

Abstract
Genetically tractable model organisms from phages to mice have taught us invaluable lessons about fundamental biological processes and disease-causing mutations. Owing to technological and computational advances, human biology and the causes of human diseases have become accessible as never before. Progress in identifying genetic determinants for human diseases has been most remarkable for Mendelian traits. In contrast, identifying genetic determinants for complex diseases such as diabetes, cancer, and cardiovascular and neurological diseases has remained challenging, despite the fact that these diseases cluster in families. Hundreds of variants associated with complex diseases have been found in genome-wide association studies (GWAS), yet most of these variants explain only a modest amount of the observed heritability, a phenomenon known as “missing heritability.” The missing heritability has been attributed to many factors, mainly inadequacies in genotyping and phenotyping. We argue that lessons learned about complex traits in model organisms offer an alternative explanation for missing heritability in humans. In diverse model organisms, phenotypic robustness differs among individuals, and those with decreased robustness show increased penetrance of mutations and express previously cryptic genetic variation. We propose that phenotypic robustness also differs among humans and that individuals with lower robustness will be more responsive to genetic and environmental perturbations and hence susceptible to disease. Phenotypic robustness is a quantitative trait that can be accurately measured in model organisms, but not as yet in humans. We propose feasible approaches to measure robustness in large human populations, proof-of-principle experiments for robustness markers in model organisms, and a new GWAS design that takes differences in robustness into account.
Introduction
Complex diseases such as diabetes, cancer, and cardiovascular and neurological diseases are the predominant causes of morbidity and mortality in the developed world. As they tend to cluster in families, these diseases are thought to involve genetic factors in addition to environmental ones. Although genome-wide association studies (GWAS) have identified hundreds of common variants associated with complex diseases, and although susceptibility loci have been reported for many disorders, the overall genetic risk explained by these loci remains modest. Thus, only a small proportion of heritability (proportion of phenotypic variance explained by genetic variants) has been accounted for [1], [2]. This discrepancy, termed “missing heritability,” has been attributed to many factors. First, rare variants of large effect size (odds ratio >2) may account for some of the unexplained genetic risk, as observed in neuropsychiatric disorders such as autism, schizophrenia, and developmental delay [1]–[4]. By their nature, rare variants are difficult to detect and to associate with phenotype using statistics. Second, highly repetitive structural and sequence variants have remained inaccessible to large-scale genotyping [1], [2], [5]. Third, heritability estimates may be artificially inflated due to interactions between genes, to shared environments in families, and to imprecise diagnoses of complex disorders [1], [2], [5], [6]. Consequently, current research addresses the problem of missing heritability with more comprehensive genotyping of genetic variants in statistically well-powered cohorts of individuals that are better characterized for disease phenotypes, genetic background, and environmental exposure [1], [2]. This approach is rooted in the prevalent hypothesis that some combination of rare variants of large effect, common variants of small effect, and environmental factors will translate into disease [1],[2],[5].
An alternative view posits that cryptic genetic variation accounts for a substantial fraction of disease-associated risk [7]. In robust individuals, cryptic genetic variation will not contribute to disease and will elude detection by GWAS. In contrast, in individuals with decreased overall phenotypic robustness, formerly cryptic genetic variants will contribute to disease, and disease-related variants will increase in penetrance, resulting in increased heritability. This hypothesis draws on findings from diverse model organisms including yeast, worms, flies, plants, and fish: Decreased phenotypic robustness significantly increases heritability of complex traits due to revealed, formerly cryptic genetic variation and increased penetrance of genetic variants [8]–[16]. In this review, we describe the causes and consequences of decreased phenotypic robustness in model organisms, relate these findings to complex disease phenotypes, and propose an alternative GWAS approach that accounts for differences in robustness among humans.
What Is Phenotypic Robustness?
Phenotypic robustness is the ability of a given genotype to produce a constant phenotype, even when the organism is faced with genetic or environmental perturbations. The remarkable phenotypic robustness of wild-type organisms is commonly attributed to features of the underlying genetic networks, such as modularity, feedback loops, gene redundancy, connectivity, degeneracy, and the presence of activity-modulating microRNAs [9], [11], [17]–[33]. In model organisms, targeted perturbation of any of these features can decrease phenotypic robustness and release cryptic genetic variation [8], [10], [14], [15], [17], [24]–[26], [32], [34], [35].
Phenotypic robustness is a measurable quantitative trait. Traditionally, robustness of individuals has been measured as the degree of symmetry in morphological features [36]. A high degree of symmetry is thought to be associated with high fitness and even with the perception of beauty for human faces [37]–[39]. In most organisms, objective and high-throughput analysis of symmetry is complicated by the complexity of morphological features and their profound changes throughout development. Another measure of robustness is the degree of accuracy with which a genotype produces a particular phenotype across many isogenic siblings [36]. By this measure, phenotypic robustness, like any other quantitative trait, shows a distribution among genetically divergent individuals of a species and can be mapped to distinct genetic loci [13]. The ability to buffer mutations can vary among isogenic individuals, suggesting that nongenetic mechanisms significantly affect robustness [15], . These nongenetic robustness determinants will elude genetic approaches. None of the robustness measures that have been used in model organisms are applicable in humans; however, they have proven useful to identify master regulators or network hubs that may contribute to robustness in humans.
The best-characterized master regulator of robustness is the molecular chaperone HSP90 [8]–[10], [12]–[16], [21], [34], [40]–[43]. HSP90 assists the proper folding and function of many key enzymes and transcription factors that govern growth and development [43]. The chaperone is essential in eukaryotes, is evolutionarily conserved, is highly connected, and plays a crucial role in integrating environmental signals [43]. HSP90's function is of even greater importance under environmental stress that compromises protein folding [43]. In genetically divergent plant, fly, yeast, and fish populations, HSP90 inhibition significantly increases heritability due to increased penetrance of known genetic variants and revealed cryptic genetic variants [8], [10], [14], [34]. In worms, naturally varying HSP90 levels predict mutation penetrance: lower HSP90 levels result in greater penetrance of mutations [15], [16]. In plants, yeast, and flies, HSP90-dependent variants are common in natural strains and often affect complex traits [12], [13], [34], [44].
Decreased Phenotypic Robustness Is Associated with Genome Instability
Loss of robustness may be associated with an increased mutation rate. In flies, HSP90 inhibition increases transposon transcription and mobility [40], [42]. In human cells, HSP90 inhibition compromises repair of DNA damage in response to radiation [45] and increases the mutation rate of microsatellites [46]. In yeast, severe HSP90 inhibition induces aneuploidy [47]. In plants, HSP90 inhibition increases the rate of homologous recombination (Figure 1). HSP90 inhibition appears to interfere broadly with genome stability by affecting transposon silencing, DNA repair, microsatellite stability, chromosome segregation, and homologous recombination. Given the extent of standing variation that responds to HSP90 inhibition [12], [13], [34], [44], newly arising mutations probably play a minor role in HSP90-dependent phenotypes, yet genome instability may be a hallmark of decreased robustness. These HSP90-specific results are consistent with stress-induced increases in mutation rate in bacteria, yeast, and plants [47]–[52]. Environmental stress also decreases robustness in diverse organisms, supporting an association of robustness with genome stability [8], [10], [14], [53], [54].
Figure 1. HSP90 inhibition increases homologous recombination in plants.
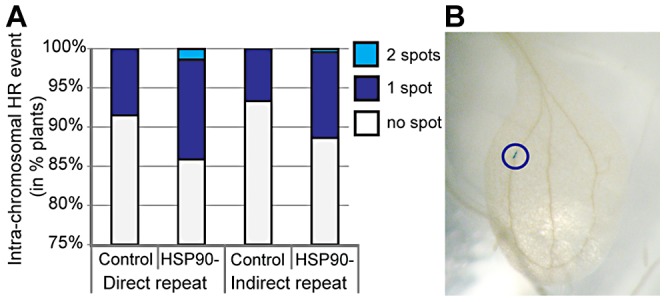
(A) Transgenic plants carrying reporter constructs that monitor somatic homologous recombination (HR) events [48] were grown with and without HSP90 inhibition. HR restores a functional GUS gene, allowing for detection of somatic HR events. Intra-chromosomal HR events were significantly increased in plants grown with the HSP90 inhibitor geldanamycin for direct and indirect repeat reporter constructs (Poisson regression p = 0.0078). A total of 925 seedlings were analyzed with 381 seedlings for the direct repeat reporter line and 544 for the indirect repeat reporter line. (B) Example of somatic HR event in plant leaf (blue circle around GUS spot) (unpublished data by K. Carlson, A. Nuttle, and C. Queitsch).
HSP90 is only one of several “robustness” master regulators. In yeast, 60–300 genes, all network hubs, have been identified for which deletion of any one decreases robustness [24]. In worms, decreased function of highly connected hub genes enhances the phenotypic consequences of decreased function in many other genes [35]. It remains untested how many of these yeast and worm network hubs reveal cryptic genetic variation in divergent populations or decrease genome stability when perturbed. However, effects on genome instability seem likely as both worm and yeast network hub genes are strongly enriched for chromatin regulators [24], [35]. In plants, a novel master regulator of robustness has been found, in which loss of function decreases phenotypic robustness and releases cryptic genetic variation [55]. Consistent with an association of robustness and genome stability, inhibition of this master regulator increases transposon mobility [56].
Release of Cryptic Genetic Variation May Contribute to Rise of Complex Diseases
While both rare variants of large effect and many common variants of small effect contribute to complex diseases [5], neither model satisfactorily explains the significant rise of complex diseases [5], [7]. In the last century, dramatic changes in lifestyle and environment included diet changes, refrigeration, departure from natural circadian rhythm through artificial lighting, modern hygiene, and urban living, to name a few [7], [57]–[59]. In particular, changes in diet and refrigeration have led to the fast evolution and changing composition of the human gut microbiome [60]–[66]. Greg Gibson has suggested that these environmental perturbations may alter the genetic contributions to phenotype by revealing cryptic genetic variation, especially among individuals with reduced phenotypic robustness [5], [7].
Gibson's hypothesis that cryptic genetic variation contributes to disease susceptibility is supported by the properties of disease risk variants and their distribution among human populations. A surprising number of the single nucleotide polymorphisms (SNPs) associated with disease risk are ancestral, indicating that the protective variant arose in the human lineage. Thus, disease susceptibility cannot be easily explained by acquisition of deleterious mutations in the human lineage. Further, disease risk alleles vary dramatically in frequency and effect size in human populations, indicating extensive population structure and the importance of environmental factors for developing disease [7].
Features of Certain Complex Diseases Are Consistent with Decreased Robustness
As common SNPs fail to confer significant disease risk for disorders such as autism, schizophrenia, and mental retardation, rare variants of potentially large effect have been examined [1], [2]. The human genome contains regions that are predisposed to copy number variation (CNV) due to their repeated architecture [3]. Several of these rare, recurrent CNVs are associated with schizophrenia, autism, cardiac and renal anomalies, epilepsy, obesity, diabetes, and mental retardation [3], [4], [67]–[72]. However, many of these CNVs are also found in control populations and unaffected family members. Moreover, the same CNV can be associated with a large spectrum of disorders [3], [4]. For example, del17q12 is associated with renal cysts, maturity-onset diabetes, developmental delay, brain malformations, seizures, schizophrenia, and autism [3], [4]. This variability in expressivity is thought to be due to additional rare events: the classical genetic modifier hypothesis [3], [4], [68], [69], [73]. Alternatively, such CNV lesions may not be causal but rather reflect decreased genome stability [74] and decreased robustness.
If decreased robustness correlates with or causes genome instability, patients with complex diseases should carry a higher burden of CNVs. This burden (or mutational load) may be inherited or may arise de novo through environmental stress in early development. Indeed, schizophrenia patients show a significantly higher global burden of rare CNVs [72], [75]. Most importantly, private CNVs—that is, CNVs specific to a particular individual—are highly enriched in schizophrenia patients [75]. A similar increase of CNV burden is found in autism patients [73]. Patients with the recurrent CNV on chromosome 16p12.1, which is associated with severe developmental delay, are also more likely to carry additional CNVs than matched controls [69]. Moreover, patients with second CNV hits—possibly less robust individuals—show distinct and more severe clinical features [69]. Consistent with decreased robustness, the facial symmetry of some patients is visibly perturbed [69]. In nine other genomic disorders, additional CNV hits occur more frequently in patients than controls [69]. This enrichment of CNV hits in patients is particularly strong for disorders with variable penetrance and expressivity [3], [4], [69]. In multiplex autism families (families with multiple occurrences), CNVs in affected siblings are fourfold enriched compared to unaffected siblings [76]. This observation led to the hypothesis that multiplex autism is due to an inherited predisposition in addition to other co-occurring mutations, including CNVs [3], [76]. We speculate that this predisposition may be decreased robustness.
The observation of additional, mostly private CNVs in patients is consistent with the existence of an extraordinarily large number of distinct genetic modifiers leading to disease. While certainly possible, this explanation is not consistent with our basic knowledge of genetic networks and their robustness to mutations. In yeast and worms, for which systematic analyses of single and double mutant phenotypes have been conducted, most loss-of-function mutations, even if combined, do not show a phenotype unless they occur in network hubs or master regulators such as HSP90 [24], [35], [77]–[79]. The number of network hubs in humans is large but not infinitely large. Under our hypothesis, the increased CNV burden is an expression of a generally less robust and therefore sensitized background rather than a cause of disease. This explanation is consistent with the fact that the same CNV can be associated with different disorders. The different expressivity of a particular disorder may be due to different degrees of robustness loss or different revealed, formerly cryptic genetic variants in patients. In short, we propose that phenotypic robustness differs among humans as it does in model organisms and that those with lower robustness will be more susceptible to genetic and environmental perturbations and hence disease. This proposal is akin to assertions from physicians of generations past that certain people have a robust constitution whereas others have weak ones.
How Can We Assess Robustness in Humans to Increase the Heritability of Complex Diseases?
Currently, researchers attempt to identify unifying patterns of genetic variants, lifestyle choices, and environmental factors in affected individuals (cases, Figure 2). In contrast, we would like to assess all individuals first for their level of phenotypic robustness (robust versus not robust, Figure 2). Individuals with significantly decreased robustness would then be analyzed for genetic variants associated with disease. This analysis will identify formerly cryptic variants that were unknown to influence disease risk. In addition, previously identified predisposing variants will increase in penetrance because healthy individuals carrying these variants will be robust and therefore not contribute to associations among the group with decreased robustness. In addition, this analysis will facilitate the identification of rare causal variants of large effect, which should be enriched in robust but affected individuals. Increased penetrance of common variants together with revealed, formerly cryptic variants and causative rare alleles of large effect will significantly increase heritability for at least some complex diseases.
Figure 2. Current and suggested GWAS approaches.
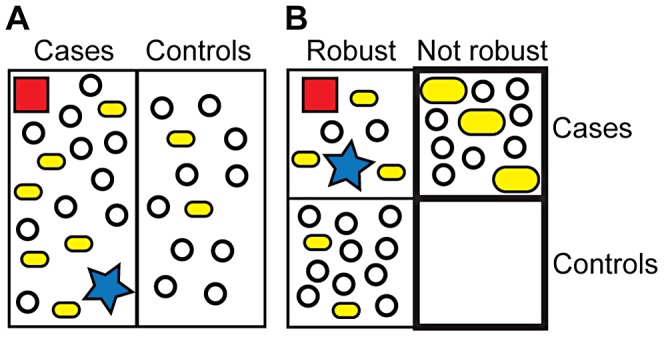
(A) Current approach. GWAS identify variants that are overrepresented in cases. Rare variants of large effect (red square, blue star) may escape detection, thereby contributing to missing heritability. Common variants that are overrepresented in cases (small yellow bar, 6 versus 2) do not contribute strongly to disease risk. A cryptic disease-related variant does not show significant overrepresentation in cases (open circle). (B) Suggested approach. Individuals are first analyzed for phenotypic robustness (bold box) and then for variants associated with disease. Rare variants of large effect will be enriched in robust cases, although they may also be present in nonrobust cases. Variants that are overrepresented in all cases (robust, nonrobust) will show higher penetrance in nonrobust individuals (large yellow bars). The formerly cryptic, disease-related variant (open circle) is significantly enriched in nonrobust cases versus nonrobust controls (and robust cases) and can therefore be identified. Together, heritability significantly increases. The formerly cryptic genetic variant and higher penetrance variant can be thought of as “disease-specifiers” as they determine the specific disease phenotype of individuals carrying them. Note symbols represent highly simplified frequencies of specific variant in indicated groups and not individuals carrying certain variants.
This approach hinges critically on the identification of reliable markers for phenotypic robustness that can be readily assessed in large human populations. The functionality of master regulators such as HSP90 could potentially provide such a robustness measure. In yeast, however, the group of master regulators that affect robustness is large and functionally diverse (about 1%–5% of all nonessential genes) [24]. Assaying the diverse functions of hundreds of proteins is not a suitable high-throughput test.
Because decreased robustness correlates with and produces genome instability at several levels—an increase in microsatellite mutations, transposon mobility, recombination rates, base-substitution mutation rate, and large duplications and deletions—we suggest that these different genome instability events can serve as readouts for decreased robustness. Recent technological advances have made most of these features easily accessible in large populations of humans and model organisms. The individual events may be rare, as observed for CNV variation; hence, they need to be investigated on a genome-wide scale. As microsatellites show by far the greatest mutation rate [80], somatic microsatellite variation may be the most sensitive robustness marker. In fact, microsatellite variation has a long history as a marker for deficient DNA repair in certain cancers [81], [82]. At this point, however, assessment of microsatellite variation requires high-quality, expensive Sanger-sequencing. Neither the more cost-effective next-generation sequencing nor array-based genotyping can accurately determine variation of small microsatellites, but given the pace of current technology development, this technical hurdle should soon disappear.
Before investing in human studies, the suitability of molecular robustness markers is easily testable in model organisms. We envision the following proof-of-principle experiments: First, a diverse wild-type population, ideally freshly collected, is phenotyped for robustness, using traditional robustness measures such as symmetry or quantitative trait variation in genetically identical offspring (such as exists in plants and worms). We expect a distribution of robustness. Second, the least robust and most robust individuals are assessed for genome instability events at several levels. We expect to observe a higher frequency of these events in less robust individuals. If a significant correlation between a traditional robustness measure and any genome instability event is found, this type of genome instability is a robustness marker that is applicable to humans. Third, if our hypothesis is correct, less robust individuals should be more susceptible to environmental stresses and will show higher expressivity of mutations and genetic variation. This assumption can be tested by exposing the least robust and most robust individuals to environmental stress and mutagenesis.
If genome instability events fail to predict robustness, alternatives exist. For humans, DNA-, RNA-, or cell-based assays are preferable, as the necessary material can be obtained with relative ease. Indeed, several cell-based, high-throughput assays for somatic mutations already exist for humans [83]. In principle, these assays monitor allele loss resulting in an altered phenotypic output such as fluorescence. Another, potentially more promising, cell-based approach for determining robustness in humans would be to assess cell population variance for a given individual in gene expression, genome methylation, or chromatin states. Cell population variance in shape and other morphological features could also serve as a robustness marker [24]. In yeast, this approach identified robustness master regulators by calculating the variance of 70 phenotypic traits among individual cells stained for nuclei, actin, and a cell wall marker [24]. Given this arsenal of possible robustness measures, high-throughput molecular or cell-based robustness markers seem feasible in the near future.
Our hypothesis is testable even in the absence of human robustness markers. If decreased robustness predisposes to complex diseases, we can test whether individuals suffering from one complex disease are more likely to suffer from another, as we would predict. Another test would compare variants between cohorts of patients suffering from two different complex diseases. This comparison would filter out shared variants that are not causative and possibly related to decreased robustness and reveal statistically enriched causative variants that are specific to each disease.
Our hypothesis that robustness differences among individuals contribute to the missing heritability of disease is akin to prior propositions that epistasis (i.e., genetic interactions) accounts for the missing heritability [5], [6]. Epistatically interacting loci could certainly be detected through GWAS, yet this will require very large sample sizes to find sufficient individuals of each genotype combination. In contrast, our approach reduces the intractable complexity of possible genetic interactions to robustness, which we propose is a universal disease and trait modifier that can be feasibly measured in large human populations. Furthermore, not all instances of disease may involve genetic interactions; some may arise from interactions of risk alleles with nongenetic mechanisms or environmental factors. Unlike traditional GWAS, our approach might capture these instances because robustness differences can arise through nongenetic mechanisms and environmental perturbations. Taking robustness into account has the potential to free us from disentangling the multitude of factors contributing to specific instances of disease. If successful, this approach might render complex disease more deterministic and predictable, allowing us to better identify the contributing lifestyle choices and environmental exposures and ultimately decrease the severity and incidence of these devastating diseases.
Acknowledgments
We thank Elhanan Borenstein, Evan Eichler, Stanley Fields, Greg Gibson, Harmit Malik, Ray Monnat, and Maynard Olson for helpful discussions and comments. We thank Alexander Nuttle for sharing unpublished data.
Funding Statement
This work was supported by grants from the National Institute of Health Training Program in Genetics (T32-GM07735) (KDC) and the National Institute of Health (DP2OD008371) (CQ). The funders had no role in the preparation of the article.
References
- 1. Eichler EE, Flint J, Gibson G, Kong A, Leal SM, et al. (2010) Missing heritability and strategies for finding the underlying causes of complex disease. Nat Rev Genet 11: 446–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Manolio TA, Collins FS, Cox NJ, Goldstein DB, Hindorff LA, et al. (2009) Finding the missing heritability of complex diseases. Nature 461: 747–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Girirajan S, Campbell CD, Eichler EE (2011) Human copy number variation and complex genetic disease. Annu Rev Genet 45: 203–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Girirajan S, Eichler EE (2010) Phenotypic variability and genetic susceptibility to genomic disorders. Hum Mol Genet 19: R176–R187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Gibson G (2011) Rare and common variants: twenty arguments. Nat Rev Genet 13: 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Zuk O, Hechter E, Sunyaev SR, Lander ES (2012) The mystery of missing heritability: genetic interactions create phantom heritability. Proc Natl Acad Sci U S A 109: 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Gibson G (2009) Decanalization and the origin of complex disease. Nat Rev Genet 10: 134–140. [DOI] [PubMed] [Google Scholar]
- 8. Queitsch C, Sangster TA, Lindquist S (2002) Hsp90 as a capacitor of phenotypic variation. Nature 417: 618–624. [DOI] [PubMed] [Google Scholar]
- 9. Rutherford S, Hirate Y, Swalla BJ (2007) The Hsp90 capacitor, developmental remodeling, and evolution: the robustness of gene networks and the curious evolvability of metamorphosis. Crit Rev Biochem Mol Biol 42: 355–372. [DOI] [PubMed] [Google Scholar]
- 10. Rutherford SL, Lindquist S (1998) Hsp90 as a capacitor for morphological evolution. Nature 396: 336–342. [DOI] [PubMed] [Google Scholar]
- 11. Sangster TA, Lindquist S, Queitsch C (2004) Under cover: causes, effects and implications of Hsp90-mediated genetic capacitance. Bioessays 26: 348–362. [DOI] [PubMed] [Google Scholar]
- 12. Sangster TA, Salathia N, Lee HN, Watanabe E, Schellenberg K, et al. (2008) HSP90-buffered genetic variation is common in Arabidopsis thaliana. Proc Natl Acad Sci U S A 105: 2969–2974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sangster TA, Salathia N, Undurraga S, Milo R, Schellenberg K, et al. (2008) HSP90 affects the expression of genetic variation and developmental stability in quantitative traits. Proc Natl Acad Sci U S A 105: 2963–2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yeyati PL, Bancewicz RM, Maule J, van Heyningen V (2007) Hsp90 selectively modulates phenotype in vertebrate development. PLoS Genet 3: e43 doi:10.1371/journal.pgen.0030043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Burga A, Casanueva MO, Lehner B (2011) Predicting mutation outcome from early stochastic variation in genetic interaction partners. Nature 480: 250–253. [DOI] [PubMed] [Google Scholar]
- 16. Casanueva MO, Burga A, Lehner B (2012) Fitness trade-offs and environmentally induced mutation buffering in isogenic C. elegans. Science 335: 82–85. [DOI] [PubMed] [Google Scholar]
- 17. Bergman A, Siegal ML (2003) Evolutionary capacitance as a general feature of complex gene networks. Nature 424: 549–552. [DOI] [PubMed] [Google Scholar]
- 18. Ciliberti S, Martin OC, Wagner A (2007) Innovation and robustness in complex regulatory gene networks. Proc Natl Acad Sci U S A 104: 13591–13596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. de Visser JA, Hermisson J, Wagner GP, Ancel Meyers L, Bagheri-Chaichian H, et al. (2003) Perspective: evolution and detection of genetic robustness. Evolution Int J Org Evolution 57: 1959–1972. [DOI] [PubMed] [Google Scholar]
- 20. Hornstein E, Shomron N (2006) Canalization of development by microRNAs. Nat Genet 38 Suppl: S20–S24. [DOI] [PubMed] [Google Scholar]
- 21. Jarosz DF, Taipale M, Lindquist S (2010) Protein homeostasis and the phenotypic manifestation of genetic diversity: principles and mechanisms. Annu Rev Genet 44: 189–216. [DOI] [PubMed] [Google Scholar]
- 22. Leclerc RD (2008) Survival of the sparsest: robust gene networks are parsimonious. Mol Syst Biol 4: 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lehner B (2008) Selection to minimise noise in living systems and its implications for the evolution of gene expression. Mol Syst Biol 4: 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Levy SF, Siegal ML (2008) Network hubs buffer environmental variation in Saccharomyces cerevisiae. PLoS Biol 6: e264 doi:10.1371/journal.pbio.0060264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li X, Cassidy JJ, Reinke CA, Fischboeck S, Carthew RW (2009) A microRNA imparts robustness against environmental fluctuation during development. Cell 137: 273–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manu, Surkova S, Spirov AV, Gursky VV, Janssens H, et al. (2009) Canalization of gene expression and domain shifts in the Drosophila blastoderm by dynamical attractors. PLoS Comput Biol 5: e1000303 doi:10.1371/journal.pcbi.1000303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Masel J, Siegal ML (2009) Robustness: mechanisms and consequences. Trends Genet 25: 395–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Raser JM, O'Shea EK (2005) Noise in gene expression: origins, consequences, and control. Science 309: 2010–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wagner A (2000) Robustness against mutations in genetic networks of yeast. Nat Genet 24: 355–361. [DOI] [PubMed] [Google Scholar]
- 30. Wagner A (2008) Gene duplications, robustness and evolutionary innovations. Bioessays 30: 367–373. [DOI] [PubMed] [Google Scholar]
- 31. Salathia N, Queitsch C (2007) Molecular mechanisms of canalization: Hsp90 and beyond. J Biosci 32: 457–463. [DOI] [PubMed] [Google Scholar]
- 32. Baggs JE, Price TS, DiTacchio L, Panda S, Fitzgerald GA, et al. (2009) Network features of the mammalian circadian clock. PLoS Biol 7: e52 doi:10.1371/journal.pbio.1000052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Whitacre JM (2012) Biological robustness: paradigms, mechanisms, and systems principles. Frontiers in Genetics 3: 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jarosz DF, Lindquist S (2010) Hsp90 and environmental stress transform the adaptive value of natural genetic variation. Science 330: 1820–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lehner B, Crombie C, Tischler J, Fortunato A, Fraser AG (2006) Systematic mapping of genetic interactions in Caenorhabditis elegans identifies common modifiers of diverse signaling pathways. Nat Genet 38: 896–903. [DOI] [PubMed] [Google Scholar]
- 36. Debat V, David P (2001) Mapping phenotypes:canalization, plasticity and developmental stability. TRENDS in Ecology & Evolution 16: 555–561. [Google Scholar]
- 37. Gangestad SW, Thornhill R, Garver-Apgar CE (2005) Women's sexual interests across the ovulatory cycle depend on primary partner developmental instability. Proc Biol Sci 272: 2023–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scheib JE, Gangestad SW, Thornhill R (1999) Facial attractiveness, symmetry and cues of good genes. Proc Biol Sci 266: 1913–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Thornhill R, Gangestad SW (1999) Facial attractiveness. Trends Cogn Sci 3: 452–460. [DOI] [PubMed] [Google Scholar]
- 40. Gangaraju VK, Yin H, Weiner MM, Wang J, Huang XA, et al. (2011) Drosophila Piwi functions in Hsp90-mediated suppression of phenotypic variation. Nat Genet 43: 153–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sollars V, Lu X, Xiao L, Wang X, Garfinkel MD, et al. (2003) Evidence for an epigenetic mechanism by which Hsp90 acts as a capacitor for morphological evolution. Nat Genet 33: 70–74. [DOI] [PubMed] [Google Scholar]
- 42. Specchia V, Piacentini L, Tritto P, Fanti L, D'Alessandro R, et al. (2010) Hsp90 prevents phenotypic variation by suppressing the mutagenic activity of transposons. Nature 463: 662–665. [DOI] [PubMed] [Google Scholar]
- 43. Taipale M, Jarosz DF, Lindquist S (2010) HSP90 at the hub of protein homeostasis: emerging mechanistic insights. Nat Rev Mol Cell Biol 11: 515–528. [DOI] [PubMed] [Google Scholar]
- 44. Carey CC, Gorman KF, Rutherford S (2006) Modularity and intrinsic evolvability of Hsp90-buffered change. PLoS ONE 1: e76 doi:10.1371/journal.pone.0000076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Dote H, Burgan WE, Camphausen K, Tofilon PJ (2006) Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer Res 66: 9211–9220. [DOI] [PubMed] [Google Scholar]
- 46. Mittelman D, Sykoudis K, Hersh M, Lin Y, Wilson JH (2010) Hsp90 modulates CAG repeat instability in human cells. Cell Stress Chaperones 15: 753–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Chen G, Bradford WD, Seidel CW, Li R (2012) Hsp90 stress potentiates rapid cellular adaptation through induction of aneuploidy. Nature 482: 246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Molinier J, Ries G, Zipfel C, Hohn B (2006) Transgeneration memory of stress in plants. Nature 442: 1046–1049. [DOI] [PubMed] [Google Scholar]
- 49. Boyko A, Kovalchuk I (2011) Genome instability and epigenetic modification–heritable responses to environmental stress? Curr Opin Plant Biol 14: 260–266. [DOI] [PubMed] [Google Scholar]
- 50. Ponder RG, Fonville NC, Rosenberg SM (2005) A switch from high-fidelity to error-prone DNA double-strand break repair underlies stress-induced mutation. Mol Cell 19: 791–804. [DOI] [PubMed] [Google Scholar]
- 51. Shee C, Gibson JL, Darrow MC, Gonzalez C, Rosenberg SM (2011) Impact of a stress-inducible switch to mutagenic repair of DNA breaks on mutation in Escherichia coli. Proc Natl Acad Sci U S A 108: 13659–13664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cooley MB, Carychao D, Nguyen K, Whitehand L, Mandrell R (2010) Effects of environmental stress on stability of tandem repeats in Escherichia coli O157:H7. Appl Environ Microbiol 76: 3398–3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dworkin I (2005) A study of canalization and developmental stability in the sternopleural bristle system of Drosophila melanogaster. Evolution; International Journal of Organic Evolution 59: 1500–1509. [PubMed] [Google Scholar]
- 54. Parsons PA (1992) Fluctuating asymmetry: a biological monitor of environmental and genomic stress. Heredity 68 Pt 4: 361–364. [DOI] [PubMed] [Google Scholar]
- 55. Lempe J, Sullivan A, Alexandre C, Skelly DA, Lachowiec J, et al. (submitted) RNA polymerase V as a fast-evolving capacitor in A. thaliana . [Google Scholar]
- 56. Ito H, Gaubert H, Bucher E, Mirouze M, Vaillant I, et al. (2011) An siRNA pathway prevents transgenerational retrotransposition in plants subjected to stress. Nature 472: 115–119. [DOI] [PubMed] [Google Scholar]
- 57. Hugot JP, Alberti C, Berrebi D, Bingen E, Cezard JP (2003) Crohn's disease: the cold chain hypothesis. Lancet 362: 2012–2015. [DOI] [PubMed] [Google Scholar]
- 58. Strachan DP (2000) The role of environmental factors in asthma. Br Med Bull 56: 865–882. [DOI] [PubMed] [Google Scholar]
- 59. Strachan DP (2000) Family size, infection and atopy: the first decade of the “hygiene hypothesis.”. Thorax 55 Suppl 1: S2–S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Greenblum S, Turnbaugh PJ, Borenstein E (2012) Metagenomic systems biology of the human gut microbiome reveals topological shifts associated with obesity and inflammatory bowel disease. Proc Natl Acad Sci U S A 109: 594–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ley RE, Hamady M, Lozupone C, Turnbaugh PJ, Ramey RR, et al. (2008) Evolution of mammals and their gut microbes. Science 320: 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Ley RE, Lozupone CA, Hamady M, Knight R, Gordon JI (2008) Worlds within worlds: evolution of the vertebrate gut microbiota. Nat Rev Microbiol 6: 776–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Malekzadeh F, Alberti C, Nouraei M, Vahedi H, Zaccaria I, et al. (2009) Crohn's disease and early exposure to domestic refrigeration. PLoS ONE 4: e4288 doi:10.1371/journal.pone.0004288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Prentice AM, Gershwin ME, Schaible UE, Keusch GT, Victora CG, et al. (2008) New challenges in studying nutrition-disease interactions in the developing world. J Clin Invest 118: 1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Turnbaugh PJ, Backhed F, Fulton L, Gordon JI (2008) Diet-induced obesity is linked to marked but reversible alterations in the mouse distal gut microbiome. Cell Host Microbe 3: 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, et al. (2009) A core gut microbiome in obese and lean twins. Nature 457: 480–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bailey JA, Gu Z, Clark RA, Reinert K, Samonte RV, et al. (2002) Recent segmental duplications in the human genome. Science 297: 1003–1007. [DOI] [PubMed] [Google Scholar]
- 68. Girirajan S, Brkanac Z, Coe BP, Baker C, Vives L, et al. (2011) Relative burden of large CNVs on a range of neurodevelopmental phenotypes. PLoS Genet 7: e1002334 doi:10.1371/journal.pgen.10b02334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Girirajan S, Rosenfeld JA, Cooper GM, Antonacci F, Siswara P, et al. (2010) A recurrent 16p12.1 microdeletion supports a two-hit model for severe developmental delay. Nat Genet 42: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, et al. (2007) Strong association of de novo copy number mutations with autism. Science 316: 445–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sharp AJ, Hansen S, Selzer RR, Cheng Z, Regan R, et al. (2006) Discovery of previously unidentified genomic disorders from the duplication architecture of the human genome. Nat Genet 38: 1038–1042. [DOI] [PubMed] [Google Scholar]
- 72. Walsh T, McClellan JM, McCarthy SE, Addington AM, Pierce SB, et al. (2008) Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science 320: 539–543. [DOI] [PubMed] [Google Scholar]
- 73. Pinto D, Pagnamenta AT, Klei L, Anney R, Merico D, et al. (2010) Functional impact of global rare copy number variation in autism spectrum disorders. Nature 466: 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Heng HH (2010) Missing heritability and stochastic genome alterations. Nat Rev Genet 11: 813. [DOI] [PubMed] [Google Scholar]
- 75. The Schizophrenia Consortium (2008) Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature 455: 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Itsara A, Wu H, Smith JD, Nickerson DA, Romieu I, et al. (2010) De novo rates and selection of large copy number variation. Genome Res 20: 1469–1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Boone C, Bussey H, Andrews BJ (2007) Exploring genetic interactions and networks with yeast. Nat Rev Genet 8: 437–449. [DOI] [PubMed] [Google Scholar]
- 78. Davierwala AP, Haynes J, Li Z, Brost RL, Robinson MD, et al. (2005) The synthetic genetic interaction spectrum of essential genes. Nat Genet 37: 1147–1152. [DOI] [PubMed] [Google Scholar]
- 79. Giaever G, Chu AM, Ni L, Connelly C, Riles L, et al. (2002) Functional profiling of the Saccharomyces cerevisiae genome. Nature 418: 387–391. [DOI] [PubMed] [Google Scholar]
- 80. Legendre M, Pochet N, Pak T, Verstrepen KJ (2007) Sequence-based estimation of minisatellite and microsatellite repeat variability. Genome Res 17: 1787–1796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Li GM (2008) Mechanisms and functions of DNA mismatch repair. Cell Res 18: 85–98. [DOI] [PubMed] [Google Scholar]
- 82. Preston BD, Albertson TM, Herr AJ (2010) DNA replication fidelity and cancer. Semin Cancer Biol 20: 281–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Albertini RJ, Nicklas JA, Fuscoe JC, Skopek TR, Branda RF, et al. (1993) In vivo mutations in human blood cells: biomarkers for molecular epidemiology. Environ Health Perspect 99: 135–141. [DOI] [PMC free article] [PubMed] [Google Scholar]