

Abstract
Phage display library technology is a common method to produce human antibodies. In this technique, the immunoglobulin variable regions are displayed in a bacteriophage in a way that each filamentous virus displays the product of a single antibody gene on its surface. From the collection of different phages, it is possible to isolate the virus that recognizes specific targets. The most common form in which to display antibody variable regions in the phage is the single chain variable fragment format (scFv), which requires assembly of the heavy and light immunoglobulin variable regions in a single gene.
In this work, we describe a simple and efficient method for the assembly of immunoglobulin heavy and light chain variable regions in a scFv format. This procedure involves a two-step reaction: (1) DNA amplification to produce the single strand form of the heavy or light chain gene required for the fusion; and (2) mixture of both single strand products followed by an assembly reaction to construct a complete scFv gene. Using this method, we produced 6-fold more scFv encoding DNA than the commonly used splicing by overlap extension PCR (SOE-PCR) approach. The scFv gene produced by this method also proved to be efficient in generating a diverse scFv phage display library. From this scFv library, we obtained phages that bound several non-related antigens, including recombinant proteins and rotavirus particles.
Keywords: SOE-PCR, independent strand amplification, phage display library, scFv assembly
Introduction
Antibodies are the most common biological reagents used in pharmaceutical and medical research, and they are used to treat human pathologies, including cancer, autoimmunity and infectious diseases.1-6 Mouse monoclonal antibodies (mAbs) were used as therapeutic agents, but the immune reaction against the murine protein,7-9 which causes rapid elimination of the mAb and the possibility of anaphylaxis reaction, encouraged scientists to reduce the most immunogenic murine regions of the antibody structure using recombinant DNA technology. Using this approach, murine immunoglobulin regions were replaced by the corresponding human sequences to produce first chimeric antibodies, and then humanized antibodies.1,10 More recently, transgenic mouse11 and phage display technologies have been used to obtain human mAbs.12,13 The phage display technology presents several advantages comparing to the classical mAb production system, including (1) the avoidance of animal use; (2) the possibility to improve antibody properties using mutagenesis14-18 and (3) the combination of different immunoglobulin variable domains to produce engineered antibodies.19-21
Phage display technology is a highly flexible method for the isolation of a single gene product with specific binding properties.22 Therefore, it could be easily combined with antibody recombinant technology to produce a very effective method to isolate human antibodies against specific targets.23,24 Using this methodology, the immunoglobulin variable regions are fused to a structural phage protein in such a way that each filamentous virus displays the product of a single antibody coding sequence on its surface. This allows the production of a wide collection of phages that are able to interact with different antigens. Two types of these phage display libraries can be produced: (1) a synthetic human variable gene library in which complementary determining regions are produced synthetically25-27 and (2) a naïve library derived from natural non-immunized human in which the complementarity-determining regions (CDRs) are obtained from rearranged genes of B lymphocytes.28-30
To display antibody variable regions in the phage, several forms have been described, with the antigen-binding fragment (Fab) and single-chain variable fragment (scFv) arrangement the most commonly used. The Fab form is composed of two chains, that must be assembled in the same phage, while scFv is a single chain in which the immunoglobulin variable heavy and light chain regions are fused using a flexible linker. The latter form has been widely used because it is more stable and better tolerated by bacteria.23
A critical step in the scFv library production process is the assembly of heavy and light chain variable regions in the same molecule. For this purpose, different strategies have been applied, including laborious methods such as independent cloning of immunoglobulin variable regions followed by the assembly of the scFv gene by cloning methods or the use of recombination systems involving bacteria carrying the immunoglobulin heavy-chain variable region (VHR) and a phage carrying the immunoglobulin light-chain variable region (VLR).31 The most common technique is the splicing by overlap extension PCR (SOE-PCR), in which VHR and VLR are amplified by PCR with primers that share an overlapping region to create a linker region by combining both variable region amplifications. Both PCR products are then mixed and incubated with external primers to produce a scFv gene.32,33
We report here that we modified the SOE-PCR to produce a simple and highly efficient method to combine antibody heavy and light chain variable regions in a scFv format by using a single strand DNA amplification strategy. In the first step, each immunoglobulin variable region is amplified independently using a single primer to produce a single strand DNA. Next, both variable regions are mixed and cycled to produce a full-length scFv gene. We used this method to produce a diverse scFv phage display library. In addition, this library was used to obtain phages capable of interacting with different non-related antigens.
Results
Amplification of Immunoglobulin Variable Regions and scFv Gene Assembly
Total RNA was obtained from peripheral blood mononuclear cells (PBMC) of 8 healthy non-immunized donors. cDNA was prepared using oligo-dT and employed as a template to amplify the immunoglobulin rearranged variable genes using the primers detailed in Table 1. These primers were designed to allow the amplification of the entire repertoire of immunoglobulin heavy-chain variable region genes described in VBASE.34 The VLR reverse primer and all VHR forward primers included an artificial region of overlap that allows the formation of a flexible segment corresponding to the linker (Gly4Ser)3. In order to avoid a biased amplification, each cDNA donor was amplified independently with each primer set. Figure 1 shows amplification products from a single donor using all different primer combinations. As expected, the PCR products using the VIgHFor6 primer produced less DNA due to an interaction with only a subset of the IGHV 2 family. The PCR products were pooled and further used for scFv assembly assays.
Table 1. Primers used to amplify the immunoglobulin rearranged variable genes.
| Name | Sequence | Chain | Ig variable family a |
|---|---|---|---|
|
VIgGHFor01 |
CTCGTCCTCTGGTGGCGGTGGCTCGGGCGGAGGTGGGSAGGTNCASCTGGTGSAGTCTGG |
Heavy |
IGHV 1,3,5 and 7 |
|
VIgGHFor02 |
CTCGTCCTCTGGTGGCGGTGGCTCGGGCGGAGGTGGGSARGTGCAGCTGGTGSAGTCTGG |
Heavy |
IGHV 1,3 and 5 |
|
VIgGHFor03 |
CTCGTCCTCTGGTGGCGGTGGCTCGGGCGGAGGTGGGSAGGTBCAGCTGGTRCAGTCTGG |
Heavy |
IGHV 1,3 and 7 |
|
VIgGHFor04 |
CTCGTCCTCTGGTGGCGGTGGCTCGGGCGGAGGTGGGCAGGTGCAGCTGCAGGAGTCGGG |
Heavy |
IGHV 4 |
|
VIgGHFor05 |
CTCGTCCTCTGGTGGCGGTGGCTCGGGCGGAGGTGGGCARGTVCAGCTGSWGSAGTCTGG |
Heavy |
IGHV 1,4 and 6 |
|
VIgGHFor06 |
CTCGTCCTCTGGTGGCGGTGGCTCGGGCGGAGGTGGGCAGRTCACCTTGARGGAGTCTGG |
Heavy |
IGHV 2 |
|
IgGrevNheI |
TTTTTTTGCTAGCGACCGATGGGCCCTTGGTGGARGC |
Heavy |
IgG. |
|
VIgKFor01 |
TTTTTGAATTCGRWRTTGTGATGACTCAGTCTCC |
Light |
All kappa |
| CIgKRev01 linker | CACCGCCACCAGAGGACGAGGATCTAGAGGAACCACCTGCAGCCACAGTTCGTTTG | Light | All kappa |
a Source VBase (http://vbase.mrc-cpe.cam.ac.uk/)
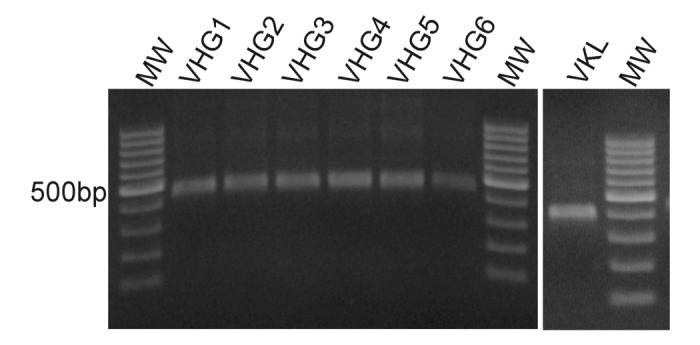
Figure 1. Heavy and Light chains variable region amplification. Total RNA of 8 healthy donors was extracted from peripheral blood mononuclear cells and cDNA was produced using oligo-dT. The cDNA was used to amplify heavy and light chains variable regions by PCR using the primers from Table 1. The figure shows representative PCR products from a single donor, resolved by electrophoresis on a 1% w/v agarose gel. MW: 100bp DNA ladder.
We hypothesized that the presence of cDNA strands in the PCR products competed with the single stranded DNA required for the scFv assembly, thereby decreasing the efficiency of SOE-PCR. To overcome this problem we decided to separate this reaction into two steps. First, we enriched the DNA strand required for scFv fusion by a separate single strand amplification using external primers for the heavy and light chain regions. Then both single strand DNA products were mixed and the assembly of the scFv gene took place by repeated cycles of denaturation annealing and extension. The different strategies used in this work for the assembly of immunoglobulin variable regions are schematically shown in Figure 2.

Figure 2. Schematic representation of different scFv gene assembly methods. Heavy and light chain variable regions were amplified by PCR and used for the assembly reaction. For the splicing by overlap extension PCR (SOE-PCR), each amplicon was mixed with VIgKFor01 (VLK) and IgGrevNheI primers and amplified for 30 cycles. In the case of the Independent Strand Amplification PCR (ISA-PCR), light chain PCR product was incubated with VLK primer while the heavy chain PCR product was incubated with IgGrevNheI primer and 30 cycles of PCR were performed. Both single strand products were mixed and the assembly reaction performed. Different numbers of denaturation annealing and extension cycles were tested during the assembly reaction.
Each independent single strand DNA amplification was performed using the VIgKFor01 and IgGrevNheI primers for the light and heavy chains, respectively, according to a PCR program as follows: 95°C for 5 min, 30 cycles at 95°C for 45 sec, 54°C for 45 sec, at 72°C for 45 sec, and an extension reaction at 72°C for 1 min. Immediately afterwards, both single strand DNA amplifications were mixed and the assembly reaction was performed by sequential rounds of annealing and extension (Fig. 2).
For comparison, we performed in parallel a SOE-PCR, with 30 cycles of amplification. This amplification included an assembly reaction of 5 cycles of denaturation annealing and extension in the absence of primers, which is reported to facilitate the assembly of chains, followed by amplification reaction where primers were present.
As shown in Figure 3A, the independent strand amplification method (ISA-PCR) produced 6-fold more scFv gene product than that obtained by SOE-PCR. Similar results were obtained when our method was compared with a variant of SOE-PCR that avoids the 5 cycles of denaturation annealing and extension in the absence of primers prior to PCR32 (data not shown). In addition, we found similar results to those described above using a VHR product amplified with an IgM-specific reverse primer (data not shown).
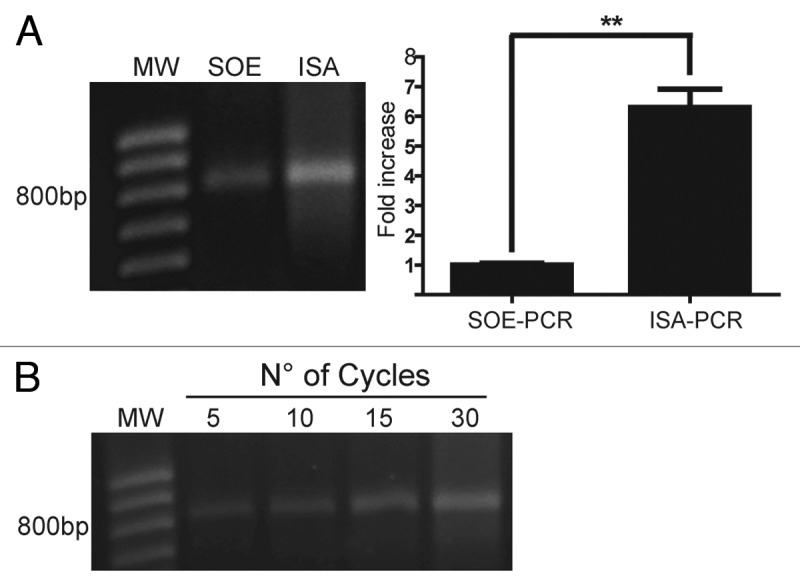
Figure 3. Assembly by ISA-PCR produces more scFv than SOE-PCR. The splicing by overlap extension PCR (SOE-PCR) and the independent strand amplification (ISA-PCR) methods were compared in terms of final scFv gene production. A) In the case of SOE-PCR, 50 ng of heavy and light variable chain amplicon were mixed with VIgKFor01 and IgGrevNheI primers and amplified for 30 cycles. For ISA-PCR, 50 ng of light chain PCR product was incubated with VIgKFor01 primer while the heavy chain PCR product was incubated with IgGrevNheI primer and 30 cycles of PCR were performed. Both single strand products were mixed and the assembly reaction performed using 30 cycles of annealing and extension. An aliquot of 10% of the assembly reaction was resolved by electrophoresis on a 1% w/v agarose gel. The amount of scFv gene produced was quantified as described in Material and Methods and the results were normalized using the mean scFv gene produced by SOE-PCR. Analysis represents the average of 3 independent assembly reactions. ** p < 0.05. B) scFv gene produced by ISA-PCR at different cycles during the assembly reaction. MW: 100bp DNA ladder.
Since the second step of ISAV-PCR (the assembly reaction) did not involve an amplification step, we decided to analyze the effect of cycle step numbers on the scFv gene production. For this purpose, we performed a single strand DNA amplification reaction, as was previously described, and the production of scFv gene at different cycle numbers during the assembly reaction was analyzed, demonstrating an increase as the cycle numbers enhanced (Fig. 3B).
scFv Library Construction and Phage Isolation
Despite the efficiency of the ISA-PCR method to produce high quantities of scFv gene, we had no information about its ability to produce diversity. To analyze this issue, we ligated 1.2 µg of scFv gene produced by this method in a phagemid to generate a pIII fusion protein. The ligation products were transformed in ER2738 bacteria to produce a phage display antibody library with a repertoire size of 2.8 × 107. To assess the efficiency of the procedure, 14 clones were randomly picked from the library and the scFv gene was amplified by PCR. We observed that all tested clones carried the scFv gene with the expected size and showed no misassembly during the ISA-PCR (Fig. 4). The PCR products were digested with MvaI (BstNI), an enzyme commonly used to analyze immunoglobulin variable region diversity. The presence of a particular restriction pattern characterizes an individual clone; however, it is possible that two different clones can exhibit the same pattern. In our analysis, we observed 12 different fingerprinting patterns (Fig. 4), which indicated the presence of multiple different clones in the library. In addition, 21 clones were randomly chosen and sequenced, revealing that all clones were unique and correctly assembled. The germline origin of the sample was analyzed using IgBlast program.35 We observed that VH3 and VL kappa 2 families were the most predominant in our library, as shown in Figure 5. This bias in amplification partially reflects the differential use of variable heavy genes in human repertoire.36 To analyze the production of functional scFv displayed in the phage, the library was infected with the helper phage M13KO7 to produce scFv display phages that were used to perform bio-panning against different antigens, including the recombinant human proteins ErbB3 ectodomain, tumor necrosis factor α (TNFα), vimentin and MHC class I-related chain A (MICA) ectodomain, as well as bovine serum albumin (BSA) and the viral particle of rotavirus. In parallel, bio-panning was performed using only blocking protein to quantify unspecific bound phages. The numbers of clones obtained after subtracting the unspecific bound phages from the first two rounds of selection are shown in Table 2.
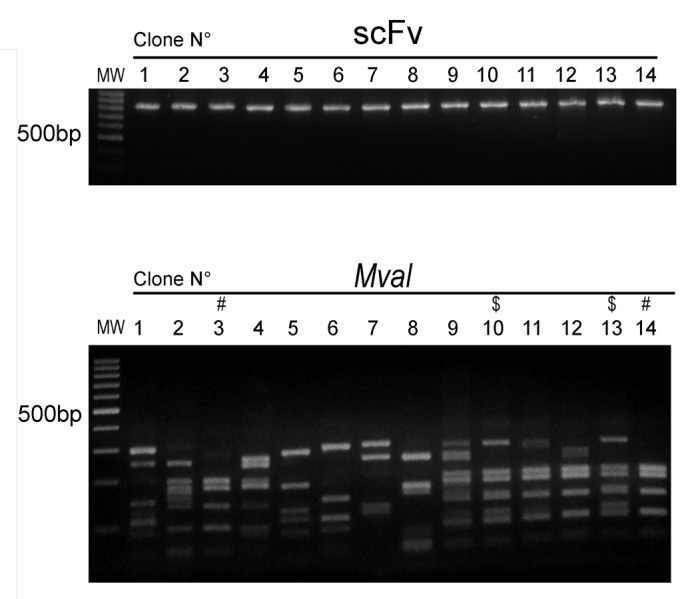
Figure 4. Sequence diversity of scFv obtained by ISA-PCR Plasmids were obtained from 14 random clones from the phage display library constructed using the scFv gene produced by ISA-PCR. Top panel shows the scFv genes produced by PCR resolved by electrophoresis on a 1% w/v agarose gel. Low panel shows digested scFv gene products with MvaI enzyme at 37°C for 3 h, resolved by electrophoresis on a 2% w/v agarose gel. $ and # indicates similar pattern. MW: 100bp DNA ladder.

Figure 5. Frequency of heavy and light chain families. Plasmids obtained from 21 random clones from the phage display library, constructed using the scFv gene produced by ISA-PCR, were sequenced and analyzed using IgBlast.35 (A) Heavy and (B) Kappa light family frequencies observed in the library.
Table 2. Results of binding selection using different targets.
| Antigen | Number of clones after 1st round of panninga | Number of clones after 2nd round of panninga | Number of binder clones after 2nd round of panninga | Number of different scFv |
|---|---|---|---|---|
|
Bovine serum albumin |
3.0x102 |
1.8x103 |
1/15 |
1/1 |
|
Human ErbB3 ectodomain |
2.3x104 |
3.7 x 106 |
3/12 |
3/3 |
|
Human tumor necrosis factor |
3.0x102 |
3.0x103 |
2/15 |
2/2 |
|
Human vimentin |
6.0x102 |
2.4x103 |
3/15 |
1/3 |
|
Human MHC class I-related chain A(MICA) |
6.6x103 |
1.0 x104 |
6/15 |
3/6 |
| Rotavirus | 6.0x102 | 1.5 x103 | 2/15 | 1/2 |
a Phage titer obtained after subtraction of the unspecific phage retrieved in controls.
After the second round of selection with a particular antigen, up to 15 clones were picked and their binding specificity determinate by phage ELISA. We were able to obtain at least one specific clone for each antigen. In the case of MICA, ErbB3 and TNFα, two or more different clones were isolated. Figure 6 indicates the phage screening of scFv clones that interact with ErbB3 and TNFα.

Figure 6. Phages selected against ErbB3. (A) To analyze the diversity displayed by phages binding ErbB3 and TNF-α, 12 clones for ErbB3 and 15 clones for TNF-α, all obtained in the second panning assay, were isolated and analyzed by phage ELISA, as described in Materials and Methods. A helper phage was used as negative control. Solid-colored bars indicate positive clones. (B) Phagemids were obtained from positives clones, sequenced and analyzed using IgBlast.35
Discussion
Phage display antibody technology is a potent tool to recover the recognition potential of the human humoral immune system. Although several protocols can be obtained from the literature,23,32,38-40 the use of this technology can be complex and not always successful, mostly due to the several steps it involves: (1) generation or recovery of the humoral diversity (synthetic or naïve library, respectively); (2) assembly of the binding domain into a single gene; and (3) isolation of phages against specific targets. The construction of a diverse antibody phage library able to recognize several antigens is strongly dependent on the first two factors.
Several methods have been described for the assembly of immunoglobulin variable regions in the scFv format. The assembly by cloning procedures involves a separate amplification of VHR and VLR by PCR and their cloning either into separate vectors or combining them by a cloning procedure.30 Another method involves a recombination system in which the VHR is cloned into a plasmid with lox sites flanking the gene, while the VLR is cloned into a phagemid with a dummy VHR flanked by lox sites. Bacteria containing the VHR plasmid and the Cre recombinase enzyme are infected with the phage containing the VLR to produce recombination.31 Despite their efficiency, these methods are laborious and time-consuming.
The most common method used for the assembly of heavy and light variable regions is the SOE-PCR. In this work, we modified this technique to produce a fast and simple method for the assembly of scFv gene that produces six-fold more scFv gene than SOE-PCR (Fig. 3A). To achieve this, we separated the reaction into two steps. Initially, we enriched the single-stranded DNA required for scFv gene assembly by an independent DNA strand amplification using a single primer, which minimizes the inhibitory effect produced by the presence of complementary chains in the assembly reaction. In the second step, we produced the assembly reaction by mixing both single strand products (Fig. 2). We observed that multiple cycling between denaturation; annealing and extension strongly increases scFv gene production (Fig. 3B). This process probably facilitates the annealing of both single strand products, but we cannot rule out possible amplification of the assembled scFv gene due to the presence of primers remaining from the single strand production reaction.
The diversity produced by the ISA-PCR method was analyzed by a restriction analysis using the enzyme MvaI (Fig. 4); this showed at least 12 different patterns in 14 clones analyzed, which is in agreement with previous observations.28,37,41 It is noteworthy that all clones possess an amplification for full-length scFv gene, showing no misassembly during the ISA-PCR and the absence of vector background in the library.
The DNA sequence analysis of 21 randomly selected scFv genes revealed that all clones were correctly assembled. We observed a bias in the variable gene usage with the over-representations of the VH3 and VL kappa 2 families (Fig. 5). The VH3 family bias has been previously observed during the construction of different scFv libraries.29,42 In addition, this bias reflects the variable gene used in resting B cells repertoire.36 The bias in the use of VL kappa gene family is not as common as the VH gene. Sheets et al. identified the VL kappa 1 as the most frequent family in library developed by sequential cloning,29 but Glanville et al. found the VL kappa 3 as the most frequent.42 The identification of the VL kappa 2 as the most frequent VL family in our library could be produced by the diversity in primer annealing for different families rather than a preferential assembly induced by ISA-PCR.
The scFv genes produced by our method were used to construct an antibody phage display library generated by the amplification of immunoglobulin variable regions from the blood of 8 healthy donors and obtained in a single transformation. Regardless of the limited number of donors and the limited library size (our library possesses 2.8 × 107 clones, while other libraries are in the range 108 to 1010.clones29,30,37), we were able to isolate phages binders to all tested antigens (Table 2). These included different recombinant proteins and rotavirus particles. The frequency of positive clones observed in the second panning was in agreement with previous results using different assembly methods, including SOE-PCR.29,30,37 Moreover, we were able to identify different scFv sequences of binding phages obtained from the proteins TNFα, ErbB3 and MICA (Table 2 and Figure 6).
The results obtained from random clone sequencing and phage binders isolations suggest that the ISA-PCR method is efficient in generating antibody diversity.
The success in the use of antibodies as therapeutic agents43 and the advent of antibodies in high-throughput research have dramatically increased their demand.44 Antibody phage display technology appears to be a suitable source of candidates. According to our experience, the use of ISA-PCR method would facilitate the development of antibody phage display libraries. It is noteworthy that the simplicity and the minimal equipment required for the ISA-PCR method could facilitate the construction of scFv libraries in all types of institutions, including small research facilities, making antibody phage display technology more accessible.
Considering the characteristics of this technique, the use of ISA-PCR is not restricted to the construction of scFv libraries because it could also be used in the construction of any fusion protein with DNA from diverse origin, including evolutionary studies, modular protein constructions and synthetic protein development.
In this study, we developed a fast, simple and highly efficient method to assemble the immunoglobulin light and heavy variable regions in scFv coding genes. The methodology is based on the specific enrichment of DNA strands required for the scFv gene assembly by independent single strand amplification. The ISA-PCR produced high amounts of scFv genes together to the production of sequence diversity and phages that interact with a wide range of different non-related antigens. Taking advantage of its efficiency, the method requires fewer assembly reactions to produce an antibody phage display library, minimizing time and resources, and could be used in the construction of fusion protein.
Material and Methods
scFv Gene Production
Immunoglobulin Variable Regions Amplification
PBMC were obtained from12.5 mL of blood from 8 healthy volunteers by separation with Ficoll-Hypaque (Axis-Shield). All volunteers signed an informed consent approved by the Ethical Committee of the Faculty of Medicine, University of Chile, in the report N°044–2009.Total RNA extraction was performed with Trizol as recommended by the manufacturer (Life Technologies). Full-length cDNAs were generated from 2 µg of total RNA using 200U of Moloney Murine Leukemia Virus reverse transcriptase, according to the manufacturer’s instructions (Promega).
VHR genes were amplified using each of the 6 primers described in Table 1, as well as the reverse primer IgGrevNheI. Briefly, a 20 µL PCR reaction was performed containing 2 µL of cDNA, 0.4 µM of each primer and GoTaq Green Master Mix (Promega). The amplification was performed using the following program: initial denaturation at 94°C for 5 min, followed by 30 cycles at 94°C for 45 sec, 56°C for 45 sec and 72°C for 45s, and a final extension reaction for 10 min at 72°C. To amplify the kappa light variable gene, a PCR reaction was performed using the primers described in Table 1, along with the same reaction mixture previously used for the heavy variable gene amplification. The PCR reaction was performed by heating at 95°C for 2 min followed by 30 cycles at 95°C for 45 sec, 54°C for 45 sec and 72°C for 45 sec, and a final extension reaction for 5 min at 72°C. An independent amplification reaction using each primer set was performed for each donor. After checking the amplification, all heavy or light chain PCR products were pooled, resolved by electrophoresis on a 1% w/v agarose gel and purified using QIAQuick gel extraction kit (QIAGEN).
ISA-PCR Assembly Reaction
The ISA-PCR method is composed by two independent reactions: The single strand DNA amplification and the assembly reaction (Fig. 2). For the single strand amplification reaction 50 ng of the PCR product of the heavy or light chain variable genes were incubated with 0.4 µM of the corresponding primer (VIgKFor01 in the case of VLC and IgGrevNheI for VHC) and Paq DNA polymerase (Stratagene) in 25 µL reaction. Heavy or light chain single strand amplification reactions were performed using the following program: 95°C for the initial denaturation; 30 cycles at 95°C for 45 sec, 54°C for 45 sec and 72°C for 45s, and a final extension reaction of 1 min at 72°C. Both single strand amplifications products were mixed and 10 µM of dNTPs and 5 units of Paq polymerase were added. The assembly reaction was perform by the following conditions; 95°C for the initial denaturation; different number of cycles (5, 10, 15 and 30) at 95°C for 45 sec, 60°C for 45 sec and 72°C for 1 min, and a final extension for 10 min at 72°C. The products were resolved by electrophoresis on 1% w/v agarose gels and purified as previously using QIAQuick gel extraction kit (QIAGEN).
To quantify scFv production, an aliquot of 10% of ISA-PCR product was resolved by electrophoresis on 1% w/v agarose gels. Digital images were captured and the signal intensity of each band was quantified. A region in the upper part of the lane was quantified and subtracted from the corresponding band to eliminate background. The total scFv gene produced was calculated using the ladder band of 800 bp from the 100bp ladder (Fermentas) as mass reference.
SOE-PCR assembly reaction
SOE-PCR was performed as previously described.32,37 Briefly, 50 ng of VHC and VLC PCR products were mixed with dNTPs and Paq DNA polymerase (Stratagene) without primers. The reaction was cycled 5 times (94°C for 45 sec, 60°C for 50 sec, and 72°C for 60 sec). Primers were added to a final concentration of 0.4 µM and amplification was perform by the subsequent program: An initial denaturation at 94°C for 2 min, followed by 30 cycles at 94°C for 45 sec, 54°C for 45 sec and 72°C for 1min, and a final extension reaction for 10 min at 72°C. To quantify scFv production, an aliquot of 10% of ISA-PCR product was resolved by electrophoresis on 1% w/v agarose gels. Digital images were captured and the signal intensity of each band was quantified. A region in the upper part of the lane was quantified and subtracted from the corresponding band to eliminate background. The total scFv gene produced was calculated using the band of 800 bp from the 100bp ladder (Fermentas) as mass reference.
A modification that avoids an initial assembly step was also performed as previously described32; 50 ng of VHC and VLC PCR products were mixed with 0.4 µM of each primer and Paq DNA polymerase (Stratagene). The amplification reaction was then performed by the subsequent program: An initial denaturation step at 94°C for 2 min, followed by 30 cycles at 94°C for 45 sec, 54°C for 45 sec and 72°C for 1 min, and a final extension reaction for 10 min at 72°C.
scFv Library Construction and Phage Antibody Selection
scFv Phage Library Construction
The scFv gene produced by ISA-PCR was digested with EcoRI and NheI restriction enzymes (Fermentas) at 37°C for 16 h; the products were resolved by electrophoresis on a 1% w/v agarose gel and purified using QIAQuick gel extraction kit (QIAGEN). A total of 1.2 µg of digested scFv gene was ligated with 600 ng of pUCH1 using T4 DNA ligase (NEB) and incubated overnight at 16°C. The ligation product was precipitated using glycogen (Fermentas) following the manufacturer’s instructions and used to electroporate E. coli ER2738 (NEB). Later, cells were resuspended in 6 mL of SOC and incubated 1h at 37°C. To quantify the number of transformants, 10-fold serial dilutions were made and plated in LB ampicillin (100µg/mL). The remaining bacteria were centrifuged for 5 min at 5000 rpm, resuspended in LB/ampicillin (100µg/mL) and infected with 1x1010 helper phage M13KO7 (NEB) for 1h at 37°C. After cells were transferred to a flask with 50 mL of LB containing ampicillin (100 µg/mL) and kanamycin (50 µg/mL) and incubated overnight at 37°C, phages were precipitated using PEG and stored at –80°C.
DNA Fingerprinting
Isolated clones obtained from the library were grown overnight at 37°C and plasmids were purified using Wizard Plus SV Minipreps DNA Purification Systems (Promega). The 1:30 plasmid dilution was used as a template to amplify the scFv gene by PCR. The 25 µL reactions were composed of 3 µL of the corresponding plasmid 0.4 µM of primers VIgKFor01and IgGrevNheI and GoTaq Green Master Mix (Promega). The PCR was performed using the following program: 95°C for the initial denaturation; 30 cycles at 95°C for 45 sec, 54°C for 45 sec and 72°C for 45 sec, and a final extension reaction of 1 min at 72°C. Five units of MvaI were added to 10 µL PCR product and incubated at 37°C for 3 h. Digestion products were resolved by electrophoresis on 2% w/v agarose gels.
Selection of Phages
Selection of phages was performed with immobilized antigens by ELISA. 96 NUNC Maxisorb flat wells (Nunc) were coated with antigens (2–5 µg/well) diluted in carbonate buffer (15 mM of NaCO3, 35 mM of NaHCO3,pH 9.6) for 2h at 37°C and blocked with 1% BSA in TBS (Tris 20 mM, NaCl 150 mM, pH 7.6), except in the case of BSA panning assay, when 0.05% of soybean extract protein in TBS was used as a blocking agent.45 The following antigens were used: Human TNFα was purchased from R&D; BSA was purchased from Rockland, Rotavirus particles and human vimentin were kindly provided by Dr. Jonas Chnaiderman (University of Chile, Chile) and Dr Diego Catalan (University of Chile, Chile), respectively. The ectodomains of human ErbB3 and human MICA were cloned in pETb15 (Novagen) transformed in E. coli BL21 DE3 and purified in agarose Ni-NTA columns (Invitrogen).
109 phages from the library were added to the coated wells and incubated for 2 h at 37°C. Phages were also incubated with wells coated with the blocking protein, to identify unspecific binders. Unbound phages were removed by washing 10–20 times with TBS containing 0.5% Tween 20 and the bound phages were eluted incubating with 100 µL of 100 mM glycine-HCl pH 2.2 solution for 10 min at room temperature. Immediately afterwards, the solution was neutralized with 6 µL of 2M Tris-Base, and used to infect exponentially growing E.coli ER2738 for 1 h at 37°C. To quantify phage binders, a 1:100 bacteria dilution was plated on LB-Agar containing ampicillin (100 µg/mL); phages from control wells were plated in the same form. Next, the remaining bacterial culture was infected with 4 × 1010 helper phage M13KO7 and allowed to grow overnight at 37°C followed by phages purification using PEG, which were used to a new panning procedure.
ELISA Screening of Selected Clones
Individual clones were picked and allowed to grow in LB ampicillin overnight at 37°C. After wards, a 50 µL aliquot of bacteria was infected with 450µL of LB carrying 5 × 108 M13KO7 helper phage and incubated overnight at 37°C. Later, the phage clones were precipitated using the PEG protocol and resuspended in 200 µL TBS 1% BSA. 96 NUNC Maxisorb flat wells (Nunc) were coated with 3 µg of the recombinant protein in carbonate buffer for 2h at 37°C and blocked with 1% BSA in TBS, except in the case of BSA panning assay, when 0.05% of soybean extract protein in TBS was used as a blocking agent. As a control, wells were coated only with the corresponding blocking protein. 50 µL of purified phage solution was added to coated plates and incubated for 2h at 37°C. Plates were then washed with TBS 0.05% Tween20 and incubated with 1:5000 dilution of a mouse anti-M13 peroxidase conjugated antibody (GE Healthcare) in 1% BSA in TBS for 2 h at room temperature. Plates were washed as previously described. ABTS substrate was added and the absorbance was read at 405nm using a Sunrise absorbance reader (TECAN). Absorbance from the control wells were subtracted from test wells to obtain the total absorbance.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr. Jonas Chnaiderman for providing the Rotavirus particles, Karina Kramm for helping us in protein purification, and Nancy Fabres for her technical assistance. We also would like to thank Mr. Herve Camus for the technical support during ErbB3 diversity analysis. This study was supported by FONDEF-Chile D06I1005, Millennium Nucleus on Immunology and Immunotherapy (P07/088-F) and Millennium Institute on Immunology and Immunotherapy P09–016-F.
Glossary
Abbreviations:
- PCR
polymerase chain reaction
- Ig
immunoglobulin
- scFv
single chain variable fragment
- Fab
fragment, antigen binding region
- SOE-PCR
splicing by overlap extension PCR
- HAMA
human anti-murine antibody
- CDR
complementary determining regions
- VHR
immunoglobulin heavy-chain variable region
- VLR
immunoglobulin light-chain variable region
- PBMC
peripheral blood mononuclear cells
- ISA-PCR
independent strand amplification method
- BSA
bovine serum albumin
- TNF-α
human tumor necrosis factor alpha
- MICA
human MHC class I-related chain A
Footnotes
Previously published online: www.landesbioscience.com/journals/mabs/article/20653
References
- 1.Brekke OH, Sandlie I. Therapeutic antibodies for human diseases at the dawn of the twenty-first century. Nat Rev Drug Discov. 2003;2:52–62. doi: 10.1038/nrd984. [DOI] [PubMed] [Google Scholar]
- 2.Brennan DJ, O’Connor DP, Rexhepaj E, Ponten F, Gallagher WM. Antibody-based proteomics: fast-tracking molecular diagnostics in oncology. Nat Rev Cancer. 2010;10:605–17. doi: 10.1038/nrc2902. [DOI] [PubMed] [Google Scholar]
- 3.Chan CE, Chan AH, Hanson BJ, Ooi EE. The use of antibodies in the treatment of infectious diseases. Singapore Med J. 2009;50:663–72, quiz 673. [PubMed] [Google Scholar]
- 4.Huber M, Olson WC, Trkola A. Antibodies for HIV treatment and prevention: window of opportunity? Curr Top Microbiol Immunol. 2008;317:39–66. doi: 10.1007/978-3-540-72146-8_2. [DOI] [PubMed] [Google Scholar]
- 5.Plosker GL, Figgitt DP. Rituximab: a review of its use in non-Hodgkin’s lymphoma and chronic lymphocytic leukaemia. Drugs. 2003;63:803–43. doi: 10.2165/00003495-200363080-00005. [DOI] [PubMed] [Google Scholar]
- 6.Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A, et al. Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med. 2001;344:783–92. doi: 10.1056/NEJM200103153441101. [DOI] [PubMed] [Google Scholar]
- 7.Aybay C, Ozel S, Aybay C. Demonstration of specific antibodies against infliximab induced during treatment of a patient with ankylosing spondylitis. Rheumatol Int. 2006;26:473–80. doi: 10.1007/s00296-005-0085-0. [DOI] [PubMed] [Google Scholar]
- 8.Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods. 2005;36:3–10. doi: 10.1016/j.ymeth.2005.01.001. [DOI] [PubMed] [Google Scholar]
- 9.Tjandra JJ, Ramadi L, McKenzie IF. Development of human anti-murine antibody (HAMA) response in patients. Immunol Cell Biol. 1990;68:367–76. doi: 10.1038/icb.1990.50. [DOI] [PubMed] [Google Scholar]
- 10.Gavilondo JV, Larrick JW. Antibody engineering at the millennium. Biotechniques 2000; 29:128-32, 34-6, 38 passim. [DOI] [PubMed]
- 11.Lonberg N. Human antibodies from transgenic animals. Nat Biotechnol. 2005;23:1117–25. doi: 10.1038/nbt1135. [DOI] [PubMed] [Google Scholar]
- 12.Bradbury AR, Sidhu S, Dübel S, McCafferty J. Beyond natural antibodies: the power of in vitro display technologies. Nat Biotechnol. 2011;29:245–54. doi: 10.1038/nbt.1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rau R. Adalimumab (a fully human anti-tumour necrosis factor alpha monoclonal antibody) in the treatment of active rheumatoid arthritis: the initial results of five trials. Ann Rheum Dis. 2002;61(Suppl 2):ii70–3. doi: 10.1136/ard.61.suppl_2.ii70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Finlay WJ, Cunningham O, Lambert MA, Darmanin-Sheehan A, Liu X, Fennell BJ, et al. Affinity maturation of a humanized rat antibody for anti-RAGE therapy: comprehensive mutagenesis reveals a high level of mutational plasticity both inside and outside the complementarity-determining regions. J Mol Biol. 2009;388:541–58. doi: 10.1016/j.jmb.2009.03.019. [DOI] [PubMed] [Google Scholar]
- 15.Gram H, Marconi LA, Barbas CF, 3rd, Collet TA, Lerner RA, Kang AS. In vitro selection and affinity maturation of antibodies from a naive combinatorial immunoglobulin library. Proc Natl Acad Sci U S A. 1992;89:3576–80. doi: 10.1073/pnas.89.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jung S, Honegger A, Plückthun A. Selection for improved protein stability by phage display. J Mol Biol. 1999;294:163–80. doi: 10.1006/jmbi.1999.3196. [DOI] [PubMed] [Google Scholar]
- 17.Lippow SM, Wittrup KD, Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–6. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Beuerlein G, Nie Y, Smith H, Lee BA, Hensler M, et al. Stepwise in vitro affinity maturation of Vitaxin, an alphav beta3-specific humanized mAb. Proc Natl Acad Sci U S A. 1998;95:6037–42. doi: 10.1073/pnas.95.11.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–36. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 20.Le Gall F, Kipriyanov SM, Moldenhauer G, Little M. Di-, tri- and tetrameric single chain Fv antibody fragments against human CD19: effect of valency on cell binding. FEBS Lett. 1999;453:164–8. doi: 10.1016/S0014-5793(99)00713-9. [DOI] [PubMed] [Google Scholar]
- 21.Müller D, Kontermann RE. Bispecific antibodies for cancer immunotherapy: Current perspectives. BioDrugs. 2010;24:89–98. doi: 10.2165/11530960-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 22.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–7. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 23.Bradbury AR, Marks JD. Antibodies from phage antibody libraries. J Immunol Methods. 2004;290:29–49. doi: 10.1016/j.jim.2004.04.007. [DOI] [PubMed] [Google Scholar]
- 24.McCafferty J, Griffiths AD, Winter G, Chiswell DJ. Phage antibodies: filamentous phage displaying antibody variable domains. Nature. 1990;348:552–4. doi: 10.1038/348552a0. [DOI] [PubMed] [Google Scholar]
- 25.Griffiths AD, Williams SC, Hartley O, Tomlinson IM, Waterhouse P, Crosby WL, et al. Isolation of high affinity human antibodies directly from large synthetic repertoires. EMBO J. 1994;13:3245–60. doi: 10.1002/j.1460-2075.1994.tb06626.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Knappik A, Ge L, Honegger A, Pack P, Fischer M, Wellnhofer G, et al. Fully synthetic human combinatorial antibody libraries (HuCAL) based on modular consensus frameworks and CDRs randomized with trinucleotides. J Mol Biol. 2000;296:57–86. doi: 10.1006/jmbi.1999.3444. [DOI] [PubMed] [Google Scholar]
- 27.Söderlind E, Strandberg L, Jirholt P, Kobayashi N, Alexeiva V, Aberg AM, et al. Recombining germline-derived CDR sequences for creating diverse single-framework antibody libraries. Nat Biotechnol. 2000;18:852–6. doi: 10.1038/78458. [DOI] [PubMed] [Google Scholar]
- 28.Marks JD, Hoogenboom HR, Bonnert TP, McCafferty J, Griffiths AD, Winter G. By-passing immunization. Human antibodies from V-gene libraries displayed on phage. J Mol Biol. 1991;222:581–97. doi: 10.1016/0022-2836(91)90498-U. [DOI] [PubMed] [Google Scholar]
- 29.Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, et al. Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens. Proc Natl Acad Sci U S A. 1998;95:6157–62. doi: 10.1073/pnas.95.11.6157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, et al. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nat Biotechnol. 1996;14:309–14. doi: 10.1038/nbt0396-309. [DOI] [PubMed] [Google Scholar]
- 31.Sblattero D, Bradbury A. Exploiting recombination in single bacteria to make large phage antibody libraries. Nat Biotechnol. 2000;18:75–80. doi: 10.1038/71958. [DOI] [PubMed] [Google Scholar]
- 32.Barbas CF Phage display: a laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press, 2001. [Google Scholar]
- 33.Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–8. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- 34.Retter I, Althaus HH, Münch R, Müller W. VBASE2, an integrative V gene database. Nucleic Acids Res. 2005;33(Database issue):D671–4. doi: 10.1093/nar/gki088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Altschul SF, Madden TL, Schäffer AA, Zhang J, Zhang Z, Miller W, et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davidkova G, Pettersson S, Holmberg D, Lundkvist I. Selective usage of VH genes in adult human B lymphocyte repertoires. Scand J Immunol. 1997;45:62–73. doi: 10.1046/j.1365-3083.1997.d01-376.x. [DOI] [PubMed] [Google Scholar]
- 37.Pansri P, Jaruseranee N, Rangnoi K, Kristensen P, Yamabhai M. A compact phage display human scFv library for selection of antibodies to a wide variety of antigens. BMC Biotechnol. 2009;9:6. doi: 10.1186/1472-6750-9-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Aitken R Antibody phage display: methods and protocols. Dordrecht; New York: Humana Press, 2009. [Google Scholar]
- 39.Clackson T. Lowman HB Phage display: a practical approach. Oxford: Oxford University Press, 2004. [Google Scholar]
- 40.O'Brien PM. Aitken R Antibody phage display: methods and protocols. Totowa, N.J.: Humana Press, 2002. [Google Scholar]
- 41.Okamoto T, Mukai Y, Yoshioka Y, Shibata H, Kawamura M, Yamamoto Y, et al. Optimal construction of non-immune scFv phage display libraries from mouse bone marrow and spleen established to select specific scFvs efficiently binding to antigen. Biochem Biophys Res Commun. 2004;323:583–91. doi: 10.1016/j.bbrc.2004.08.131. [DOI] [PubMed] [Google Scholar]
- 42.Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta GR, et al. Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc Natl Acad Sci U S A. 2009;106:20216–21. doi: 10.1073/pnas.0909775106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nelson AL, Dhimolea E, Reichert JM. Development trends for human monoclonal antibody therapeutics. Nat Rev Drug Discov. 2010;9:767–74. doi: 10.1038/nrd3229. [DOI] [PubMed] [Google Scholar]
- 44.Baldwin JP, Boseley PG, Bradbury EM, Ibel K. The subunit structure of the eukaryotic chromosome. Nature. 1975;253:245–9. doi: 10.1038/253245a0. [DOI] [PubMed] [Google Scholar]
- 45.Aguillón J, Ramos R, Chiong M, Vallejos P, Ferreira A. Soybean proteins: alternative blocking agents for immunoassays using nitrocellulose or plastic solid phases. Biol Res. 1992;25:79–83. [Google Scholar]