

Abstract
β-catenin is a multifunctional protein that is involved in cellular structure and the Wnt/β-catenin signaling pathway. Wnt/β-catenin signaling is believed to be an inducer of cell proliferation in different tumors. However, in certain physiological contexts β-catenin also promotes apoptosis. High levels of β-catenin are found in a number of cancer cell types. Recent studies have shown that β-catenin may be correlated with carcinogenesis. Its effects and interaction with interferon (IFN)γ signaling in hepatocellular carcinoma (HCC) cells remains unknown. In the present study, high levels of β-catenin did not induce antiproliferative effects or apoptosis and did not lead to changes in the levels of caspases or activated STATs. However, high levels of β-catenin did cause positive p53 accumulation and Bcl-XL downregulation in HepG2 cells, a HCC cell line. When treated with IFNγ, apoptosis was induced more rapidly compared with cells with low β-catenin levels (P<0.05), whereas caspases 3, 8 and 9 were markedly activated. The caspase inhibitor Z-VAD-FMK and the STAT3 inhibitor blocked this IFNγ-induced apoptosis. Therefore, we report that high levels of β-catenin promote IFNγ-induced apoptosis in HCC in a caspase- and STAT3-dependent manner, and facilitate the activation of executor caspases, possibly via regulation of p53 and Bcl-XL levels. These findings may provide foundations for the development of new IFN-based therapies against liver cancer.
Keywords: β-catenin, interferon-γ, apoptosis, hepatocellular carcinoma
Introduction
There are three types of interferons (IFNs): type I, II and III (1,2). The signaling pathway induced by all IFNs is the JAK/STAT pathway, in which different STAT proteins play critical roles (3). IFNγ is the only type II IFN and is involved in a broad spectrum of immune regulations, including antiviral and antitumor activities. One of the dominant mechanisms of these activities is facilitating the induction of apoptosis in the affected cells (4).
Apoptosis, also known as programmed cell death, is one of the most important mechanisms for antiviral and antitumor activities, and is induced by a number of cytokines (5). It is primarily executed by caspases, the cysteine aspartate-specific proteases (6,7). There are two major pathways, one mediated by mitochondria (intrinsic pathway) and another mediated by death receptors (extrinsic pathway), both of which lead to activation of caspases (5,8,9). Activated caspases cleave different cellular proteins causing genomic DNA fragmentation, cell morphology changes and, eventually, cell death. Other proteins, including p53 and Bcl-XL, have also been reported to be critical in the apoptosis signaling pathway (10). Regulations between these proteins and the β-catenin pathway have been reported in several cancer cell types, but not in hepatocellular carcinoma (HCC) cells (11–14).
β-catenin is a key component of the Wnt/β-catenin signaling pathway, and a mediator for the Ras/phosphatidylinositol 3-kinase (PI3K) pathways (15,16). Active β-catenin interacts with transcription factors such as T cell factor/lymphoid enhancer (TCF/LEF), CBP and p300, leading to target gene transcription. The downstream biological activities mediated by active β-catenin include differentiation, survival and proliferation. In addition, active β-catenin also binds to cadherins in the cell membrane to provide structural support for adhesion (17,18). The Wnt signaling pathway is also involved in the carcinogenesis of a number of types of cancer and is commonly believed to be a survival pathway. There are only a few reports of its contribution to apoptosis induction (12–14,19,20). Nothing has been reported concerning the effects of high levels of β-catenin on IFNγ signaling in HCC cells.
Previously, we studied the regulation of IFNγ and the β-catenin/Wnt signaling pathway in human astrocytes (21). In the present study, we intended to investigate the effect of upregulated β-catenin on IFNγ-induced apoptosis in human liver carcinoma cells, the molecular mechanisms by which this occurs.
Materials and methods
Reagents and antibodies
FITC-conjugated mouse anti-human caspase 3, caspase 8 and p53 antibodies, APC-conjugated mouse anti-human caspase 9 antibody and mouse anti-human β-catenin antibody were purchased from BD Biosciences (San Jose, CA, USA). FITC-conjugated goat anti-mouse antibody was purchased from Jackson ImmunoResearch Laboratories, Inc. (West Grove, PA, USA). Mouse anti-human Bcl-XL antibody was purchased from MBL International Corporation (Woburn, MA, USA). The STAT1 inhibitor fludarabine (FLUD) was purchased from Sigma-Aldrich (St. Louis, MO, USA. The STAT3 inhibitor S3I was purchased from Calbiochem/EMD Biosciences, Inc. (Gibbstown, NJ, USA). The pancaspase inhibitor Z-VAD-FMK was purchased from Calbiochem/EMD Biosciences.
Cell lines, DNA constructs and transfection
HepG2 cells, a human HCC cell line (PriCell Research Institute, Wuhan, China) were maintained in DMEM (Sigma-Aldrich) with 10% heat-inactivated fetal bovine serum (FBS; Sigma-Aldrich). HepG2 cells were transfected with a constitutively active β-catenin construct or its cognate vector using TransIT transfection kit (Mirus Bio LLC, Madison, WI, USA) following manufacturer’s instruction. The constitutively active β-catenin plasmid contains a serine-to-tyrosine mutation at position 33 that protects the protein from proteosomal degradation.
Immunofluorescence staining and flow cytometry analysis
Flow cytometry was performed as described previously (21). To detach HepG2 cells without cleaving surface proteins, they were incubated with 1 mM EDTA for 5 min and then washed and suspended in 1X PBS. Cells were stained with appropriate target antibodies and isotype antibodies using conventional surface- and/or intracellular-staining methods. When both surface and intracellular staining was desired, cells were first fixed and made permeable using BD Cytofix/Cytoperm Fixation and Permeating Solution (BD Pharmingen; San Diego, CA, USA), followed by staining for intracellular proteins. Cells were then washed extensively with 1X PBS to remove excess antibodies, stained for extracellular targets, and fixed with 2% formaldehyde. Fluorescence was evaluated with a FACS Caliber flow cytometer, and data analyzed using FlowJo software (Tree Star, Inc., Ashland, OR, USA).
Proliferation and cell viability assays
Cell viability assays were performed as previously described (22,23). Briefly, to determine cell viability, equal amount of cells (105 cells/well) were plated in 6-well plates and transfected and/or treated, as indicated in the text. Dead cells lost their attachment and were washed away by 1X PBS. Viable (adherent) cells were released from the wells by trypsinization prior to cell counting.
TUNEL assay
TUNEL assay to determine DNA fragmentation in apoptotic cells was performed according to the manufacturer’s instructions (Promega Corporation, Madison, WI, USA). Briefly, 3–5x106 cells were trypsinized, washed twice with cold PBS, fixed in 4% paraformaldehyde at 4˚C for 20 min, washed again with PBS and made permeable with 0.5 ml 0.5% saponin at 22˚C for 5 min. The cells were washed with PBS, incubated with 80 μl equilibration buffer at 22˚C for 5 min, washed with PBS, re-suspended in 50 μl Nucleotide Mix and incubated in the dark at 37˚C for 1 h. Cells were washed again with PBS then analyzed by fluorescence microscopy.
Statistical analysis
Statistical analyses were performed using Prism software (GraphPad Prism). Untreated and treated groups were compared using the Student’s t-test when the data were normally distributed. When the data showed abnormal distribution, the two groups were compared using the nonparametric Mann-Whitney U test. All tests were two-tailed. P<0.05 was considered to indicate a statistically significant difference.
Results
Excess β-catenin promotes IFNγ-induced apoptosis in HepG2 cells
To upregulate β-catenin, we transfected HepG2 HCC cells with a constitutively active construct of β-catenin (β-catenin pcDNA). Being controls, equal amount of HepG2 cells were transfected with cognate vector (Mock) and GFP construct (GFP) respectively. To test the efficiency of the transfection, the level of GFP and active β-catenin was determined by flow cytometry, and the results are shown in Fig. 1A and B, respectively. Compared with Mock- and GFP-transfected cells, the levels of β-catenin in β-catenin pcDNA-transfected cells were significantly elevated (P<0.05).
Figure 1.
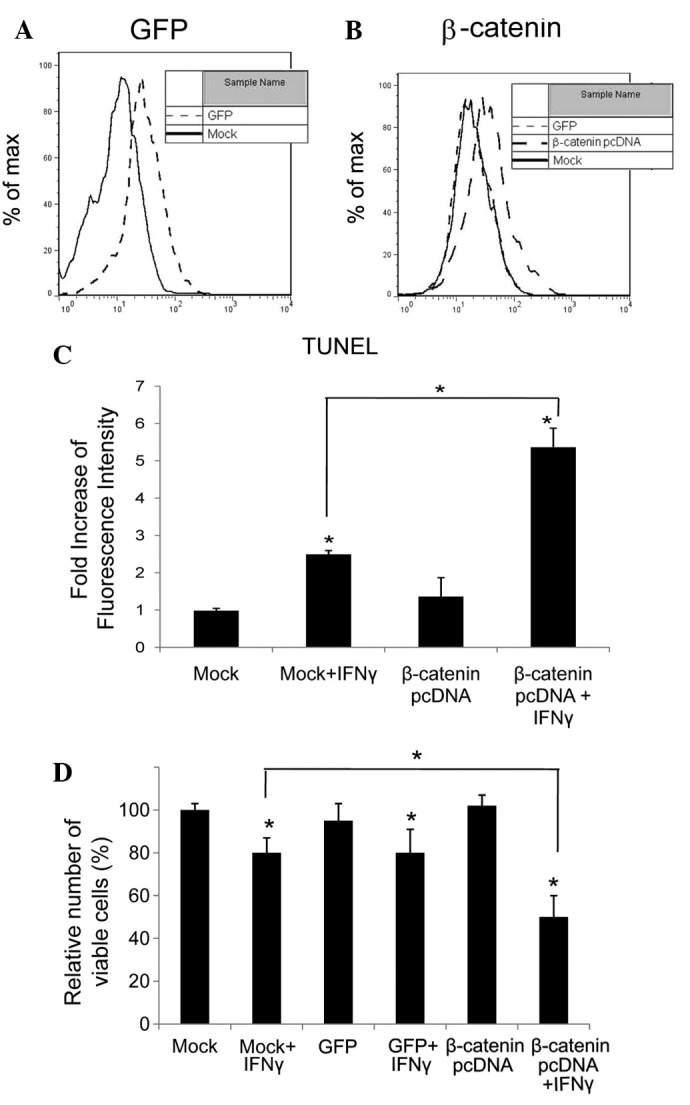
Overexpression of β-catenin in HepG2 cells. (A) HepG2 cells were transfected with plasmid expressing GFP (GFP) or its cognate vector (Mock), and GFP expression was tested by flow cytometry. (B) HepG2 cells were transfected with plasmid expressing GFP (GFP), active β-catenin (β-catenin pcDNA) or its cognate vector (Mock), and β-catenin levels were determined by flow cytometry. (C) HepG2 cells were transfected with plasmid expressing GFP (GFP), active β-catenin (β-catenin pcDNA), or its cognate vector (Mock). The transfected cells were then treated with or without IFNγ (100 ng/ml) for 48 h, and viable cells were counted. (D) HepG2 cells were transfected with plasmid expressing active β-catenin (β-catenin pcDNA) or its cognate vector (Mock). The transfected cells were then treated with or without IFNγ (100 ng/ml) for 48 h, and TUNEL assays were performed to detect apoptosis. *P<0.05 compared with corresponding untreated cells. IFN, interferon.
HepG2 cells transfected with β-catenin pcDNA, cognate vector (Mock) or GFP construct (GFP) were left untreated and treated with IFNγ (100 ng/ml) for 72 h, and viable cells were counted under a microscope. The results demonstrated that, compared with untreated controls, IFNγ reduced viable cell counts in all three groups of transfected cells, but most significantly in cells which expressed excess β-catenin (P<0.05). Upregulated β-catenin alone in HepG2 cells did not affect cell proliferation (P<0.05; Fig. 1C).
To determine whether apoptosis was induced by IFNγ in HepG2 cells, TUNEL assay was used to detect DNA fragmentation in apoptosis, and was performed on β-catenin pcDNA- and cognate vector (Mock)-transfected HepG2 cells, treated with or without IFNγ for 72 h. The fluorescence was elevated by 2.5-fold in the IFNγ-treated Mock-transfected cells and 5.5-fold in cells expressing excess β-catenin (Fig. 1D). IFNγ-induced apoptosis was promoted in HepG2 cells with high levels of β-catenin.
β-catenin upregulation leads to changes in signaling components in apoptosis pathway
We next investigated whether levels of signaling components in the apoptosis pathway were changed due to β-catenin upregulation. Levels of activated caspase 3 (Fig. 2A), 8 (Fig. 2B) and 9 (Fig. 2C) were tested, and found to be increased 2-, 2- and 1.5-fold in HepG2 cells transfected with cognate vector (Mock) or GFP and 4-, 4- and 2-fold in HepG2 cells with excess β-catenin, when treated with IFNγ (100 ng/ml) for 72 h, compared with untreated cells (P<0.05), respectively.
Figure 2.
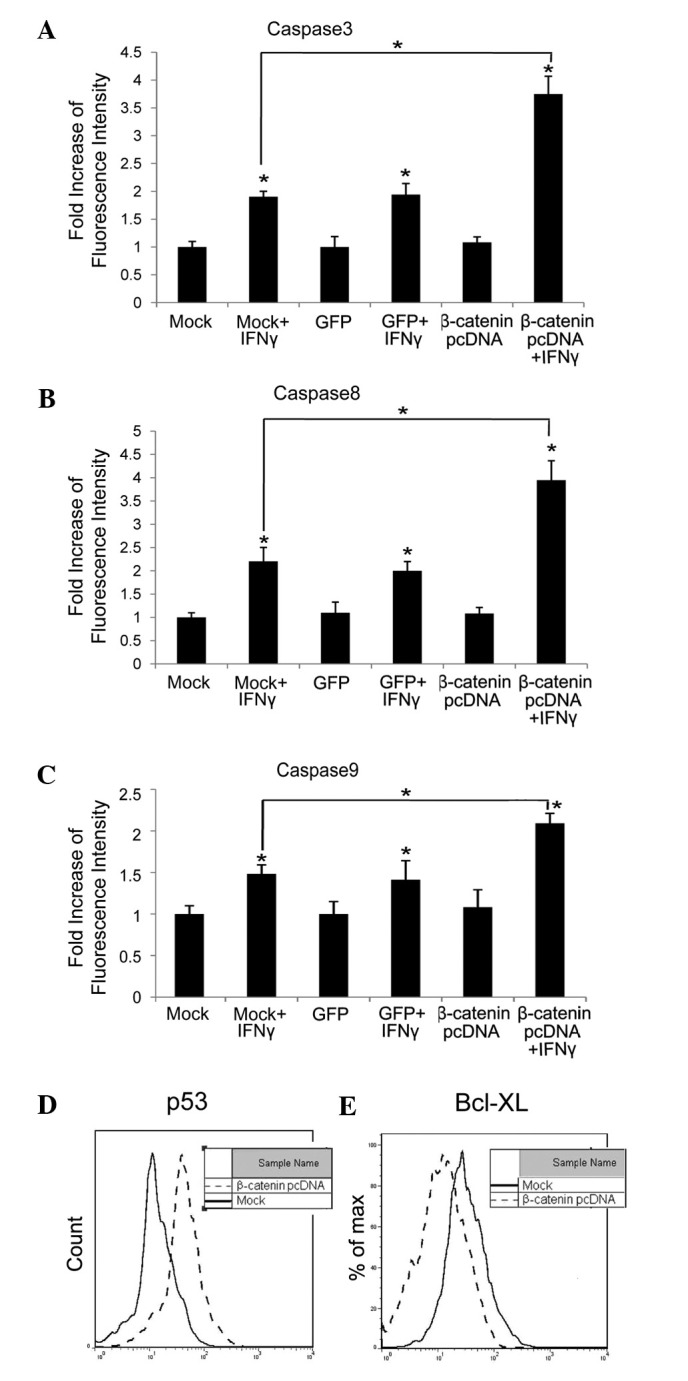
Excess β-catenin promotes IFNγ-induced apoptosis. HepG2 cells were transfected with plasmid expressing GFP (GFP), active β-catenin (β-catenin pcDNA)or its cognate vector (Mock). The transfected cells were then treated with or without IFNγ (100 ng/ml) for 48 h, and levels of activated (A) caspase 3, (B) caspase 8 and (C) caspase 9 were tested by flow cytometry. In HepG2 cells overexpressing active β-catenin, (D) p53 and (E) Bcl-XL levels were determined by flow cytometry. *P<0.05 compared with corresponding untreated cells. IFN, interferon.
Previous studies have reported that, due to high levels of β-catenin, p53 was accumulated and the Bcl-XL level was decreased (11–14). p53 is a proapoptotic cellular protein, while Bcl-XL is antiapoptotic. Both are vital components in carcinogenesis (10).
In the present study we tested p53 and Bcl-XL levels in HepG2 cells transfected with β-catenin pcDNA or its cognate vector (Mock). The results indicated that, when β-catenin was upregulated, the p53 level was elevated and the Bcl-XL level was reduced (Fig. 2D and E).
Roles of STATs and caspases in IFNγ-induced apoptosis in HepG2 cells with excess β-catenin
To further identify the key signaling components for IFNγ to induce apoptosis in HepG2 cells that express excess β-catenin, we used inhibitors for STAT1, STAT3 and caspases. In a previous study, we demonstrated that STAT1 and STAT3 were induced by IFNγ in human astroglioma cells, in which STAT3 played a key role in the regulation of the β-catenin pathway (21). In the present study, we confirmed the STAT1 and STAT3 activation induced by IFNγ, and the effects of STAT1 inhibitor, FLUD, and STAT3 inhibitor, S3I, in HepG2 cells with high levels of β-catenin by flow cytometry (Fig. 3A and B). Apoptosis induction in the presence or absence of FLUD or S3I was then tested by TUNEL assay, and results showed that FLUD partially blocked IFNγ-induced apoptosis, while S3I completely suppressed it (Fig. 3C and D). These results demonstrated that STAT3 is critical for IFNγ-induced apoptosis in HepG2 cells with high levels of β-catenin.
Figure 3.
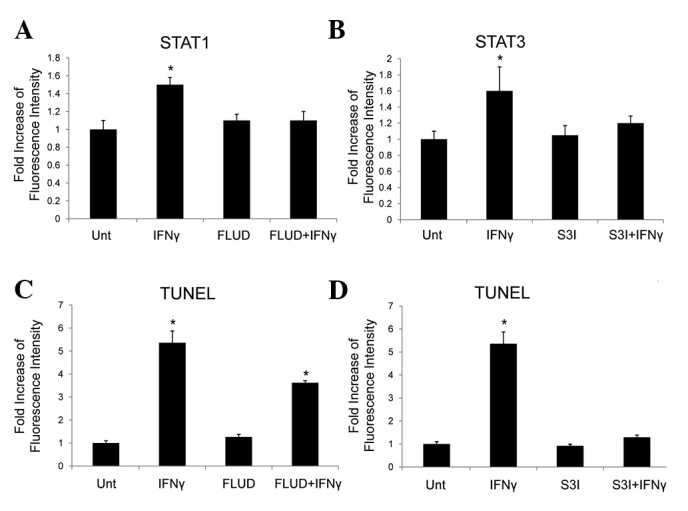
Apoptosis induced by IFNγ in HepG2 cells with excess β-catenin is STAT3-dependent. HepG2 cells were transfected with plasmid expressing active β-catenin. The transfected cells were then treated with or without IFNγ (100 ng/ml) in the presence or absence of (A) STAT1 inhibitor FLUD or (B) STAT3 inhibitor S3I, and active STAT1 and STAT3 levels were determined by flow cytometry, respectively. The transfected cells were treated with or without IFNγ (100 ng/ml) in the presence or absence of (C) FLUD or (D) S3I for 48 h, and apoptosis induction was evaluated by TUNEL assay. *P<0.05 compared with corresponding untreated cells. IFN, interferon; FLUD, fludarabine.
We have demonstrated that caspases were induced by IFNγ in HepG2 cells with upregulated β-catenin. We then tested their necessity. The pancaspase inhibitor Z-VAD-FMK was used. Caspases 3, 8 and 9 were blocked by Z-VAD-FMK (Fig. 4A–C) and IFNγ-induced apoptosis was also inhibited, as demonstrated by the results of the TUNEL assay (Fig. 4D). These data illustrate that the apoptosis induced by IFNγ in HepG2 cells with high levels of β-catenin is caspase-dependent.
Figure 4.
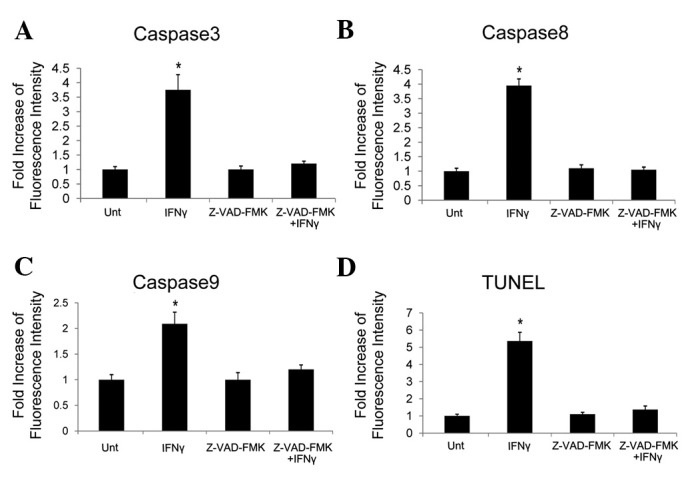
Apoptosis induced by IFNγ in HepG2 cells with excess β-catenin is caspase-dependent. HepG2 cells were transfected with plasmid expressing active β-catenin. The transfected cells were treated with or without IFNγ (100 ng/ml) in the presence or absence of the pancaspase inhibitor Z-VAD-FMK for 48 h. The levels of active caspase (A) 3, (B) 8 and (C) 9 were determined by flow cytometry. (D) Apoptosis was evaluated by TUNEL assay. *P<0.05 compared with corresponding untreated cells. IFN, interferon.
Discussion
Although β-catenin pathway is recognized as a well-known enhancer of proliferation and survival in tumor cells, its over-expression or accumulation has also been reported to induce apoptosis in fibroblasts and multiple myeloma cells, as well as several other tumor cell lines (12–14,19,20,24). Raab et al demonstrated that inhibition of PKC led to accumulation of active β-catenin, which contributes to enzastaurin-induced cell death in multiple myeloma cells (20). Other studies have shown that constitutively active β-catenin triggered p53-dependent growth arrest in fibroblasts and endometrial carcinoma cells (12,13). However, Kim et al reported the induction of apoptosis independent of p53 status and LEF-1 activation by β-catenin, when it was overexpressed in colon cancer or HeLa cells (14). Overexpression of a stable form of β-catenin or inhibited endogenous β-catenin degradation has been reported to lead to G2 cell cycle arrest and apoptosis in epidermal keratinocytes (24). Nevertheless, the ability of β-catenin to induce apoptosis has been discovered but not well characterized. In the present study, we found that overexpression of β-catenin alone did not promote apoptosis in liver carcinoma cells. However, when combined with IFNγ stimulation, apoptosis was markedly induced compared with Mock-transfected liver carcinoma cells. In addition, we aimed to identify key modulators in this regulation. Studies concerning the regulation of the β-catenin pathway by IFNγ have been published (21,25). We have shown that the β-catenin pathway regulates IFNγ signaling.
In this study, we showed the proapoptotic effect of accumulated β-catenin in IFNγ-treated liver carcinoma cells. The active β-catenin was upregulated by transfection of a plasmid containing sequence of a constitutively active β-catenin (β-catenin pcDNA). High levels of β-catenin alone did not affect the proliferation of transfected HepG2 cells, but promoted IFNγ-induced apoptosis compared with data of Mock-transfected cells, confirmed by TUNEL assay (Fig. 1). In other studies, upregulated β-catenin alone led to apoptosis in specific cell lines (11,12,14), which is different from the results of the present study in HepG2 cells.
We next found that excess β-catenin further promoted the IFNγ-induced activation of caspases 3, 8 and 9, upregulated the p53 level and downregulated Bcl-XL, compared with Mock-transfected cells (Fig. 2). It is known that IFNγ induces caspases 3, 8 and 9 in certain cell lines, including glioblastoma and conjunctival epithelial cells (26,27). In the present study, we demonstrated that IFNγ induced these caspases in HCC cells, and that their activation was enhanced by β-catenin overexpression. It has been reported that excess β-catenin results in p53 accumulation (11–13), which is consistent with our findings. We also showed that excess β-catenin down-regulated Bcl-XL in HCC cells, which is in accordance with the study by Kim et al, where Bcl-XL inhibited the apoptotic effects of excess β-catenin (14).
We further investigated the importance of several key signaling components in IFNγ-induced apoptosis. We used STAT1, STAT3 and caspase inhibitors (FLUD, S3I and Z-VAD-FMK, respectively) to inhibit specific signaling proteins, and observed their effects on IFNγ-induced apoptosis in cells expressing stable β-catenin. STAT1 and STAT3 are induced by all IFNs, including IFNγ, and are critical signaling components in the JAK/STAT pathway (3). Z-VAD-FMK has been reported to be able inhibit most caspases to block IFNγ-induced apoptosis in HT29 colorectal carcinoma cells (28). We found that STAT3 and caspases, but not STAT1, were indispensible for apoptosis induction (Figs. 3 and 4). This is consistent with the results of our previous study in human astroglioma cells, in which IFNγ regulates the β-catenin pathway in a STAT3-dependent manner, in which STAT1 it is not necessarily involved (21).
The β-catenin pathway is generally considered a survival signaling pathway, but the results of the present study, along with several others, clearly describe its positive roles in apoptosis induction (11–14,20). It remains unclear as to which mechanisms it employs to trigger apoptosis. There may be a molecular ‘detector’ to monitor β-catenin levels, which may be extremely high in cancer cells. When the level of β-catenin reaches a certain threshold level, the detector triggers apoptosis, with or without additional stimulation, for example, by IFNγ. This hypothesis requires further investigation. IFNγ is a strong immune modulator, and has a broad effect on the immune system (4). New findings on the interaction between the β-catenin and IFNγ pathways may aid the understanding of the cellular signaling network, the identification of the potentials of β-catenin and IFNγ signaling and the development of approaches to manage different types of cancer.
We have identified the potential of the β-catenin pathway in promoting apoptosis induction. It is possible that the upregulation of β-catenin in cancer cells may induce apoptosis and eliminate cancer cells. Further studies are required to test this hypothesis. There are chemicals, such as DKK1 neutralizing antibody, that upregulate β-catenin (21) and which may be used to promote IFNγ-induced apoptosis in liver cancer cells.
In conclusion, we have revealed the regulation of the IFNγ signaling pathway by the β-catenin pathway in liver cancer cells. We have shown in this study that the overexpression of β-catenin in HCC cells promoted IFNγ-induced apoptosis, possibly via the regulation of p53 and Bcl-XL levels. The apoptosis was STAT3- and caspase-dependent. These findings extend our knowledge of the Wnt/β-catenin pathway and its interaction with the IFN signaling pathway, which may aid the development of new strategies to manage liver cancer.
Reference
- 1.Parmar S, Platanias LC. Interferons. Cancer Treat Res. 2005;126:45–68. doi: 10.1007/0-387-24361-5_3. [DOI] [PubMed] [Google Scholar]
- 2.Kelly C, Klenerman P, Barnes E. Interferon lambdas: the next cytokine storm. Gut. 2011;60:1284–1293. doi: 10.1136/gut.2010.222976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol. 2008;19:311–318. doi: 10.1016/j.semcdb.2008.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Boehm U, Klamp T, Groot M, Howard JC. Cellular responses to interferon-gamma. Annu Rev Immunol. 1997;15:749–795. doi: 10.1146/annurev.immunol.15.1.749. [DOI] [PubMed] [Google Scholar]
- 5.Clemens MJ. Interferons and apoptosis. J Interferon Cytokine Res. 2003;23:277–292. doi: 10.1089/107999003766628124. [DOI] [PubMed] [Google Scholar]
- 6.Thornberry NA. Caspases: key mediators of apoptosis. Chem Biol. 1998;5:R97–R103. doi: 10.1016/s1074-5521(98)90615-9. [DOI] [PubMed] [Google Scholar]
- 7.Stennicke HR, Salvesen GS. Caspase assays. Methods Enzymol. 2000;322:91–100. doi: 10.1016/s0076-6879(00)22010-7. [DOI] [PubMed] [Google Scholar]
- 8.Kalvakolanu DV. The GRIMs: a new interface between cell death regulation and interferon/retinoid induced growth suppression. Cytokine Growth Factor Rev. 2004;15:169–194. doi: 10.1016/j.cytogfr.2004.01.002. [DOI] [PubMed] [Google Scholar]
- 9.Hengartner MO. The biochemistry of apoptosis. Nature. 2000;407:770–776. doi: 10.1038/35037710. [DOI] [PubMed] [Google Scholar]
- 10.Elmore S. Apoptosis: a review of programmed cell death. Toxicol Pathol. 2007;35:495–516. doi: 10.1080/01926230701320337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Damalas A, Ben-Ze’ev A, Simcha I, et al. Excess beta-catenin promotes accumulation of transcriptionally active p53. EMBO J. 1999;18:3054–3063. doi: 10.1093/emboj/18.11.3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Damalas A, Kahan S, Shtutman M, Ben-Ze’ev A, Oren M. Deregulated beta-catenin induces a p53- and ARF-dependent growth arrest and cooperates with Ras in transformation. EMBO J. 2001;20:4912–4922. doi: 10.1093/emboj/20.17.4912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Saegusa M, Hashimura M, Kuwata T, Hamano M, Okayasu I. Beta-catenin simultaneously induces activation of the p53-p21WAF1 pathway and overexpression of cyclin D1 during squamous differentiation of endometrial carcinoma cells. Am J Pathol. 2004;164:1739–1749. doi: 10.1016/s0002-9440(10)63732-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim K, Pang KM, Evans M, Hay ED. Overexpression of beta-catenin induces apoptosis independent of its transactivation function with LEF-1 or the involvement of major G1 cell cycle regulators. Mol Biol Cell. 2000;11:3509–3523. doi: 10.1091/mbc.11.10.3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Espada J, Pérez-Moreno M, Braga VM, Rodriguez-Viciana P, Cano A. H-Ras activation promotes cytoplasmic accumulation and phosphoinositide 3-OH kinase association of beta-catenin in epidermal keratinocytes. J Cell Biol. 1999;146:967–980. doi: 10.1083/jcb.146.5.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willert K, Shibamoto S, Nusse R. Wnt-induced dephosphorylation of axin releases beta-catenin from the axin complex. Genes Dev. 1999;13:1768–1773. doi: 10.1101/gad.13.14.1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Moon RT, Brown JD, Torres M. WNTs modulate cell fate and behavior during vertebrate development. Trends Genet. 1997;13:157–162. doi: 10.1016/s0168-9525(97)01093-7. [DOI] [PubMed] [Google Scholar]
- 18.Miller JR, Moon RT. Signal transduction through beta-catenin and specification of cell fate during embryogenesis. Genes Dev. 1996;10:2527–2539. doi: 10.1101/gad.10.20.2527. [DOI] [PubMed] [Google Scholar]
- 19.Ghosh JC, Altieri DC. Activation of p53-dependent apoptosis by acute ablation of glycogen synthase kinase-3beta in colorectal cancer cells. Clin Cancer Res. 2005;11:4580–4588. doi: 10.1158/1078-0432.CCR-04-2624. [DOI] [PubMed] [Google Scholar]
- 20.Raab MS, Breitkreutz I, Tonon G, et al. Targeting PKC: a novel role for beta-catenin in ER stress and apoptotic signaling. Blood. 2009;113:1513–1521. doi: 10.1182/blood-2008-05-157040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li W, Henderson LJ, Major EO, Al-Harthi L. IFN-gamma mediates enhancement of HIV replication in astrocytes by inducing an antagonist of the beta-catenin pathway (DKK1) in a STAT 3-dependent manner. J Immunol. 2011;186:6771–6778. doi: 10.4049/jimmunol.1100099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li W, Lewis-Antes A, Huang J, Balan M, Kotenko SV. Regulation of apoptosis by type III interferons. Cell Prolif. 2008;41:960–979. doi: 10.1111/j.1365-2184.2008.00558.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li W, Huang X, Liu Z, Wang Y, Zhang H, Tong H, Wu H, Lin S. Type III interferon induces apoptosis in human lung cancer cells. Oncol Rep. 2012;28:1117–1125. doi: 10.3892/or.2012.1901. [DOI] [PubMed] [Google Scholar]
- 24.Olmeda D, Castel S, Vilaro S, Cano A. Beta-catenin regulation during the cell cycle: implications in G2/M and apoptosis. Mol Biol Cell. 2003;14:2844–2860. doi: 10.1091/mbc.E03-01-0865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nava P, Koch S, Laukoetter MG, et al. Interferon-gamma regulates intestinal epithelial homeostasis through converging beta-catenin signaling pathways. Immunity. 2010;32:392–402. doi: 10.1016/j.immuni.2010.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Janardhanan R, Banik NL, Ray SK. N-(4-Hydroxyphenyl) retinamide induced differentiation with repression of telomerase and cell cycle to increase interferon-gamma sensitivity for apoptosis in human glioblastoma cells. Cancer Lett. 2008;261:26–36. doi: 10.1016/j.canlet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang X, Chen W, De Paiva CS, et al. Interferon-gamma exacerbates dry eye-induced apoptosis in conjunctiva through dual apoptotic pathways. Invest Ophthalmol Vis Sci. 2011;52:6279–6285. doi: 10.1167/iovs.10-7081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang MC, Liu HP, Demchik LL, Zhai YF, Yang DJ. LIGHT sensitizes IFN-gamma-mediated apoptosis of HT-29 human carcinoma cells through both death receptor and mitochondria pathways. Cell Res. 2004;14:117–124. doi: 10.1038/sj.cr.7290210. [DOI] [PubMed] [Google Scholar]