on some of the material related to this manuscript on behalf of Dr Philibert. Drs Beach and Brody do not have any conflicts to disclose.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/epigenetics/article/22520
Footnotes
Previously published online: www.landesbioscience.com/journals/epigenetics/article/22520
References
- 1.Centers for Disease Control and Prevention (CDC) Vital signs: current cigarette smoking among adults aged ≥18 years--United States, 2005-2010. MMWR Morb Mortal Wkly Rep. 2011;60:1207–12. [PubMed] [Google Scholar]
- 2.Khuder SA, Dayal HH, Mutgi AB. Age at smoking onset and its effect on smoking cessation. Addict Behav. 1999;24:673–7. doi: 10.1016/S0306-4603(98)00113-0. [DOI] [PubMed] [Google Scholar]
- 3.Heron J, Hickman M, Macleod J, Munafò MR. Characterizing patterns of smoking initiation in adolescence: comparison of methods for dealing with missing data. Nicotine Tob Res. 2011;13:1266–75. doi: 10.1093/ntr/ntr161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Colder CR, Mehta P, Balanda K, Campbell RT, Mayhew KP, Stanton WR, et al. Identifying trajectories of adolescent smoking: an application of latent growth mixture modeling. Health Psychol. 2001;20:127–35. doi: 10.1037/0278-6133.20.2.127. [DOI] [PubMed] [Google Scholar]
- 5.Chassin L, Presson CC, Sherman SJ, Edwards DA. The natural history of cigarette smoking: predicting young-adult smoking outcomes from adolescent smoking patterns. Health Psychol. 1990;9:701–16. doi: 10.1037/0278-6133.9.6.701. [DOI] [PubMed] [Google Scholar]
- 6.Tyas SL, Pederson LL. Psychosocial factors related to adolescent smoking: a critical review of the literature. Tob Control. 1998;7:409–20. doi: 10.1136/tc.7.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J-C, Kapoor M, Goate AM. The genetics of substance dependence. Annu Rev Genomics Hum Genet. 2012;13:241–61. doi: 10.1146/annurev-genom-090711-163844. [null.] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Saccone SF, Hinrichs AL, Saccone NL, Chase GA, Konvicka K, Madden PA, et al. Cholinergic nicotinic receptor genes implicated in a nicotine dependence association study targeting 348 candidate genes with 3713 SNPs. Hum Mol Genet. 2007;16:36–49. doi: 10.1093/hmg/ddl438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bryant J, Bonevski B, Paul C, Lecathelinais C. Assessing smoking status in disadvantaged populations: is computer administered self report an accurate and acceptable measure? BMC Med Res Methodol. 2011;11:153. doi: 10.1186/1471-2288-11-153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramo DE, Hall SM, Prochaska JJ. Reliability and validity of self-reported smoking in an anonymous online survey with young adults. Health Psychol. 2011;30:693–701. doi: 10.1037/a0023443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.McBride CM, Curry SJ, Lando HA, Pirie PL, Grothaus LC, Nelson JC. Prevention of relapse in women who quit smoking during pregnancy. Am J Public Health. 1999;89:706–11. doi: 10.2105/AJPH.89.5.706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vasankari T, Jousilahti P, Knekt P, Marniemi J, Heistaro S, Lppo K, et al. Serum cotinine predicts bronchial obstruction regardless of self-reported smoking history. Scand J Public Health. 2011;39:547–52. doi: 10.1177/1403494811401474. [DOI] [PubMed] [Google Scholar]
- 13.Dwoskin LP, Teng L, Buxton ST, Crooks PA. (S)-(-)-Cotinine, the major brain metabolite of nicotine, stimulates nicotinic receptors to evoke [3H]dopamine release from rat striatal slices in a calcium-dependent manner. J Pharmacol Exp Ther. 1999;288:905–11. [PubMed] [Google Scholar]
- 14.Florescu A, Ferrence R, Einarson T, Selby P, Soldin O, Koren G. Methods for quantification of exposure to cigarette smoking and environmental tobacco smoke: focus on developmental toxicology. Ther Drug Monit. 2009;31:14–30. doi: 10.1097/FTD.0b013e3181957a3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wells AJA, English PBP, Posner SFS, Wagenknecht LEL, Perez-Stable EJE. Misclassification rates for current smokers misclassified as nonsmokers. Am J Public Health. 1998;88:1503–9. doi: 10.2105/AJPH.88.10.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jarvis MJ, Russell MA, Saloojee Y. Expired air carbon monoxide: a simple breath test of tobacco smoke intake. Br Med J. 1980;281:484–5. doi: 10.1136/bmj.281.6238.484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sandberg A, Sköld CM, Grunewald J, Eklund A, Wheelock ÅM. Assessing recent smoking status by measuring exhaled carbon monoxide levels. PLoS One. 2011;6:e28864. doi: 10.1371/journal.pone.0028864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Launay J-M, Del Pino M, Chironi G, Callebert J, Peoc’h K, Mégnien JL, et al. Smoking induces long-lasting effects through a monoamine-oxidase epigenetic regulation. PLoS One. 2009;4:e7959. doi: 10.1371/journal.pone.0007959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Philibert RA, Gunter TD, Beach SR, Brody GH, Madan A. MAOA methylation is associated with nicotine and alcohol dependence in women. Am J Med Genet B Neuropsychiatr Genet. 2008;147B:565–70. doi: 10.1002/ajmg.b.30778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Philibert RA, Sears RA, Powers LS, Nash E, Bair T, Gerke AK, et al. Coordinated DNA methylation and gene expression changes in smoker alveolar macrophages: specific effects on VEGF receptor 1 expression. J Leukoc Biol. 2012;92:621–31. doi: 10.1189/jlb.1211632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Monick MM, Beach SR, Plume JT, Sears R, Gerrard M, Brody GH, et al. Coordinated Changes in AHRR Methylation in Lymphoblasts and Pulmonary Macrophages from Smokers American Journal of Medical Genetics. Part B Neuropsychiatric Genetics. 2012;159:141–51. doi: 10.1002/ajmg.b.32021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Breitling LP, Yang R, Korn B, Burwinkel B, Brenner H. Tobacco-smoking-related differential DNA methylation: 27K discovery and replication. Am J Hum Genet. 2011;88:450–7. doi: 10.1016/j.ajhg.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zudaire E, Cuesta N, Murty V, Woodson K, Adams L, Gonzalez N, et al. The aryl hydrocarbon receptor repressor is a putative tumor suppressor gene in multiple human cancers. J Clin Invest. 2008;118:640–50. doi: 10.1172/JCI30024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Opitz CA, Litzenburger UM, Sahm F, Ott M, Tritschler I, Trump S, et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature. 2011;478:197–203. doi: 10.1038/nature10491. [DOI] [PubMed] [Google Scholar]
- 25.Joubert BR, Håberg SE, Nilsen RM, Wang X, Vollset SE, Murphy SK, et al. 450K Epigenome-Wide Scan Identifies Differential DNA Methylation in Newborns Related to Maternal Smoking during Pregnancy. Environ Health Perspect. 2012;120:1425–31. doi: 10.1289/ehp.1205412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Philibert RA, Beach SR, Gunter TD, Brody GH, Madan A, Gerrard M. The effect of smoking on MAOA promoter methylation in DNA prepared from lymphoblasts and whole blood. Am J Med Genet B Neuropsychiatr Genet. 2010;153B:619–28. doi: 10.1002/ajmg.b.31031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, et al. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Girolami F, Spalenza V, Carletti M, Perona G, Sacchi P, Rasero R, et al. Gene expression and inducibility of the aryl hydrocarbon receptor-dependent pathway in cultured bovine blood lymphocytes. Toxicol Lett. 2011;206:204–9. doi: 10.1016/j.toxlet.2011.07.014. [DOI] [PubMed] [Google Scholar]
- 29.Kasai A, Hiramatsu N, Hayakawa K, Yao J, Maeda S, Kitamura M. High levels of dioxin-like potential in cigarette smoke evidenced by in vitro and in vivo biosensing. Cancer Res. 2006;66:7143–50. doi: 10.1158/0008-5472.CAN-05-4541. [DOI] [PubMed] [Google Scholar]
- 30.Sherry ST, Ward M-H, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11. doi: 10.1093/nar/29.1.308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rosenbloom KR, Dreszer TR, Pheasant M, Barber GP, Meyer LR, Pohl A, et al. ENCODE whole-genome data in the UCSC Genome Browser. Nucleic Acids Res. 2010;38(Database issue):D620–5. doi: 10.1093/nar/gkp961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thierry-Mieg D, Thierry-Mieg J. AceView: a comprehensive cDNA-supported gene and transcripts annotation. Genome Biol. 2006;7(Suppl 1):S12–, 1-14. doi: 10.1186/gb-2006-7-s1-s12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brody GH, Murry VM, Kogan SM, Gerrard M, Gibbons FX, Molgaard V, et al. Journal of Consulting and Clinical Psychology The Strong African American Families Program: a cluster-randomized prevention trial of long-term effects and a mediational model. J Consult Clin Psychol. 2006;74:356–66. doi: 10.1037/0022-006X.74.2.356. [DOI] [PubMed] [Google Scholar]
- 34.Kilaru V, Barfield R, Schroeder JW, Smith AK, Conneely KN. MethLAB: A GUIpackage for the analysis of array-based DNA methylation data. Epigenetics. 2012 doi: 10.4161/epi.7.3.19284. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Philibert RA, Plume JM, Gibbons FX, Brody GH, Beach SR. The impact of recent alcohol use on genome wide DNA methylation signatures. Front Genet. 2012;3:54. doi: 10.3389/fgene.2012.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benjamini Y, Hochberg Y. Controlling the false discovery rate: A practical and powerful approach to multiple testing. J R Stat Soc, B. 1995;57:289–300. [Google Scholar]
- 37.Fleiss JL. Statistical Methods for Rates and Proportions. New York, NY: John Wiley & Sons Inc, 1981. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.