

Abstract
Castration-resistant prostate cancer (PCa) is refractory to hormone therapy and new strategies for treatment are urgently needed. We found that androgen-insensitive (AI) PCa cells, LNCaP-AI, are reprogrammed to upregulate the mitotic kinase Plk1 and other M phase cell cycle proteins, which may underlie AI PCa growth. In androgen-depleted media, LNCaP-AI cells showed exquisite sensitivity to growth inhibition by subnanomolar concentrations of a small molecule inhibitor of Plk1, BI2536, suggesting that these cells are dependent on Plk1 for growth. In contrast, the androgen-responsive parental LNCaP cells showed negligible responses to BI2536 treatment under the same condition. BI2536 treatment of LNCaP-AI cells resulted in an increase in cell death marker PARP-1 but did not activate caspase-3, an apoptosis marker, suggesting that the observed cell death was caspase-independent. BI2536-treated LNCaP-AI cells formed multinucleated giant cells that contain clusters of nuclear vesicles indicative of mitotic catastrophe. Live-cell time-lapse imaging revealed that BI2536-treated giant LNCaP-AI cells underwent necroptosis, as evidenced by “explosive” cell death and partial reversal of cell death by a necroptosis inhibitor. Our studies suggest that LNCaP-AI cells underwent reprogramming in both their cell growth and cell death pathways, rendering them highly sensitive to Plk1 inhibition that induces necroptosis. Harnessing necroptosis through Plk1 inhibition may be explored for therapeutic intervention of castration-resistant PCa.
Keywords: Plk1, BI2536, mitotic catastrophe, necroptosis, autophagy, prostate cancer
Introduction
Prostate cancer (PCa) is the most common malignant cancer in men and is a major cause of cancer-related deaths in men in the United States (1). Although androgen-ablation therapy has resulted in initial response, castration-resistant PCa invariably occurs. Understanding the basis of this lethal progression may uncover potential targets for therapeutic intervention for castration-resistant PCa.
A recent genomics study showed that androgen-insensitive (AI) PCa cells have undergone a genetic reprogramming to selectively upregulate the expression of M phase cell cycle genes (2). Many of the reprogrammed genes encode proteins that are involved in spindle checkpoint regulation (2). Because mitotic progression is a highly regulated process, these aberrantly expressed M phase cell cycle proteins may confer a new mechanism for cell growth and are potential targets to inhibit the growth of castration-resistant PCa cells.
Polo-like kinase 1 (Plk1) (67-kDa) is a serine/threonine kinase that is critical for mitotic progression (3). Plk1 regulates centrosome maturation, bipolar spindle formation, chromosome architecture, chromosome congression, sister chromatid separation, cleavage furrow formation, and completion of cytokinesis (3, 4). Whether Plk1 expression has undergone “reprogramming” in the castration-resistant PCa is not known. In this study, we examined the expression of Plk1 in an androgen-insensitive PCa cell line LNCaP-AI. LNCaP-AI is a subline of LNCaP cells generated after long-term androgen deprivation (5). The parental LNCaP cells, an androgen-responsive PCa cell line derived from lymph node metastasis (6), were used as a control. Both LNCaP and LNCaP-AI cells express the androgen receptor and are p53 positive (5).
Here, we identified that Plk1 is upregulated only in the LNCaP-AI cells. We showed that the aberrantly upregulated Plk1 plays a role in LNCaP-AI cell growth under androgen-deprivation conditions as cells undergo cell death when treated with a small molecule inhibitor of Plk1, BI2536 (7). In contrast, the parental LNCaP cells showed negligible responses to the drug under the same condition. Live-cell imaging analysis revealed that BI2536-treated LNCaP-AI cells underwent cell death by necroptosis rather than apoptosis. Because apoptosis is often blocked in cancer cells, which causes many drugs to be ineffective, induction of necroptosis may circumvent cellular defenses in castration-resistant PCa tumor growth. Our findings support that Plk1 represents a unique target for the treatment of castration-resistant PCa.
Results
Plk1 is overexpressed in LNCaP-AI cells compared with LNCaP cells
We first characterized the growth of LNCaP and LNCaP-AI cells. As previously shown, LNCaP cells grew well in full media (10% FBS) but very poorly in androgen-depleted media (10% csFBS) (Figure 1a). In contrast, LNCaP-AI cells grew in either media, confirming that LNCaP-AI cells are not dependent on androgens for growth. Western blot analysis showed that both cell lines express the androgen receptor (AR) as reported (5). When grown in full media, we found that the mitotic kinase Plk1 is elevated about 3-fold in LNCaP-AI cells relative to LNCaP cells (Figure 1b and c). In addition, LNCaP-AI cells expressed higher levels of the M phase cell cycle proteins Cdc25 (for mitotic entry) (3), BubR1 (for spindle checkpoint) (8) and Sgo2 (for sister chromosome cohesion) (9) compared to LNCaP cells (Figure 1b). Because the levels of mitotic kinases are highly regulated for mitosis regulation, the aberrant high-level expression of mitosis-related proteins may be part of the reprogramming process to allow cell growth under the androgen-depletion condition.
Figure 1.
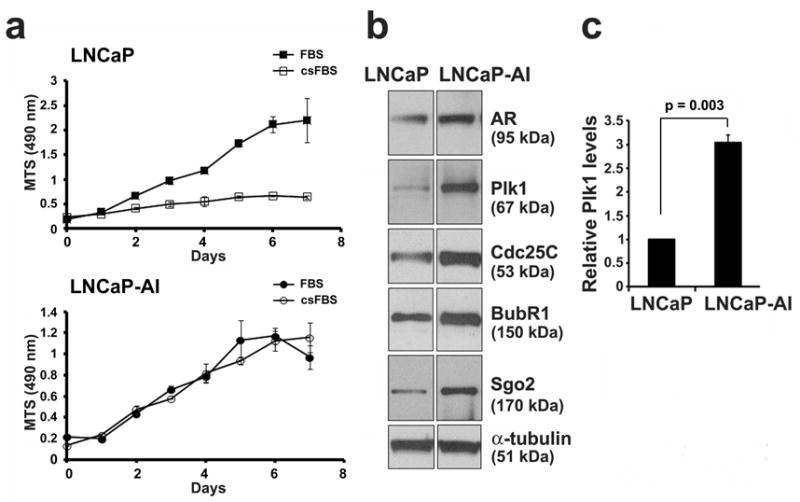
The mitotic kinase Plk1 is overexpressed in LNCaP-AI cells. (a) LNCaP and LNCaP-AI PCa cells were cultured in full media (FBS) or androgen-depleted media (csFBS). Cell growth over 7 days was determined by an MTS (OD 490 nm) growth assay. Results from triplicate samples (mean ± SD) in a representative experiment from n = 3 independent experiments are shown. (b) Lysates (20 μg) from LNCaP and LNCaP-AI cells grown in full media were immunoblotted for the androgen receptor (AR) and mitotic proteins as shown. α-tubulin was used as a loading control. (c) Relative Plk1 protein levels were determined (Plk1/tubulin and normalized against the value in LNCaP cells) (mean ± SD, n = 3 experiments).
We further analyzed the cell cycle profile of LNCaP-AI cells relative to LNCaP cells. FACS analysis of cells cultured in full media showed that more LNCaP-AI cells (36%) than LNCaP cells (14%) contained a 4N DNA content, suggesting an enrichment of LNCaP-AI cells in either G2 phase or mitotic phase (Figure 2a). To examine the basis of the higher level of Plk1 expression in LNCaP-AI cells, we examined cells costained for Plk1 and a well-known mitotic marker phospho-histone H3 (pH3) by FACS analysis. In the non-mitotic pH3(-) LNCaP-AI cells, Plk1 levels were indeed found to be higher than those in pH3(-) LNCaP cells (Figure 2b). In the mitotic pH3(+) pool, Plk1 levels increased to similar peak levels in both LNCaP-AI and LNCaP cells, but there were more cells undergoing mitosis in the LNCaP-AI cell population. These findings elucidate that the higher levels of Plk1 expression in LNCaP-AI cells are due to not only more cells undergoing mitosis, but also higher Plk1 levels in non-mitotic cells. These results raise the interesting possibility that Plk1 may have novel functions outside of mitosis that remain to be elucidated.
Figure 2.
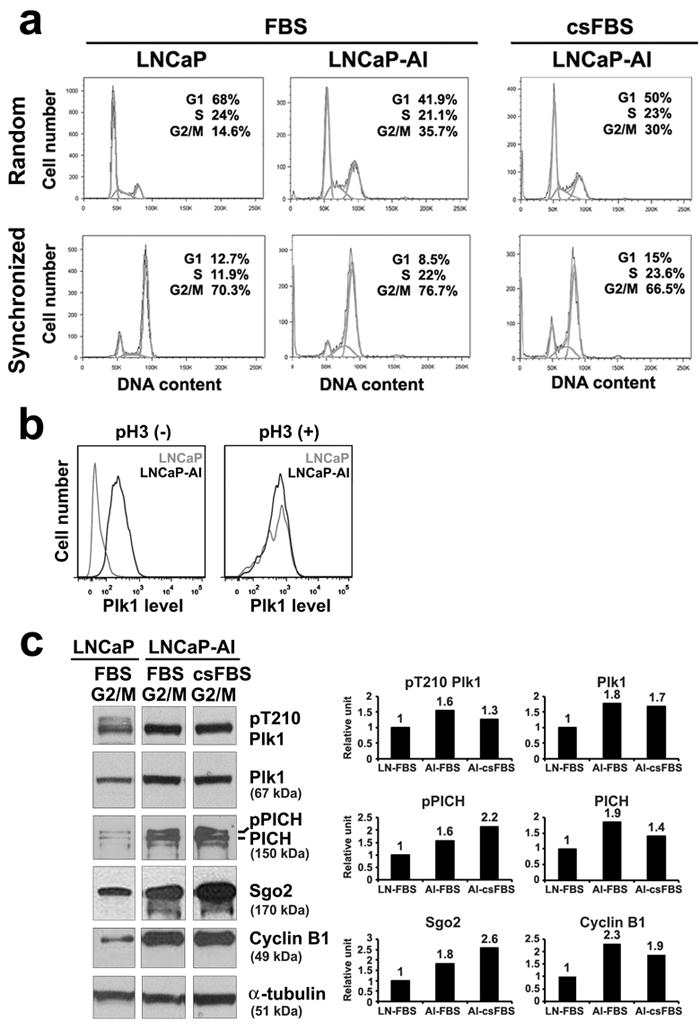
Plk1 is elevated and active in LNCaP-AI cells. Cells were cultured in either full media (FBS) or androgen-depleted media (csFBS). (a) FACS analysis of randomly growing cells and cells synchronized by nocodazole treatment to enrich for G2/M phase population. (b) FACS analysis of cells costained with Plk1 and a mitotic marker, phospho-histone H3 (pH3), in randomly cycling cells and in cells synchronized by a nocodazole treatment. pH3(-) represent non-mitotic cells; pH3(+) represent mitotic cells. (c) Lysates (20 μg) from cells enriched in G2/M by nocodazole treatment were analyzed by immunoblotting with antibodies shown. After scanning the blots (NIH ImageJ), protein levels were measured (protein/tubulin and normalized against the value in LNCaP cells). Similar data were obtained in n = 3 experiments.
When cells were synchronized by a 16 h incubation with 100 ng/ml nocodazole, a microtubule deploymerizing drug that enriches for cells in G2/M phase (10, 11), both LNCaP and LNCaP-AI cells showed a similar degree of enrichment to 70 – 80% of cells in the G2/M phase (Figure 2a). By western blot analysis of the G2/M-enriched cells, the expression of Plk1, Sgo2 and cyclin B1 was about 2-fold higher in LNCaP-AI than LNCaP cells (Figure 2c), likely due to more cells undergoing mitosis in the LNCaP-AI population (Figure 2b), and increasing mitotic protein expression in the G2 phase as cells prepare to enter mitosis. This notion is supported by the observation that Plk1 expression begins to increase as early as in S phase, continues in G2 and peaks in M phase (12). Furthermore, Plk1 is active as shown by higher levels of phosphorylation of Plk1 on T210 in its catalytic domain (pT210 Plk1) (13) and phosphorylation of a Plk1 substrate PICH (for chromosome arm architecture) (pPICH) (11) in LNCaP-AI relative to LNCaP cells (Figure 2c).
By comparison, LNCaP-AI cells cultured in androgen-depleted (csFBS) media showed a similar cell cycle distribution profile (Figure 2a) and elevated Plk1 and Plk1 pathway protein levels (Figure 2c) as cells grown in full media, further demonstrating that LNCaP-AI cells are fully capable of cell cycle progression in androgen-depleted media. The higher levels of Plk1 raise the possibility that LNCaP-AI cells may be more dependent on Plk1 for their growth.
Plk1 inhibition results in a decrease in cell growth and viability in LNCaP-AI cells
We next tested if LNCaP-AI cells are more sensitive to growth inhibition by BI2536, a small molecule ATP-competitive kinase inhibitor of Plk1 that exhibits high potency and 1,000-fold selectivity for Plk1 when compared against 63 other kinases (7). For this and subsequent experiments, cells were cultured in androgen-depleted media to mimic the clinical setting in which PCa patients have undergone androgen-ablation therapy. LNCaP-AI cell growth was inhibited by BI2536 in a dose-dependent manner, with a half-maximal inhibitory concentration (IC50) around 0.2 nM BI2536, while LNCaP cells were essentially not sensitive to the low dose of BI2536 under this growth condition (Figure 3a). FACS analysis of LNCaP-AI cells treated with four times the IC50 concentration or 0.8 nM BI2536 showed that 62% of LNCaP-AI cells accumulated in G2/M, as compared to 21% without drug treatment (Figure 3b), suggesting an arrest in mitosis. Additionally, BI2536-treated LNCaP-AI cells also showed a population with greater than 4N DNA content indicating an increase in aneuploidy as well as a population with a sub-G1 DNA content that is consistent with DNA fragmentation and/or cell death (Figure 3b). By trypan blue exclusion analysis, LNCaP-AI cells showed a dose-dependent decrease in cell viability, reaching 30% and 40% dead cells with 0.4 and 0.8 nM BI2536 treatment, respectively (Figure 3c). Thus, under conditions of androgen deprivation, LNCaP-AI cells are highly sensitive to growth inhibition by subnanomolar concentrations of BI2536.
Figure 3.
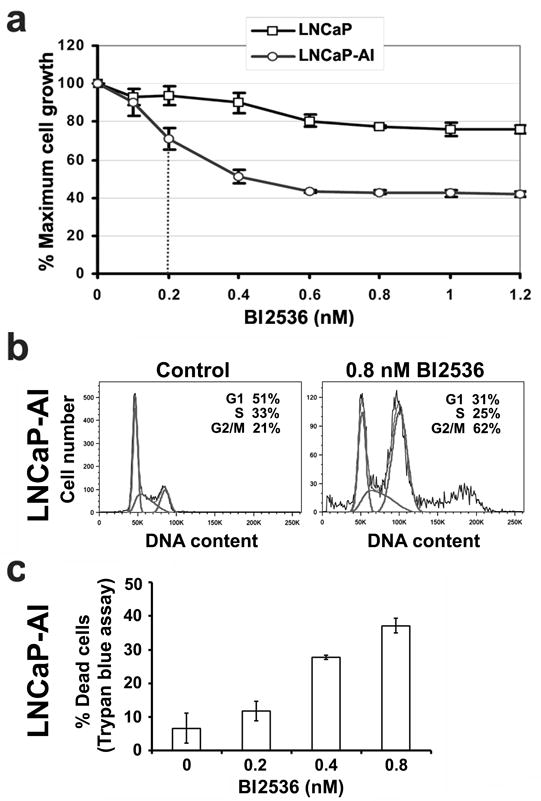
Differential response of LNCaP-AI cells to the Plk1 inhibitor BI2536. (a) Cells were cultured in androgen-depleted media and treated with increasing concentrations of BI2536 for 5 days. % Maximal growth relative to non-treated control cells (set at 100%) were analyzed in triplicates (mean ± SD) by MTS assays. Dotted line, IC50 of 0.2 nM BI2536 for LNCaP-AI cells. (b) FACS analysis of LNCaP-AI cells treated with or without 0.8 nM BI2536 for 5 days. (c) % Trypan-blue positive dead LNCaP-AI cells in (a) were counted in triplicates (mean ± SD). Similar data were obtained in n = 3 experiments.
Plk1 inhibition results in a decrease in cell growth in other androgen-insensitive prostate cancer cells
A similar sensitivity to Plk1 inhibition was also observed in three additional prostate cancer cell lines, including abl (14), C4-2B (15), and CWR22Rv1 (16), which are all androgen receptor positive but androgen-insensitive. We found that Plk1 levels are elevated in these androgen-insensitive cells relative to LNCaP cells (Figure 4a). All of these cells grew well in androgen-depleted culture conditions, although cells grew a little better in full media (Figures 4b – d). Under androgen-depleted culture conditions, the growth of abl, C4-2B and CWR22Rv1 cells was inhibited by subnanomolar concentrations of BI2536 as also observed for LNCaP-AI cells (Figures 4e – g). These results show that elevated levels of Plk1 are found not only in LNCaP-AI cells but also in other androgen-insensitive prostate cancer cells and that inhibition of Plk1 by BI2536 inhibited the growth of these androgen-insensitive prostate cancer cells. These observations raise the possibility that elevated Plk1 expression may be a shared feature in these androgen-insensitive prostate cancer cells.
Figure 4.
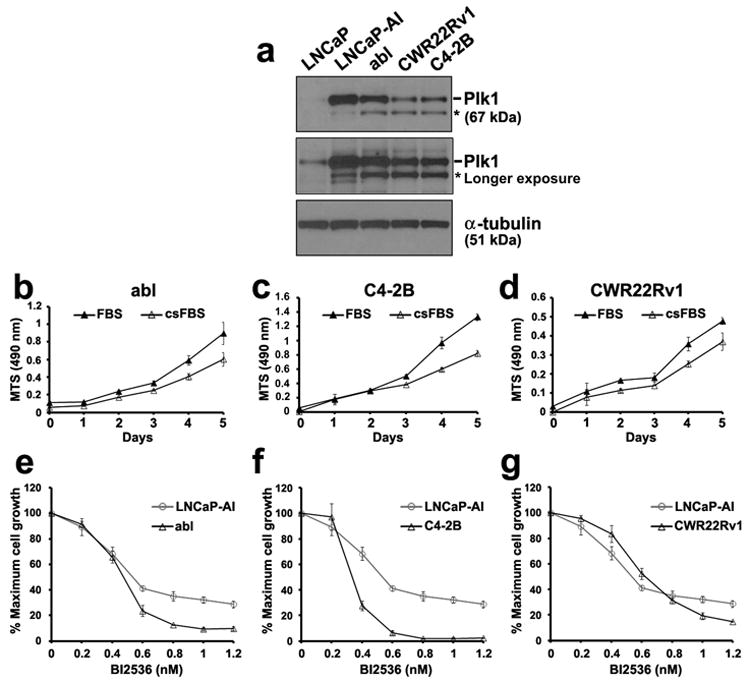
Growth inhibition of additional androgen-insensitive prostate cancer cells to the Plk1 inhibitor BI2536. (a) Lysates (20 μg) were analyzed for Plk1 levels by western blot analysis. α-tubulin was used as a loading control. *, nonspecific band. (b - d) abl, C4-2B and CWR22Rv1 cells were culture in full (FBS) or androgen-depleted (csFBS) media. Cell growth over 5 days was determined by an MTS assay. (e - g) Cells were cultured in androgen-depleted media and treated with increasing concentrations of BI2536 for 5 days. % Maximal growth relative to non-treated control cells (set at 100%) were analyzed in triplicates (mean ± SD) by MTS assays. Data in (b – g) are representative of n = 3 experiments.
Plk1 inhibition of LNCaP-AI cells does not induce classic apoptosis but does activate the autophagy pathway
We next examined the mechanism by which BI2536 mediates growth inhibition in LNCaP-AI cells. Western blot analysis showed that BI2536 treatment led to an increase in cleaved poly(ADP-ribose) polymerase-1 (PARP-1), a DNA repair enzyme whose expression is a common end point for cell death (Figure 5a), in agreement with the increase in dead cells observed above. LNCaP-AI cells treated with TNFα plus cycloheximide to induce apoptosis were used as a positive control for cell death induction (Figure 5a). These observations show that BI2536 treatment increases cell death in LNCaP-AI cells.
Figure 5.
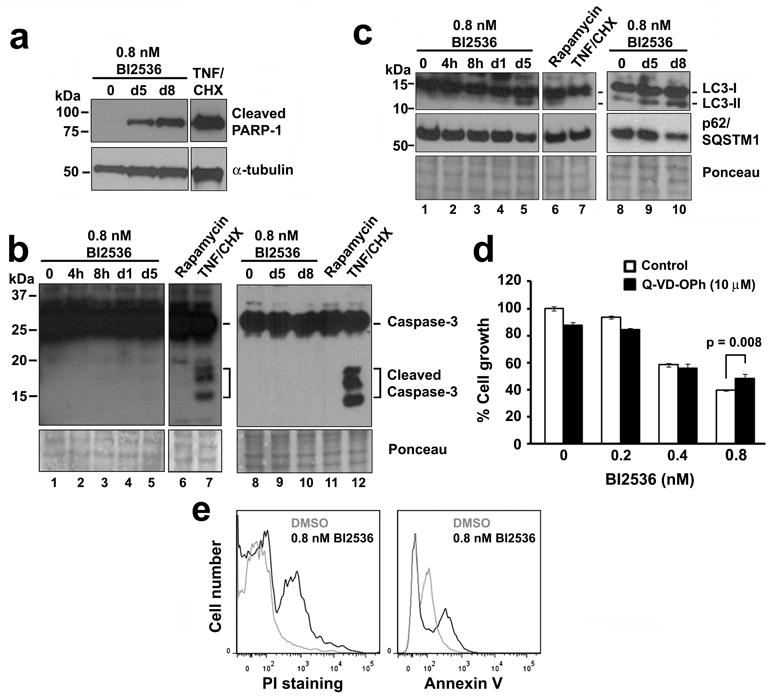
Effect of BI2536 treatment on apoptosis or autophagy pathways in LNCaP-AI cells. Cells were cultured in androgen-depleted media and treated for 5 or 8 days with 0.8 nM BI2536, 16 h with 10 ng/ml TNFα plus 10 μg/ml cycloheximide, or 24 h with 20 nM rapamycin. Cell lysates were immunoblotted for markers of (a) cell death (PARP-1); (b) apoptosis (cleaved caspase-3); and (c) autophagy (LC3-I/II and p62/sequestosome [SQSTM1]). α-tubulin or Ponceau S staining was used as a loading control. (d) % Cell growth by MTS assays of LNCaP-AI cells treated with increasing concentrations of BI2536 plus 10 μM Q-VD-OPh for 5 days. Triplicate samples were analyzed (mean ± SD). (e) FACS analysis of LNCaP-AI cells treated with or without 0.8 nM BI2536 for 5 days and stained for Annexin-V and counterstained with propidium iodide. Similar results for panels (a – d) and (e) were obtained in n = 3 and 2 experiments, respectively.
To examine whether BI2536 treatment leads to cell death by apoptosis, cells were treated with 0.8 nM BI2536 for various times up to 8 days. LNCaP-AI cells did not express cleaved caspase-3, a marker for apoptosis, even after 8 days of BI2536 treatment (Figure 5b, lanes 1 – 5, 8 – 10), even though these cells were capable of caspase-3 cleavage in response to a 16 h treatment with TNFα plus cycloheximide (Figure 5b, lanes 7 and 12). We further examined whether BI2536 treatment leads to enhanced autophagy-associated cell death by analyzing the autophagy marker light chain 3 (LC3) and the autophagy substrate p62/sequestosome1 (17). An increase in the conversion of the cytosolic-associated protein LC3I into the autophagosome-associated lipidated form LC3II and a decrease in the level of polyubiquitin-binding protein p62/sequestosome1 were observed when LNCaP-AI cells were treated with BI2536 for 5 and 8 days (Figure 5c, lanes 5, 9 and 10), but little in those treated for 1 day or less (Figure 5c, lanes 1 – 4 and 8). Cells treated with rapamycin for 24 h was used as a positive control for autophagy induction (Figure 5c, lane 6). These results suggest that autophagy induction may be involved in BI2536-induced cell death in LNCaP-AI cells.
To further confirm that the cell death observed was caspase independent, LNCaP-AI cells were incubated with BI2536 together with Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties (18), for 5 days. Q-VD-OPh did not prevent BI2536-mediated growth inhibition in LNCaP-AI cells, except at 0.8 nM BI2536 where Q-VD-OPh slightly but significantly attenuated growth inhibition (Figure 5d). We further employed FACS analysis using Annexin V and PI costaining to examine apoptotic cells. Increased PI staining was observed in BI2536-treated LNCaP-AI cells relative to DMSO-treated control cells indicating the induction of cell death in response to BI2536 treatment (Figure 5e). However, less than 15% of the BI2536-treated cells were found to die via apoptosis, as indicated by the small increase in Annexin V staining. These results suggest that cell death induced by BI2536 treatment in LNCaP-AI cells was not mediated via classic caspase-dependent apoptosis but was associated with autophagy induction. These features suggest an alternative form of cell death in BI2536-treated LNCaP-AI cells.
Plk1 inhibition results in an increase in aneuploidy in LNCaP-AI cells
To further investigate BI2536-mediated cell death modality, we examined the morphology of LNCaP-AI cells. Cells were immunostained for tubulin and counterstained with DAPI to visualize DNA. Control LNCaP-AI cells contained single round nuclei, with some cells undergoing mitosis (Figure 6a, arrow, cell in telophase). Following a 5-day treatment with 0.8 nM BI2536, all of the LNCaP-AI cells formed large multinucleated giant cells that contained up to 30 vesicles containing DAPI-positive nuclear material (Figures 6a and b). The multinucleation could explain the greater than 4N DNA profile observed by FACS analysis while the clusters of nuclear vesicles may contain missegregated chromosomes and/or fragmented chromatin that appeared as the sub-G1 population in the FACS profile (Figure 3b). Thus, Plk1 inhibition led to the formation of multinucleated giant LNCaP-AI cells with abnormal nuclear morphology.
Figure 6.
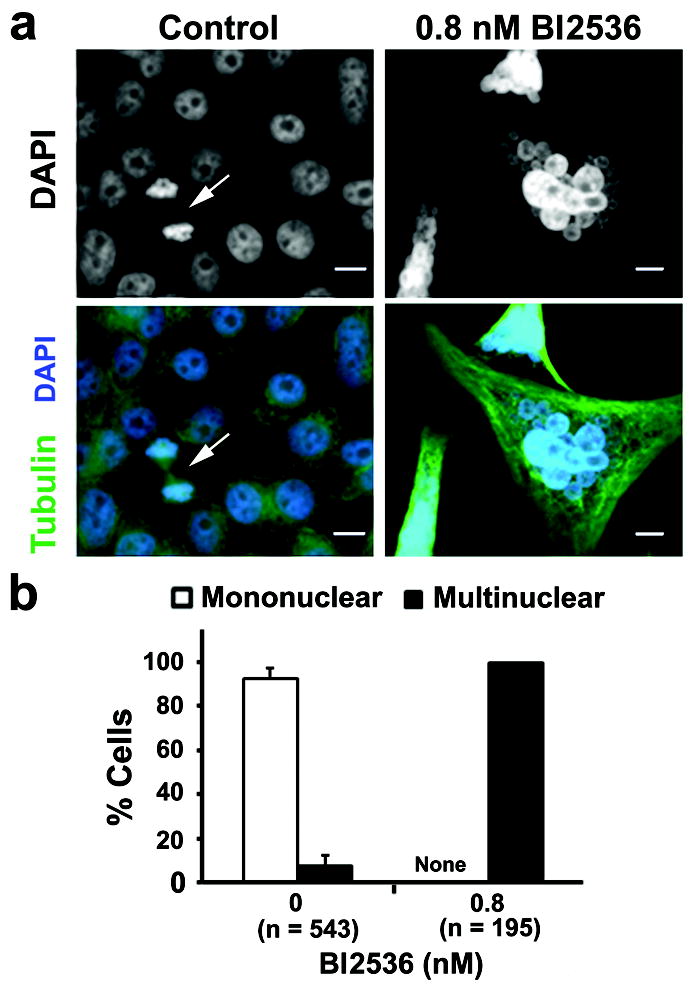
Plk1 inhibition by BI2536 resulted in distinct changes in nuclear morphology and an increase in aneuploidy in LNCaP-AI cells. (a) Cells were treated with 0.8 nM BI2536 for 5 days in androgen-depleted media, immunostained for α-tubulin (green) and counterstained with DAPI (blue) for DNA. Arrow, cells in telophase. Bars, 10 μm. (b) Nuclear morphologies for cells in A were quantified as mononuclear (normal) or multinuclear (with nuclear vesicles) (mean ± SD, n = 3 experiments). N, cell number. None, no mononuclear cells were detected.
Inhibition of Plk1 leads to mitotic catastrophe in LNCaP-AI cells
The formation of nuclear envelope around missegregated chromosomes or chromatin fragments is characteristic of cells undergoing mitotic catastrophe, a “prestage” to cell death that occurs during or shortly after a failed mitosis due to defective cell cycle checkpoints and DNA damage (19). Mitotic catastrophe can lead to cell death by apoptosis or an alternative form of necrotic cell death, recently termed necroptosis (20, 21). We next examined whether BI2536 treatment of LNCaP-AI cells leads to cell death by necroptosis. To elucidate the sequence of events leading up to cell death, live-cell imaging was used to observe individual cell fates in response to 0.8 nM BI2536. We first examined control LNCaP-AI cells, with time-lapse images taken at 15 min intervals. One LNCaP-AI cell (Figure 7a, arrow) was observed to round up in mitosis, divide into two daughter cells within the next 30 min, and complete two more rounds of cell division that generated eight progeny cells over the 96 h time course (see Supplementary Figure 1).
Figure 7.
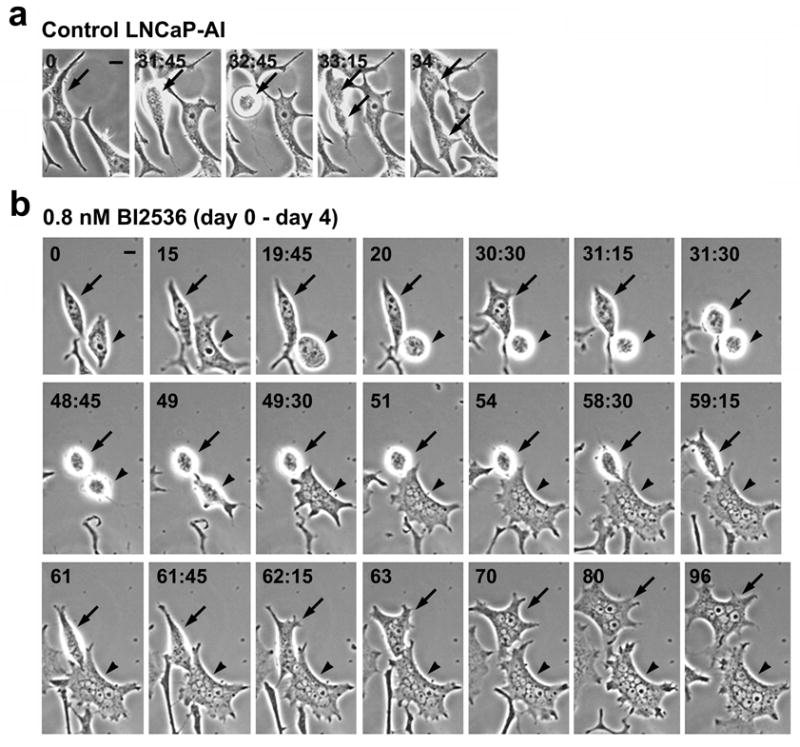
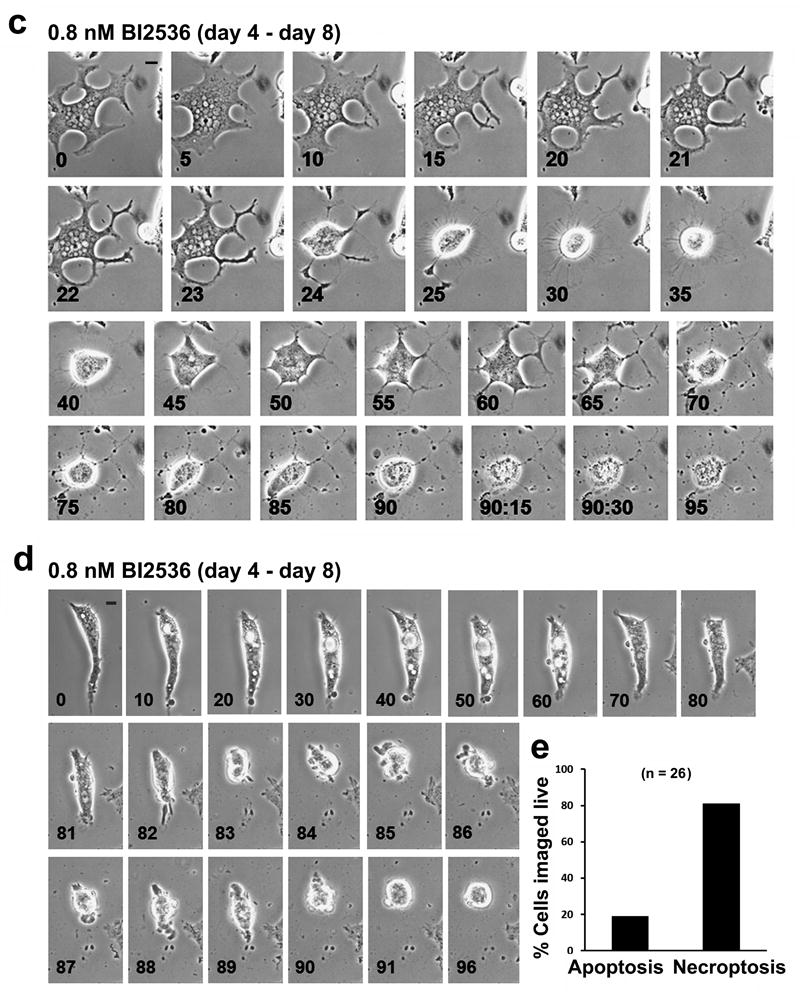
Live-cell imaging of LNCaP-AI cells in response to BI2536 treatment. Cells cultured in androgen-depleted media were treated with 0.8 nM BI2536 for 8 days. Live-cell time-lapse images were taken at 15 min intervals. (a) Control LNCaP-AI cell undergoing the first cell division (arrows). See Supplementary Figure 2 for the 96 h time course. (b) LNCaP-AI cells during days 0 – 4 of BI2536 treatment. Two cells (arrow and arrowheads) underwent cell rounding, mitotic catastrophe and turned into multinucleated giant cells. n = 83 cells were live-imaged. (c) LNCaP-AI cells during days 4 – 8 of BI2536 treatment. After a change of media on day 4, a parallel batch of cells were imaged from day 4 (T=0h) to day 8 (T=96h) of BI2536 treatment. Giant LNCaP-AI cells underwent necroptosis during this period. See Supplementary Figure 3 for additional images of the necroptotic cell death process. (d) LNCaP-AI cell undergoing apoptosis from days 4 – 8 of BI2536 treatment. All bars, 10 μm. (e) Quantification of cell death modalities in n = 26 live-imaged LNCaP-AI cells after 8 days of BI2536 treatment.
In contrast, abnormal morphologies and cellular behavior were observed after BI2536 treatment of LNCaP-AI cells. Two BI2536-treated LNCaP-AI cells (Figure 7b, arrow and arrowhead, respectively) were found to round up into mitosis, albeit with slightly different kinetics, within the first ~31 h of BI2536 treatment, which is consistent with the arrest of cells in prometaphase after Plk1 inhibition as previously reported (3, 7). The cells stayed rounded for ~30 h but failed to complete mitosis as evidenced by a lack of daughter cell production. Both cells then entered G1 and formed multinucleated giant cells containing many nuclear vesicles, indicating mitotic catastrophe (19, 22). Indeed, we followed the fate of 83 LNCaP-AI cells from day 0 to day 4 of BI2536 treatment, and found that 86% (71/83) rounded up into mitosis by 36 h; 45% (32/71) of the round cells underwent mitotic catastrophe and turned into giant cells by 72 h; and 100% (83/83) of the LNCaP-AI cells formed giant cells by 96 h of BI2536 treatment. These multinucleated giant cells also exhibited broad, pointed protrusions all around the periphery, a feature also observed in fixed cells (Figure 6).
Inhibition of Plk1 increases cell death by necroptosis in LNCaP-AI cells
To follow the fate of these multinucleated giant cells, a parallel batch of LNCaP-AI cells were imaged live from day 4 (T=0h) through day 8 (T=96h) post BI2536 treatment, after a change of media (Figure 7c). The large pointed protrusions in the giant LNCaP-AI cells became constricted and exhibited further branching along each protrusion (T=20h) (Figure 7c), suggesting extensive remodeling of the cytoskeletal network. The giant cell underwent a contraction (T=24h), likely in an attempt to enter mitosis, but remained rounded for the next 15 h, being tethered to the surface of the dish by a dense ring of thin fibrils (T=35h), which were not observed in BI2536-treated LNCaP-AI cells undergoing the first mitotic catastrophe (Figure 7b). After a prolonged mitotic arrest, the cell failed mitosis, slipped into G1, and reattached to the dish (T=40h). Remarkably, the cell flattened out along its original elaborate protrusions for the next 20 h (T=45h-60h). The cell contracted a second time (T=65h) but again failed to divide (T=90h). Within the next 15 min (T=90h:15min), there was a rapid and massive loss of plasma membrane integrity resulting in an “explosive” cell lysis that is characteristic of necroptotic cell death. A similar sequence of events was observed in another BI2536-treated giant LNCaP-AI cell (see Supplementary Figure 2). A minor population of BI2536-treated LNCaP-AI cells was observed to die by apoptosis (Figure 7d). These cells started out as a bipolar cell, formed multiple large vacuoles (T=10h-60h), underwent reduction in cell volume (T=82h-83h), exhibited extensive membrane blebbing (T=84h-87h) and formed apoptotic bodies (T=88h-90h). This minor apoptotic population likely explains the minor effect of Q-VD-OPh in preventing 0.8 nM BI2536-mediated growth inhibition in LNCaP-AI cells (Figure 5d). Of 26 live-cell recordings of single LNCaP-AI cells treated with BI2536 for 8 days, 81% were found to die by necroptosis and 19% by apoptosis (Figure 7e). These live-imaging analyses reveal that Plk1 inhibition by BI2536 in LNCaP-AI cells results in mitotic catastrophe, which activates a defined sequence of events that triggers cell death by necroptosis.
To ensure that BI2536 is targeting Plk1, we compared the growth response of LNCaP-AI cells treated with BI2536 for 5 days with cells treated with siRNA oligos for Plk1 for 5 days. LNCaP-AI cells showed reduced Plk1 expression in the presence of siPlk1 but not siLuciferase control oligos (Figure 8a). Plk1 knockdown led to a similar level of growth inhibition as compared with 0.8 nM BI2536 treatment (Figure 8b). Following Plk1 depletion, LNCaP-AI cells formed giant cells that harbored clusters of DAPI-positive nuclear vesicles (Figure 8c), a phenotype similar to that observed in BI2536-treated cells (Figures 6, 8c). This unique nuclear morphology suggests that Plk1 depletion in LNCaP-AI cells also resulted in mitotic catastrophe that can lead to necroptotic cell death (19). To further gain insight into the mechanism underlying either Plk1 depletion- or BI2536-mediated growth inhibition, we incubated LNCaP-AI cells with necrostatin-1 (Nec-1), a small molecule inhibitor of necroptosis (23), for the duration of either Plk1 depletion or BI2536 treatment for 5 days. We found that Nec-1 was able to significantly attenuate growth inhibition resulting from either Plk1 knockdown (Figure 8d) or Plk1 inhibition by 0.4 nM and 0.8 nM BI2536 treatment (Figure 8e) in LNCaP-AI cells. These results elucidate that growth inhibition of LNCaP-AI cells by either Plk1 inhibition (BI2536) or Plk1 depletion (siRNA) leads to the alternative pathway of necroptotic cell death.
Figure 8.
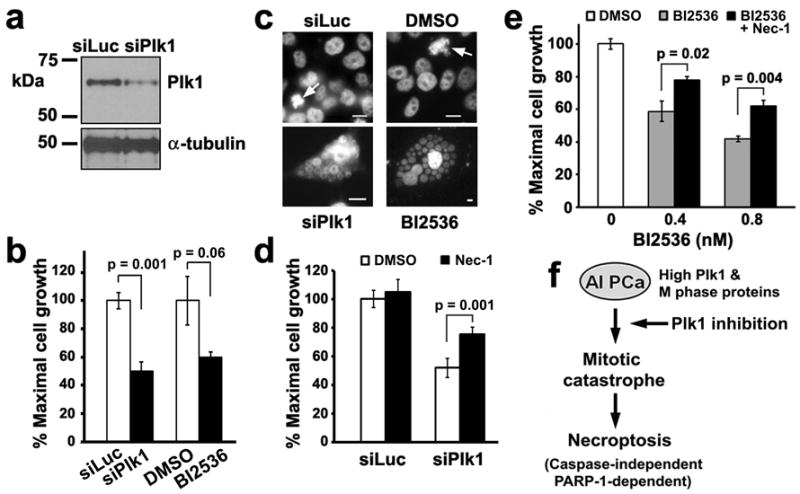
Necrostatin-1 attenuates cell death by necroptosis resulting from either Plk1 depletion or Plk1 inhibition in LNCaP-AI cells. (a) LNCaP-AI cells were transfected with either siLuciferase or Plk1 RNAi oligos for 5 days. Cell lysates (20 μg) were analyzed for Plk1 levels. α-tubulin was used as a loading control. (b) Cells prepared as in (a) or treated with 0.8 nM BI2536 were analyzed after 5 days by an MTS assay. Triplicate samples (mean ± SD) from one of n = 2 experiments are shown. (c) Cells were treated as in (b) and counterstained with DAPI to visualize DNA, shown here in black and white for contrast. Arrows, cells in mitosis in control cultures. Single giant cells containing clusters of nuclear vesicles are shown for siPlk1 and BI2536-treated LNCaP-AI cells. Bars, 10 μm. Note differences in scale. (d) Cells were transfected with siPlk1 for 5 days. 7.7 μM Necrostatin-1 (Nec-1), the necroptosis inhibitor, was added 3 h prior to siPlk1 transfection and remained throughout the experiment. Triplicate samples (mean ± SD) from MTS assays from one of n = 2 experiments are shown. (e) Cells were treated with 0.4 or 0.8 nM BI2536 for 5 days. 5 μM Nec-1 was added 3 h prior to BI2536 addition and remained throughout the experiment. Triplicate samples (mean ± SD) from MTS assays from one of n = 3 experiments are shown. (f) Working model of necroptosis induction by Plk1 inhibition in androgen-insensitive PCa cells. See text.
Discussion
Our studies demonstrate for the first time that the mitotic kinase Plk1, along with a panel of M phase cell cycle proteins, is reprogrammed for high level expression in LNCaP-AI cells. We show that LNCaP-AI cells are exquisitely sensitive to growth inhibition by subnanomolar concentrations of BI2536, a small molecule inhibitor of Plk1, which induces cell death by necroptosis. Our studies suggest that LNCaP-AI cells are reprogrammed in both their cell growth as well cell death routines. These unique properties may be explored for therapies for castration-resistant PCa.
Plk1 is a critical and versatile mitotic kinase that regulates cell cycle progression from G2 into mitosis, progression through mitosis, and the completion of cytokinesis (3, 4, 24), thus plays an essential role in the maintenance of genomic stability (25). Plk1 levels and kinase activity are tightly regulated, peaking at the onset of mitosis and declining upon mitotic exit (12). Overexpression of Plk1 has been shown to stimulate cell proliferation and oncogenic transformation in NIH 3T3 (26) while Plk1 depletion by siRNA-mediated protein knockdown leads to mitotic arrest and apoptosis in various human cancer cell lines (27 - 29). Because mitosis is a highly coordinated process, the ability of LNCaP-AI cells to adapt to a 3-fold increase in Plk1 levels may be assisted by the concomitant reprogramming of a panel of M phase proteins, including the Plk1 substrates BubR1 and PICH, and cyclin B1 identified in this study, and Cdc25 and Sgo2 as previously reported (2). Thus, the upregulation of Plk1 together with mitotic proteins that regulate cell cycle checkpoints may underlie the reprogrammed cell growth properties of LNCaP-AI cells.
We found that upregulation of Plk1 and Plk1 pathway proteins rendered LNCaP-AI cells highly sensitive to growth inhibition by BI2536 with an IC50 (0.2 nM) that is 4- to 50-fold lower than that found in other cell types (7), suggesting that LNCaP-AI cells are dependent on Plk1 for growth. BI2536 treatment not only led to defective mitosis as indicated by the accumulation of 50% of LNCaP-AI cells in G2/M, but also mitotic stress as evidenced by the appearance of multinucleated giant cells containing clusters of nuclear vesicles. Multinucleated giant cell formation is a prominent feature of mitotic catastrophe (19) and likely arose from some combinatorial failure in Plk1-regulated early mitotic functions at the centrosome (3), kinetochore (10, 30), chromosome (11) and mitotic spindle (8, 10, 24), and/or Plk1-regulated late mitotic and cytokinesis functions at the cleavage furrow (4, 31) and midzone/midbody (24, 31 - 34). The final step in mitotic catastrophe is the formation of nuclear vesicles containing missegregated chromosomes or fragmented chromatin due to chromatinolysis (19, 22, 35), which were observed in 100% of LNCaP-AI cells after 5 days of BI2536 treatment.
Our studies suggest that targeting Plk1 may render androgen-insensitive PCa cells sensitive to necroptosis as an alternative cell death modality. Necroptosis was originally thought to be passive cell death, but has since been shown to be involved in the pathogenesis of a wide range of human diseases (20). Necroptosis signals through a kinase complex consisting of receptor-interacting protein 1 (RIP1) and RIP3 (36, 37), which activates a cascade of events leading to the disintegration of mitochondrial, lysosomal and plasma membranes (20, 38). Our observation that Nec-1, a potent inhibitor of RIP1 kinase activity (23, 36), attenuated necroptotic cell death induced by either Plk1 inhibition or Plk1 depletion suggests that the RIP1/RIP3 “necrosome complex” is involved in the activation of necroptosis in LNCaP-AI cells. Further, BI2536 treatment of LNCaP-AI cells activated PARP-1, a nuclear enzyme that responds to DNA fragmentation and damage (39), but did not activate caspase-3 cleavage, which together support caspase-independent necroptotic cell death (20, 38). Live-cell imaging analysis captured not only the unique morphological features of mitotic catastrophe but also cell death by necroptosis that occurred swiftly during a failed mitosis, resulting in the explosion of the rounded up giant LNCaP-AI cells. We thus propose a model in which the disruption of Plk1 functions during mitotic progression results in mitotic catastrophe that leads to cell death by necroptosis (Figure 8f).
BI2536 treatment also led to the activation of autophagy, an intracellular degradation system that recycles cytoplasmic components and degrades proteins (17). An increase in the autophagy marker LC3II and a decrease in p62/sequestosome1 levels were observed in BI2536-treated LNCaP-AI cells. It has been suggested that autophagy may participate in necroptosis as part of a clean-up mechanism to clear away cellular debris, rather than contributing to the cell death mechanism (20, 21). How Plk1 inhibition in LNCaP-AI cells activates cellular mechanisms to induce necroptotic cell death remains to be elucidated.
One of the hallmarks of cancer cells is their general resistance to cell death by apoptosis (40, 41). Alternative means to induce cancer cell death via mechanistically distinct cell death pathway is of interest. We suggest that harnessing necroptosis through the inhibition of Plk1 may be an effective strategy for castration-resistant PCa.
Materials and methods
Antibodies and reagents
The following antibodies were used: androgen receptor (Upstate); Plk1, Cdc25C and caspase-3 (Santa Cruz Biotechnology); Plk1 (Origene), pT210-Plk1 and p62/sequestosome1 (BD Biosciences), Sgo2 and LC3 (Bethyl Laboratories), BubR1 (Novus Biologicals), PICH (Abnova), cyclin B1 (abcam), phospho-Histone H3 (Millipore), cleaved PARP-1 (Cell Signaling), α-tubulin (GeneTex), β-tubulin2.1 (Sigma-Aldrich), CREST serum (Dr. Bill Brinkley, Baylor College of Medicine), and secondary antibodies conjugated to Texas Red and Alexa Fluor 488 (Molecular Probes). Reagents used were as follows: Protease inhibitors, nocodazole, TNF-α, cycloheximide, rapamycin and insulin (Sigma-Aldrich); Q-VD-OPh (MP Biomedicals); necrostatin-1 (Enzo Life Sciences) and BI2536 (ChemieTek).
Cell culture, synchronization and growth inhibition
LNCaP cells were cultured in RPMI 1640 (GIBCO) containing 10% fetal bovine serum (FBS) (Atlanta Biologicals). LNCaP-AI cells (Dr. Anna Ferrari, New York University) (5) were cultured in RPMI 1640 containing 10% charcoal-stripped FBS (csFBS) (Hyclone Laboratories), 5 μg/ml of insulin, 2.5 mM L-glutamine and 1% Penicillin/Streptomycin (GIBCO). Cells were synchronized by incubating with 100 ng/ml nocodazole for 16 h to enrich for early mitotic cells. To inhibit Plk1 activity, cells were incubated with the small molecule inhibitor BI2536 (7), prepared in dimethyl sulfoxide (DMSO).
MTS assay
Cells were seeded at 3000/well in 96-well plate for 24 h before treatment with BI25326 as indicated. Cell growth was assessed using CellTiter 96® AQueous non-radioactive cell proliferation (MTS) assay (Promega).
Fluorescence-activated cell sorting (FACS)
Cells were resuspended in 1 ml of phosphate-buffered saline (PBS), fixed by slowly adding dropwise into 9 ml of 70% ethanol, chilled at 4°C for 2 h, resuspended in 0.5 ml containing 0.1% Triton X-100, 20 μg/ml propidium iodide (PI) and 200 μg/ml RNAse A (Sigma-Aldrich), incubated at room temperature for 30 min, and analyzed on a FACSCanto II Flow Cytometry system (BD) using FACSDiva™ software (BD), and analyzed by FlowJo software (Tree Star). For double-immunostained cells, cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, blocked in 0.2% BSA in PBS, and stained with Plk1 (mouse, Origene) and phospho-H3 (rabbit) 30 min, followed by Alexa Fluor 594 goat anti-mouse IgG (Invitrogen) and Alexa Fluor 488 goat anti-rabbit IgG (Invitrogen) for 30 min. For Annexin V and PI co-staining, non-fixed live cells were stained briefly at room temperature with Annexin V-FITC for 10 min and PI for 10 min using the Apoptosis Detection Kit (eBioscience). Samples were analyzed on an LSRFortessa Flow Cytometry system (BD) using FACSDiva™ software and analyzed by FlowJo.
Trypan blue exclusion assay
Cells were trypsinized and 0.4% trypan blue solution was added at 1:5 ratio. Cells that excluded trypan blue staining were scored as viable. Cell death was measured as % trypan blue positive cells over total cells counted on a hemocytometer.
Immunoblotting
Cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris pH 8.0, 1.5 mM EDTA, 5 mM EGTA, 0.5% Triton X-100, 5% glycerol) containing protease (mammalian protease inhibitor, 0.1 M PMSF) and phosphatase (serine/threonine and tyrosine phosphatase inhibitors, 5 mM Na3VO4, 5 mM NaF) inhibitors. Cell lysates were resolved on 4–12% gradient gels (Invitrogen), transferred to a nitrocellulose membrane (Bio-Rad), immunoblotted with a primary antibody, and detected using HRP-coupled secondary antibodies and ECL (Thermo Scientific).
Immunofluorescence microscopy
Cells were cultured on cover slips and treated with BI2536 or DMSO. After rinsing with pre-warmed PHEM (60 mM PIPES, 25 mM HEPES, 10 mM EGTA, 2 mM MgCl2, pH6.9), cells were permeabilized with 0.5% Triton X-100 for 1 – 2 min and fixed with 4% paraformaldehyde (Electron Microscopy Sciences) at room temperature for 20 min. The cover slips were washed and incubated in blocking solution (0.1 M PIPES, 1 mM MgSO4, 1 mM EGTA, 1.83% L-lysine, 1% BSA, 0.1% NaN3, pH7.2, 2% nonfat milk) for 3 h at room temperature, followed by primary antibodies overnight at 4°C and secondary antibodies for 3 h at 4°C, and mounted with ProLong Gold antifade reagent with DAPI (Invitrogen). Images were acquired using 40X magnification on a Nikon TE2000 widefield microscope system (10, 11).
Live-cell time-lapse imaging
Cells were cultured in a glass-bottom multi-experiment Hi-Q4 dish (Nikon) and let settle for 5 min in the Nikon BioStation IM chamber as described (11). Multiple image capture points were selected and images were acquired at 15 min intervals for 4 days, compiled using NIS-Elements 3.1 (Nikon) software, and presented using Adobe PhotoShop CS (Adobe Systems).
Statistical analysis
Data were confirmed in multiple independent experiments. Data quantification was performed by using the Student’s t-test and expressed as mean ± SD. p values of less than 0.05 were considered statistically significant.
Supplementary Material
Acknowledgments
We thank Dr. Anna Ferrari of New York University for the LNCaP-AI cells and Ms. Shenyan Zeng for FACS analysis. This work was supported by NIH training grants T32-AI07495 and T32-DK07696 for AD and grants from the NIH (HL080205 to NTE; CA111479 to S-HL; DK53176, AI071130 to L-yY-L), DOD (PC080847, PC093132 to S-HL), Dan L. Duncan Cancer Center (L-yY-L and S-HL), and Alkek Award in Experimental Therapeutics (L-yY-L and S-HL).
Footnotes
Supplementary Information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Wang Q, Li W, Zhang Y, Yuan X, Xu K, Yu J, et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell. 2009;138:245–256. doi: 10.1016/j.cell.2009.04.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barr FA, Sillje HH, Nigg EA. Polo-like kinases and the orchestration of cell division. Nat Rev Mol Cell Biol. 2004;5:429–440. doi: 10.1038/nrm1401. [DOI] [PubMed] [Google Scholar]
- 4.Takaki T, Trenz K, Costanzo V, Petronczki M. Polo-like kinase 1 reaches beyond mitosis - cytokinesis, DNA damage response, and development. Curr Op Cell Biol. 2008;20:650–660. doi: 10.1016/j.ceb.2008.10.005. [DOI] [PubMed] [Google Scholar]
- 5.Wang LG, Ossowski L, Ferrari AC. Androgen receptor level controlled by a suppressor complex lost in an androgen-independent prostate cancer cell line. Oncogene. 2004;23:5175–5184. doi: 10.1038/sj.onc.1207654. [DOI] [PubMed] [Google Scholar]
- 6.Horoszewicz JS, Leong SS, Kawinski E, Karr JP, Rosenthal H, Chu TM, et al. LNCaP model of human prostatic carcinoma. Cancer Res. 1983;43:1809–1818. [PubMed] [Google Scholar]
- 7.Lenart P, Petronczki M, Steegmaier M, Di Fiore B, Lipp JJ, Hoffmann M, et al. The small-molecule inhibitor BI 2536 reveals novel insights into mitotic roles of Polo-like kinase 1. Curr Biol. 2007;17:1–12. doi: 10.1016/j.cub.2006.12.046. [DOI] [PubMed] [Google Scholar]
- 8.Musacchio A, Salmon ED. The spindle assembly checkpoint in time and space. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 9.Macy B, Wang M, Yu H-G. The many faces of shugoshin, the “guardian spirit”, in chromosome segregation. Cell Cycle. 2009;8:35–37. doi: 10.4161/cc.8.1.7327. [DOI] [PubMed] [Google Scholar]
- 10.Nishino M, Kurasawa Y, Evans R, Lin S-H, Brinkley BR, Yu-Lee L-y. NudC is required for Plk1 targeting to the kinetochore and chromosome congression. Curr Biol. 2006;16:1414–1421. doi: 10.1016/j.cub.2006.05.052. [DOI] [PubMed] [Google Scholar]
- 11.Kurasawa Y, Yu-Lee L-y. PICH and cotargeted Plk1 coordinately maintain prometaphase chromosome arm architecture. Mol Biol Cell. 2010;21:1188–1199. doi: 10.1091/mbc.E09-11-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee KS, Yuan YLO, Kuriyama R, Erikson RL. Plk is an M-phase-specific protein kinase and interacts with a kinesin-like protein, CHO1/MKLP-1. Mol Cell Biol. 1995;15:7143–7151. doi: 10.1128/mcb.15.12.7143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jang Y-J, Ma S, Terada Y, Erikson RL. Phosphorylation of threonine 210 and the role of serine 137 in the regulation of mammalian polo-like kinase. Proc Natl Acad Sci. 2002;277:44115–44120. doi: 10.1074/jbc.M202172200. [DOI] [PubMed] [Google Scholar]
- 14.Culig Z, Hoffmann J, Erdel M, Eder IE, Hobisch A, Hittmair A, et al. Switch from antagonist to agonist of the androgen receptor bicalutamide is associated with prostate tumor progression in a new model system. Br J Cancer. 1999;81:242–251. doi: 10.1038/sj.bjc.6690684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thalmann GN, Anezinis PE, Chang SM, Zhau HE, Kim EE, Hopwood VL, et al. Androgen-independent cancer progression and bone metastasis in the LNCaP model of human prostate cancer. Cancer Res. 1994;54:2577–2581. [PubMed] [Google Scholar]
- 16.Sramkoski RM, Pretlow TG, II, Giaconia JM, Pretlow TP, Schwartz S, Sy M-S, et al. A new human prostate carcinoma cell line 22Rv1. In Vitro Cell Dev Biol. 1999;35:403–409. doi: 10.1007/s11626-999-0115-4. [DOI] [PubMed] [Google Scholar]
- 17.Mizushima N, Komatsu M. Autophagy: Renovation of cells and tissues. Cell. 2011;147:728–741. doi: 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 18.Caserta TM, Smith AN, Gultice AD, Reedy MA, Brown TL. Q-VD-OPh, a broad spectrum caspase inhibitor with potent antiapoptotic properties. Virology. 2003;191:589–599. [Google Scholar]
- 19.Vakifahmetoglu H, Zhivotovsky B. Death through a tragedy: mitotic catastrophe. Cell Death & Diff. 2008;15:1153–1162. doi: 10.1038/cdd.2008.47. [DOI] [PubMed] [Google Scholar]
- 20.Christofferson DE, Yuan J. Necroptosis as an alternative form of programmed cell death. Curr Op Cell Biol. 2010;22:263–268. doi: 10.1016/j.ceb.2009.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Galluzzi L, Vitale I, Abrams JM, Alnemri ES, Baehrecke EH, Blagosklonny MV, et al. Molecular definitions of cell death subroutines: recommendations of the Nomenclature Committee on Cell Death 2012. Cell Death & Diff. 2012;19:107–120. doi: 10.1038/cdd.2011.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Castedo M, Perfettini J-L, Roumier T, Andreau K, Medema RH, Kroemer G. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
- 23.Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima N, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–115. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 24.Liu XS, Song B, Liu X. The substrates of Plk1, beyond the functions in mitosis. Protein & Cell. 2011;1:999–1010. doi: 10.1007/s13238-010-0131-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Strebhardt K. Multifacted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discovery. 2010;9:643–660. doi: 10.1038/nrd3184. [DOI] [PubMed] [Google Scholar]
- 26.Smith MR, Wilson ML, Hamanaka R, Chase D, Kung H, Longo DL, et al. Malignant transformation of mammalian cells initiated by constitutive expression of the polo-like kinase. Biochem Biophys Res Comm. 1997;234:397–405. doi: 10.1006/bbrc.1997.6633. [DOI] [PubMed] [Google Scholar]
- 27.Guan R, Tapang P, Leverson JD, Albert D, Giranda VL, Luo Y. Small-interfering RNA-mediated polo-like kinse 1 depletion preferentially reduces the survival of p53-defective, oncogenic transformed cells and inhibits tumor growth in animals. Cancer Res. 2005;65:2698–2704. doi: 10.1158/0008-5472.CAN-04-2131. [DOI] [PubMed] [Google Scholar]
- 28.Reagan-Shaw S, Ahmad N. Silencing of polo-like kinase (Plk) 1 via siRNA causes induction of apoptosis and impairment of mitosis machinery in human prostate cancer cells: implications for the treatment of prostate cancer. FASEB J. 2005;19:611–613. doi: 10.1096/fj.04-2910fje. [DOI] [PubMed] [Google Scholar]
- 29.Schmit TL, Zhong W, Setaluri V, Spiegelman VS, Ahmad N. Targeted depletion of Polo-like kinase (Plk) 1 through lentiviral shRNA or a small molecule inhibitor causes mitotic catastrophe and induction of apoptosis in human melanoma cells. J Invest Dermatol. 2009;129:2843–2853. doi: 10.1038/jid.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Park JE, Erikson RL, Lee KS. Feed-forward mechanism of converting biochemical cooperativity to mitotic processes at the kinetochore plate. Proc Natl Acad Sci. 2011;108:8200–8205. doi: 10.1073/pnas.1102020108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li J, Wang J, Jiao H, Liao J, Xu X. Cytokinesis and cancer: Polo loves ROCK‘n’ Rho(A) J Genet Genomics. 2010;37:159–172. doi: 10.1016/S1673-8527(09)60034-5. [DOI] [PubMed] [Google Scholar]
- 32.Zhou T, Aumais JP, Liu X, Yu-Lee L-y, Erikson RL. A role for Plk1 phosphorylation of NudC in cytokinesis. Dev Cell. 2003;5:127–138. doi: 10.1016/s1534-5807(03)00186-2. [DOI] [PubMed] [Google Scholar]
- 33.Brennan IM, Peters U, Kapoor TM, Straight AF. Polo-like kinase controls vertebrate spindle elongation and cytokinesis. Proc Natl Acad Sci. 2007;91:5046–5050. doi: 10.1371/journal.pone.0000409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bastos RN, Barr FA. Plk1 negatively regulates Cep55 recruitment to the midbody to ensure orderly abscission. J Cell Biol. 2010;191:751–760. doi: 10.1083/jcb.201008108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Artus C, Bouhrad H, Bouharrour A, Brunelle M-N, Hoos S, Yuste VJ, et al. AIF promotes chromatinolysis and caspase-independent programmed necrosis by interacing with histone H2AX. EMBO J. 2010;29:1585–1599. doi: 10.1038/emboj.2010.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Degterev A, Hitomi J, Germscheid M, Ch’en IL, Korkina O, Teng X, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008;135:1311–1323. doi: 10.1016/j.cell.2008.10.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol. 2010;11:700–714. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 39.Alano CC, Swanson RA. Players in the PARP-1 cell death pathway: JNK1 joins the cast. Trends Biochem Sci. 2006;31:309–311. doi: 10.1016/j.tibs.2006.04.006. [DOI] [PubMed] [Google Scholar]
- 40.Hanahan DJ, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 41.Speirs CK, Hwang M, Kim S, Li W, Chang S, Varki V, et al. Harnessing the cell death pathway for targeted cancer treatment. Am J Cancer Res. 2011;1:43–61. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.