

Abstract
The putative tumor suppressor, DBC1 (deleted in breast cancer-1), was recently found to negatively regulate SIRT1 in vitro and in vivo, but the mechanism whereby DBC1 regulates SIRT1 in liver cancer remains to be elucidated. In this study, it was found that although the expression of DBC1 and SIRT1 was not aberrantly regulated in a large cohort of human hepatocellular carcinoma (HCC) patients, these proteins were highly overexpressed in a subset of HCC tissues compared with surrounding non-cancer tissues. In liver cancer, DBC1 and SIRT1 were found to be positively correlated. Inactivation of DBC1 or SIRT1 reduced SNU-182 (a liver cancer cell line) proliferation as determined by MTT viability assays. Notably, although DBC1 functions as a negative regulator of SIRT1 in A549 lung cancer cells since it suppresses the deacetylase activity of the p53 protein, it did not affect the p53 deacetylase activity of SIRT1 in SNU-182 cells. Taken together, we conclude that DBC1 is associated with SIRT1 in HCC, but that it does not inhibit SIRT1.
Keywords: deleted in breast cancer-1, silent mating type information regulation 2 homolog 1, hepatocellular carcinoma, p53
Introduction
Liver cancer is the fifth most commonly diagnosed cancer and the second most frequent cause of cancer-related mortality worldwide. Its rate is particularly high in Asia and Africa. Of the liver cancers, hepatocellular carcinoma (HCC) is the most common malignancy, and accounts for 70–85% of all liver cancers (1). Several environmental factors, such as hepatitis type B or C virus infections, aflatoxin B1 and alcohol, are considered to be the main causes of HCC, but no absolute cure for HCC has been devised (1). Many studies have suggested that the losses of several tumor suppressor genes and aberrant regulations of cell growth signaling pathways, including the ERK/MAPK pathway and the Wnt/β-catenin pathway, are involved in hepatocarcinogenesis (2–4), but the molecular mechanism underlying the pathogenesis of HCC remains elusive.
Deleted in breast cancer-1 (DBC1, KIAA1967) was originally suggested to be a putative tumor suppressor gene on chromosome 8p21, which is frequently deleted in breast cancer (5). Several researchers have shown that DBC1 regulates cell survival by modulating its diverse binding partners. DBC1 induces p53-mediated apoptosis by negatively regulating silent mating type information regulation 2 homolog 1 (SIRT1) activity (6,7), and by directly interacting with estrogen receptor-α (ERα) promotes breast cancer cell survival (8). In addition, DBC1 regulates cancer cell growth by mediating the transcriptional activation of retinoic acid receptor α (RAR α) in breast cancer or androgen receptor (AR) in prostate cancer cells (9,10). DBC1 also regulates epigenetic mechanisms by inhibiting SUV39H1 methyltransferase and histone deacetylase 3 (HDAC3) (11,12). These results indicate that DBC1 is a multifunctional protein that may be involved in a variety of cellular pathways.
Sirtuins (SIRTs) are highly conserved mammalian homologues of yeast SIR2 (silent mating type information regulation 2 homolog 1), which catalyze NAD+-dependent histone deacetylation and ADP ribosylation. Many studies have shown that SIRT1 levels are significantly elevated in prostate cancer, ovarian cancer, gastric cancer, colorectal cancer and hepatocellular carcinoma (13–17). Moreover, SIRT1 inhibition has been reported to suppress cell growth and induce cell cycle arrest or apoptosis in cancer cells (13–17). Although SIRT1 has emerged as a key regulator in various cellular pathways, the regulatory mechanisms responsible for SIRT1 activity have not been determined.
Although DBC1 modulation of SIRT1 has been suggested to be implicated in cancer cell death, its regulatory functions in liver cancer have not been elucidated. Moreover, the correlation between DBC1 and SIRT1 in liver cancer has not been studied. Therefore, in this study, we investigated the aberrant regulations of DBC1 and SIRT1 in HCC, and examined the association between DBC1 with SIRT1 in liver cancer cell lines.
Materials and methods
Tissue samples
Ten hepatocellular carcinoma tissues and corresponding normal tissues were obtained from Yonsei University, School of Medicine, Seoul. Informed consent was provided in compliance with the Declaration of Helsinki, and the study was approved by the Institutional Review of Board of Songeui Campus, College of Medicine, The Catholic University of Korea (IRB approval no. CUMC11U010).
Cell culture, transfections and treatments
The human HCC cell lines, HepG2 (wt p53) and SNU-182 (mt p53), and the non-small lung carcinoma cell line A549 (wt p53) were obtained from ATCC (American Type Culture Collection, Manassas, VA, USA). Cell lines were maintained in RPMI-1640 medium (Lonza, Walkersville, MD, USA) supplemented with 10% fetal bovine serum (Sigma, St. Louis, MO, USA) and 100 units/ml of penicillin-streptomycin (Invitrogen, Carlsbad, CA, USA).
Small interfering RNA duplexes (siRNAs) specific to SIRT1 (sense, 5′-GGAUAGAGCCUCACAUGCA-3′; anti-sense, 5′-UGCTUGUGAGGCUCUAUCC-3′) and DBC1 (sense, 5′-CAGCUUGCAUGACUACUUU-3′; antisense, 5′-AAAGUAGUCAUGCAAGCUG-3′) and negative control siRNA were purchased from Ambion (Austin, TX, USA). SIRT1 expression plasmid, pcDNA 3.1-SIRT1-myc-His was kindly donated by Dr. Kouzarides (University of Cambridge, UK). For gene silencing, siRNAs against SIRT1 and DBC1 were transfected at final concentrations of 50 and 200 nM, respectively. Two micrograms of mock (empty vector) and SIRT1 expression plasmids were used to induce SIRT1 overexpression. All transfections were performed using Lipofectamine™ 2000 reagents (Invitrogen) according to the manufacturer’s instructions. To induce DNA damage, cells were treated with 20 μM etoposide (Sigma) for 12 h.
Western blotting and co-immunoprecipitation
Tissue proteins and cell lysates were prepared using Radio Immunoprecipitation (RIPA) buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Nonidet P-40, 0.25% sodium deoxycholate) supplemented with 1 mM phenylmethane-sulfonylfluoride (PMSF, Sigma) and 1X complete protease inhibitor cocktail tablets (Roche, Mannheim, Germany). Proteins (10 μg) were separated by SDS-PAGE and transferred onto polyvinylidene difluoride membranes (PVDF, Bio-Rad Laboratories, Hercules, CA, USA). Proteins were analyzed using anti-SIRT1, -p53 and -GAPDH (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-DBC1 (Bethyl Laboratories, Montgomery, TX, USA) and anti-acetylated p53 (Cell Signaling, Danvers, MA, USA). An ECL Plus western blotting detection system (Millipore, Billerica, MA, USA) was used to detect signals. Membranes were evaluated using a LAS 3000 image analyzer (Fuji Photo Film, Japan). For immunoprecipitations, cells were lysed with sodium deoxycholate-free RIPA buffer. Cell lysates (1∼2 mg) were incubated with 1∼2 μg anti-His (Applied Biological Materials, BC, Canada) or anti-SIRT1 at 4°C overnight, and then rotated with 50 μl protein-A-agarose (Millipore) at 4°C for 4 h. Samples were analyzed by western blotting with appropriate primary antibodies.
3-(4, 5-dimethylthiazol-2-yl)-2, 5-diphenyltetrazolium bromide (MTT) assay
SNU-182 cells were seeded into 6-well plates and transfected with each siRNA. Twenty-four hours later, cells were treated with 20 μM etoposide for 12 h. After etoposide treatment, cells were incubated with 0.5 mg/ml MTT for 1 h at 37°C for the indicated times. The formazan crystals produced were dissolved with 500 μl DMSO and absorbances were read at 570 nm using a VICTOR3™ Multilabel Plate Reader (PerkinElmer, Foster City, CA, USA). All measurements were performed in triplicate and each experiment was repeated at least three times.
Results
DBC1 and SIRT1 are aberrantly overexpressed in a subset of human HCCs
It was recently suggested that SIRT1 is expressed at very low levels in normal liver tissue, but that it is overexpressed in HCC cell lines and in a subset of HCCs, and that it is essential for tumorigenesis in HCC (17). Conversely, an analysis of SIRT1 mRNA levels in HCC patients by real-time qPCR revealed that average SIRT1 mRNA levels in tumor and non-tumoral liver tissues do not differ significantly, which suggests that the tumor-specific overexpression of SIRT1 is regulated in a transcription-independent manner (17). In addition, it has been demonstrated that SIRT1 activity is negatively regulated by DBC1 by direct binding (6,7). However, how SIRT1 is regulated by DBC1 in liver cancer remains unclear. Therefore, to explore this association in liver cancer, we examined SIRT1 and DBC1 gene expressions available from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (accession numbers GSE14520 and GSE25097) in a large cohort of HCC patients. As shown previously, SIRT1 and DBC1 gene expression was not significantly different in the two large HCC cohorts (Fig. 1A and B). We then examined SIRT1 protein overexpression by using western blotting in 10 randomly selected human HCC tissues and paired non-cancerous surrounding liver tissues. As expected, SIRT1 was markedly upregulated in HCCs compared to non-cancerous tissues (Fig. 1C). Notably, we also observed that DBC1 protein was highly overexpressed in the same HCCs. Since DBC1 has been reported to function as a negative regulator of SIRT1 in other cancers, we had expected an inverse correlation between DBC1 and SIRT1. Thus, we investigated the association between DBC1 and SIRT1 expression in a liver cancer cell line by immunoprecipitation and western blot analysis.
Figure 1.

Aberrant expressions of SIRT1 and DBC1 in human HCC tissues. (A and B) Recapitulated SIRT1 and DBC1 gene expression levels in a large cohort of HCC patients. Two sets of microarray data were obtained from the National Center for Biotechnology Information (NCBI) Gene Expression Omnibus (GEO) database (accession nos. GSE14520 and GSE25097). The relative expressions of SIRT1 and DBC1 mRNAs in non-cancerous tissues (Non-Tumor) and in tumor tissues (Tumor) are illustrated by the scatter plot. The median expression level in each group is indicated by a horizontal line. Gene expression levels are shown on the ordinate (log2 intensity). The statistical analysis for differential expression of SIRT1 and DBC1 was determined using the unpaired t-test (*P<0.0001; two-tailed). (C) Western blot analysis of DBC1 and SIRT1 expression in 10 selected human HCC tissues (T) paired with histologically normal liver tissues (N). All membranes were probed for GAPDH to confirm equal protein loading. SIRT1, silent mating type information regulation 2 homolog 1; DBC1, deleted in breast cancer-1; HCC, hepatocellular carcinoma.
As shown in Fig. 2A, to elucidate whether DBC1 is associated with SIRT1, we introduced His-tagged SIRT1 expression plasmid into SNU-182 cells. In the following immunoprecipitation with anti-His antibody, endogenous DBC1 was detected by western blotting. SIRT1 was simultaneously detected in the same anti-His immunoprecipitates. In addition, endogenous DBC1 and SIRT1 were also co-immunoprecipitated in SNU-182 cells (Fig. 2B). These results demonstrated that DBC1 interacts with SIRT1 in liver cancer cells in vitro.
Figure 2.

DBC1 interacted with SIRT1 in HCC cell lines. (A) DBC1 was co-immunoprecipitated with ectopically introduced SIRT1. Cells were transfected with either 1 μg pcDNA3.1-myc-His or pcDNA3.1-SIRT1-myc-His plasmids. Cell lysates were immunoprecipitated with anti-His. Precipitated proteins were detected by western blotting using the indicated antibodies. (B) The endogenous interaction between DBC1 and SIRT1 was analyzed by co-immunoprecipitation and western blotting. Cell lysates were immunoprecipitated with anti-SIRT1 and precipitates were analyzed by western blotting with anti-DBC1. SIRT1, silent mating type information regulation 2 homolog 1; DBC1, deleted in breast cancer-1; HCC, hepatocellular carcinoma.
Targeted inhibition of DBC1 and SIRT1-mediated growth in liver cancer cell lines
Next, to explain the biological consequences of the aberrant expressions of SIRT1 and DBC1 in hepatocarcinogenesis, SIRT1 and DBC1 expression was abrogated using the RNA interference-mediated protein knockdown method in HepG2 and SNU-182 cells (two different HCC cell lines). As previously observed (17), SIRT1 depletion resulted in a significant reduction in tumor cell growth in both cell lines (Fig. 3A and B). Notably, when DBC1 protein was inactivated by its siRNA, both cell lines exhibited significantly less cell viability. However, double knockdown of SIRT1 and DBC1 did not exhibit a synergistic effect on cell viability (Fig. 3A and B). These results suggest that DBC1 does not negatively regulate SIRT1, but that it does contribute to the neoplastic property of liver cancer cells.
Figure 3.

Targeted disruptions of DBC1 or SIRT1 reduced liver cancer cell viability. HepG2 (A) and SNU-182 (B) cells were transfected with siRNAs targeting SIRT1 or DBC1 (50 or 200 nM), and then treated with 20 μM of etoposide for 12 h. Cell viabilities were determined by MTT assay as described in Materials and methods. Data are presented as means ± standard deviation of three experiments (unpaired Student’s t-test, *P<0.05 vs. si-control). SIRT1, silent mating type information regulation 2 homolog 1; DBC1, deleted in breast cancer-1.
In previous studies, inactivation of DBC1 by siRNA attenuated etoposide-induced p53 acetylation by inhibiting SIRT1 deacetylase activity in A549 and U2OS cell lines (6,7). To determine whether DBC1 inhibits p53, we analyzed acetylated p53 in the absence or presence of DBC1 protein in liver cancer cells. Initially, to confirm the suppressive effect of DBC1 on p53 acetylation by SIRT1, we performed etoposide-induced hyperacetylation of p53 in A549 cells. As shown in Fig. 4A, DBC1 silencing reduced the acetylation status of p53 in response to etoposide treatment in A549 cells (Fig. 3B). However, inactivation of DBC1 by its siRNA did not affect the etoposide-mediated p53 hyperacetylation status in SNU-182 cells (Fig. 4B). These results suggest that DBC1 does not functionally influence SIRT1 activity in liver cancer cells.
Figure 4.
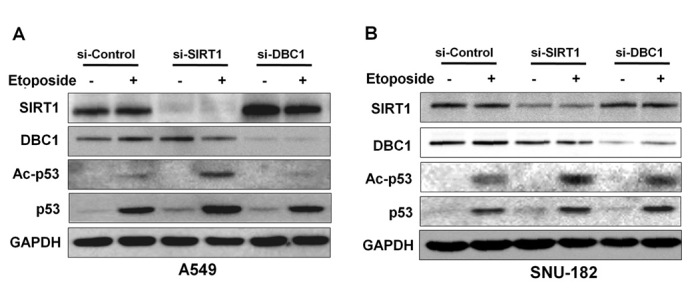
DBC1 did not affect SIRT1 activity in vitro in liver cancer cells. A549 and SNU-182 (A and B) cells were transfected with siRNAs targeting SIRT1 or DBC1 (50 or 200 nM). Etoposide (20 μM) was administered for 12 h to induce p53 hyperacetylation due to DNA damage. Knockdown of SIRT1 or DBC1 and acetylation of p53 were evaluated by western blotting with the indicated antibodies. All membranes were probed for GAPDH to confirm equal protein loading. SIRT1, silent mating type information regulation 2 homolog 1; DBC1, deleted in breast cancer-1.
Discussion
Accumulating evidence indicates that the interaction between DBC1 and SIRT1 is implicated in metabolic disease and cancers. For example, DBC1 knockout mice were found to be protected against high-fat diet-induced liver steatosis with increased SIRT1 activity (18). It has also been reported that interactions between DBC1 and SIRT1 are dysregulated in breast cancer cell lines (19). In terms of the regulation of estrogen receptor α (ERα) activity, DBC1 and SIRT1 reciprocally interacted with the cell cycle and apoptosis regulator 1 (CCAR1), and modulate its co-activator function to ERα (20). Furthermore, the inactivation of SIRT1 or the introduction of DBC1 promoted c-MYC-induced apoptosis in different cell lines (21). In the present study, we found DBC1 is associated with SIRT1, but that it does not function as a negative regulator of SIRT1 in HCC.
The functions of DBC1 and SIRT1 in tumorigenesis remain controversial. In an early publication, the pro-apoptotic and tumor-suppressive functions of DBC1 were proposed to be due to the involvement of the caspase-mediated activation of DBC1 in apoptotic induction by TNF-α (22). On the other hand, DBC1 was found to be highly upregulated and associated with poor survival in gastric carcinoma and breast cancer (15,23). Notably, and in agreement with previous results in studies of gastric carcinoma and breast cancer, our results show that both DBC1 and SIRT1 proteins are highly overexpressed in human HCC tissues (Fig. 1C). Furthermore, the mRNA expression of the SIRT1 gene in a large cohort of HCC patients in the GEO (Gene Expression Omnibus) database showed no significant differences between tumor and matched non-tumor tissues (Fig. 1A). However, it was of note to find that DBC1 overexpression was also observed in HCC tissues that showed SIRT1 overexpression. Thus, it appears that both proteins are aberrantly regulated at a post-transcriptional level in liver cancer, and positively associated within cells. Indeed, our results show that the expressions of DBC1 and SIRT1 are positively correlated in HCC cells (Fig. 2).
Many proteins positively or negatively regulate SIRT1 activity by direct binding in vitro. The active regulator of SIRT1, AROS, activates SIRT1 and enhances the p53-suppressive function of SIRT1 (24). In addition, casein kinase 2 (CK2) mediated SIRT1 phosphorylation and increased deacetylation of p53 by SIRT1 and protected cells from apoptosis following DNA damage (25). On the other hand, sentrin-specific proteinase 1 (SENP1) inhibited SIRT1 activity by interacting and desumoylating SIRT1, thereby promoting stress-induced apoptosis (26). Of these, CK2 and SENP1 have been shown to regulate p53 activity either directly or indirectly (27,28). Our results show that the inactivation of DBC1 or SIRT1 reduced HCC cell viability and that DBC1 does not negatively regulate SIRT1 activity in HCC cells (Figs. 3 and 4). Many studies have shown that SIRT1 is overexpressed in various human cancers, and that the targeted disruption of SIRT1 inhibits cancer cell growth. Recently, DBC1 was also suggested to be a new candidate gene with a potential role in colon cancer (29). These reports suggest that DBC1 acts during tumor progression or development, and that the interaction between DBC1 and SIRT1 plays a crucial role in tumorigenesis. Taken together, our results suggest that the overexpression of DBC1 is associated with HCC and that DBC1 does not function as a negative regulator of SIRT1 activity in HCC.
Acknowledgments
This study was supported by the National Research Foundation of Korea (NRF) which is funded by the Korean government (MEST) (Grant No. 2011-0010705), by the Korean Ministry of the Environment ‘The Converging-Technology Project’ (Grant No. 212 101 003), and by the Korean Science and Engineering Foundation via the ‘Cancer Evolution Research Center’ at The Catholic University of Korea.
References
- 1.Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin. 2011;61:69–90. doi: 10.3322/caac.20107. [DOI] [PubMed] [Google Scholar]
- 2.Whittaker S, Marais R, Zhu AX. The role of signaling pathways in the development and treatment of hepatocellular carcinoma. Oncogene. 2010;29:4989–5005. doi: 10.1038/onc.2010.236. [DOI] [PubMed] [Google Scholar]
- 3.Ito Y, Sasaki Y, Horimoto M, et al. Activation of mitogen-activated protein kinases/extracellular signal-regulated kinases in human hepatocellularcarcinoma. Hepatology. 1998;27:951–958. doi: 10.1002/hep.510270409. [DOI] [PubMed] [Google Scholar]
- 4.Wong CM, Fan ST, Ng IO. Beta-catenin mutation and overexpression in hepatocellular carcinoma: clinicopathologic and prognostic significance. Cancer. 2001;92:136–145. doi: 10.1002/1097-0142(20010701)92:1<136::aid-cncr1301>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 5.Hamaguchi M, Meth JL, von Klitzing C, et al. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc Natl Acad Sci USA. 2002;99:13647–13652. doi: 10.1073/pnas.212516099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature. 2008;451:583–586. doi: 10.1038/nature06500. [DOI] [PubMed] [Google Scholar]
- 7.Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature. 2008;451:587–590. doi: 10.1038/nature06515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trauernicht AM, Kim SJ, Kim NH, Boyer TG. Modulation of estrogen receptor α protein level and survival function by DBC-1. Mol Endocrinol. 2007;21:1526–1536. doi: 10.1210/me.2007-0064. [DOI] [PubMed] [Google Scholar]
- 9.Fu J, Jiang J, Li J, et al. Deleted in breast cancer 1, a novel androgen receptor (AR) coactivator that promotes AR DNA-binding activity. J Biol Chem. 2009;284:6832–6840. doi: 10.1074/jbc.M808988200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garapaty S, Xu CF, Trojer P, Mahajan MA, Neubert TA, Samuels HH. Identification and characterization of a novel nuclear protein complex involved in nuclear hormone receptor-mediated gene regulation. J Biol Chem. 2009;284:7542–7552. doi: 10.1074/jbc.M805872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li Z, Chen L, Kabra N, Wang C, Fang J, Chen J. Inhibition of SUV39H1 methyltransferase activity by DBC1. J Biol Chem. 2009;284:10361–10366. doi: 10.1074/jbc.M900956200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chini CC, Escande C, Nin V, Chini EN. HDAC3 is negatively regulated by the nuclear protein DBC1. J Biol Chem. 2010;285:40830–40837. doi: 10.1074/jbc.M110.153270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Huffman DM, Grizzle WE, Bamman MM, et al. SIRT1 is significantly elevated in mouse and human prostate cancer. Cancer Res. 2007;67:6612–6618. doi: 10.1158/0008-5472.CAN-07-0085. [DOI] [PubMed] [Google Scholar]
- 14.Jang KY, Kim KS, Hwang SH, et al. Expression and prognostic significance of SIRT1 in ovarian epithelial tumours. Pathology. 2009;41:366–371. doi: 10.1080/00313020902884451. [DOI] [PubMed] [Google Scholar]
- 15.Cha EJ, Noh SJ, Kwon KS, et al. Expression of DBC1 and SIRT1 is associated with poor prognosis of gastric carcinoma. Clin Cancer Res. 2009;15:4453–4459. doi: 10.1158/1078-0432.CCR-08-3329. [DOI] [PubMed] [Google Scholar]
- 16.Stunkel W, Peh BK, Tan YC, et al. Function of the SIRT1 protein deacetylase in cancer. Biotechnol J. 2007;2:1360–1368. doi: 10.1002/biot.200700087. [DOI] [PubMed] [Google Scholar]
- 17.Chen J, Zhang B, Wong N, et al. Sirtuin 1 is upregulated in a subset of hepatocellular carcinomas where it is essential for telomere maintenance and tumor cell growth. Cancer Res. 2011;71:4138–4149. doi: 10.1158/0008-5472.CAN-10-4274. [DOI] [PubMed] [Google Scholar]
- 18.Escande C, Chini CC, Nin V, et al. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest. 2010;120:545–558. doi: 10.1172/JCI39319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim JE, Lou Z, Chen J. Interactions between DBC1 and SIRT 1 are deregulated in breast cancer cells. Cell Cycle. 2009;8:3784–3785. doi: 10.4161/cc.8.22.10055. [DOI] [PubMed] [Google Scholar]
- 20.Ji Yu E, Kim SH, Heo K, Ou CY, Stallcup MR, Kim JH. Reciprocal roles of DBC1 and SIRT1 in regulating estrogen receptor α activity and co-activator synergy. Nucleic Acids Res. 2011;39:6932–6943. doi: 10.1093/nar/gkr347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Menssen A, Hydbring P, Kapelle K, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA. 2011 doi: 10.1073/pnas.1105304109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sundararajan R, Chen G, Mukherjee C, White E. Caspase-dependent processing activates the proapoptotic activity of deleted in breast cancer-1 during tumor necrosis factor-α-mediated death signaling. Oncogene. 2005;24:4908–4920. doi: 10.1038/sj.onc.1208681. [DOI] [PubMed] [Google Scholar]
- 23.Lee H, Kim KR, Noh SJ, et al. Expression of DBC1 and SIRT1 is associated with poor prognosis for breast carcinoma. Hum Pathol. 2010;42:204–213. doi: 10.1016/j.humpath.2010.05.023. [DOI] [PubMed] [Google Scholar]
- 24.Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell. 2007;28:277–290. doi: 10.1016/j.molcel.2007.08.030. [DOI] [PubMed] [Google Scholar]
- 25.Kang H, Jung JW, Kim MK, Chung JH. CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS One. 2009;4:e6611. doi: 10.1371/journal.pone.0006611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Fu W, Chen J, et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol. 2007;9:1253–1262. doi: 10.1038/ncb1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McKendrick L, Milne D, Meek D. Protein kinase CK2-dependent regulation of p53 function: evidence that the phosphorylation status of the serine 386 (CK2) site of p53 is constitutive and stable. Mol Cell Biochem. 1999;191:187–199. [PubMed] [Google Scholar]
- 28.Bennett R, Pan Y, Christian J, Hui T, May WS., Jr The RAX/PACT-PKR stress response pathway promotes p53 sumoylation and activation, leading to G1 arrest. Cell Cycle. 2012;11:407–417. doi: 10.4161/cc.11.2.18999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Reid JF, Gariboldi M, Sokolova V, et al. Integrative approach for prioritizing cancer genes in sporadic colon cancer. Genes Chromosomes Cancer. 2009;48:953–962. doi: 10.1002/gcc.20697. [DOI] [PubMed] [Google Scholar]