

Abstract
A substantial number of the world's population continues to smoke tobacco, even in the setting of a cancer diagnosis. Studies have shown that cancer patients with a history of smoking have a worse prognosis. Modulation of several physiologic processes involved in drug disposition has been associated with chronic exposure to tobacco smoke. The most common of these can be categorized into effects on cytochrome P450 mediated metabolism, glucuronidation, and protein binding. Perturbation in the pharmacokinetics of anticancer drugs could result in clinically significant consequences, given they are amongst the most toxic, but potentially beneficial, pharmaceuticals prescribed. Unfortunately, the effect of tobacco smoking on drug disposition has only been explored for a few marketed anticancer drugs, thus very little prescribing information is available to guide clinicians on the vast majority of compounds. The carcinogenic properties of multiple compounds found in tobacco smoke have been well studied, however relatively little attention has been given to the effects of nicotine itself on cancer growth. Emerging data are available which identify nicotine's effects on cancer cell apoptosis, tumor angiogenesis, invasion, and metastasis. The implications of such are unclear, but may lead to important questions to be addressed regarding approaches to smoking cessation in cancer patients.
Keywords: tobacco, nicotine, pharmacokinetics, cancer
Introduction
Well over 1 billion of the world's population actively smokes tobacco. There have been dramatic reductions in the proportion of U.S. citizens who are active tobacco users over the past several decades, however approximately one-third of people in some regions of the country still smoke.1 The frequency of smokers in the population of patients presenting to a cancer treatment facility or private practice site may vary greatly based on the demographics/socioeconomic status of its catchment area. Furthermore, the number of smokers at specific clinical sites may also be somewhat dependent on the types of cancer more commonly treated there. For instance, a practice that specializes in cancers more frequently associated with tobacco use (e.g. lung, head/neck, breast cancer, etc.) may have a higher percentage of smokers than those treating a more general mix of cancer diagnoses.
Studies have indicated that between one-third to one-half of smokers continue smoking following diagnosis.2 For example, recently reported data from one academic center show 56 percent of smokers continued to smoke cigarettes after being diagnosed.3 Smoking cessation is a vital goal for all cancer patients, however optimal strategies for circumvention of barriers to cessation in this population have not been elucidated. We therefore continue to see high rates of tobacco use despite attempted interventions.4
A number of studies have documented patients who are active smokers at diagnosis tend to fare poorly compared to nonsmokers. These data suggest detrimental effects on treatment efficacy, acquisition of secondary malignancies, duration of survival, and quality of life.5,6 For example, Fortin, et al found that patients who were smoking upon initiation of cisplatin + radiotherapy treatment for head & neck cancer had a reduced frequency of local control (hazard ratio 2.8; p 0.004) and shorter overall survival (hazard ratio1.4; p 0.007).7 There may be various explanations for the etiology of these effects. Most lung cancers are both rapidly proliferating and genetically unstable. For example, mutations found in cancers which result in dysfunction of DNA repair could increase sensitivity of tumor cells to become even more aggressive. Thus inhalation of additional procarcinogens and carcinogens from tobacco smoke could further accelerate these processes and contribute to worse outcomes. Facilitation of a proinflammatory state is also thought to promote tumor growth. Smoking related changes in the clinical pharmacology of anticancer drugs may have meaningful clinical implications, given the substantial data that demonstrate association of inter-patient variability in systemic exposure with pharmacodynamics.8
This review will focus on pharmacologic factors which may play a role in these effects, in addition to evaluation of the emerging data which potentially implicate nicotine as a tumor growth stimulant. A review of basic nicotine pharmacology, in particular the interesting inter-patient genetic factors which may influence addiction, is beyond the scope of this manuscript, however readers are referred to the detailed recent description of this topic by Benowitz.9
Tobacco Exposure and Anticancer Drug Disposition
The potential effects of tobacco on processes which determine the pharmacokinetic disposition of drugs is multifaceted. The mechanisms of these interactions are likely related to smoking-induced acceleration of CYP450s; induction of drug glucuronidation via uridine diphosphate glucuronosyltransferases (UGTs); and increased concentrations of circulating drug binding protein (Figure 1). In addition, tobacco may contain chemicals such as cadmium and arsenic that can induce endogenous metallothioneins, which could theoretically alter the pharmacokinetic disposition of some anti-cancer drugs.
Figure 1.
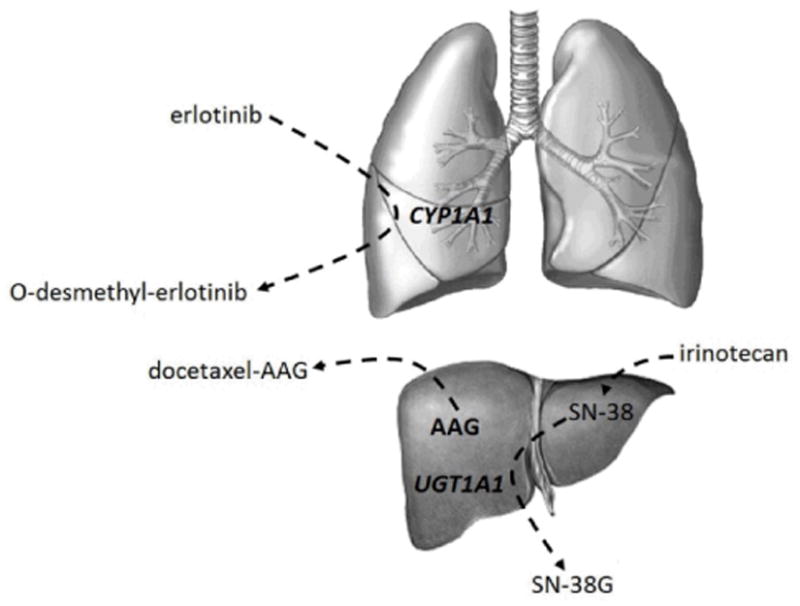
Representative pharmacokinetic processes associated with tobacco smoking and their effects on drug disposition. CYP450 enzymes such as CYP1A1 (in lung) and glucuronosyltransferases such as UGT1A1 (in liver) are induced by exposure to tobacco smoke and lead to detoxification of parent drugs or active metabolites in the examples shown. Upregulation of alpha-1-acid glycoprotein (AAG) hepatic production as a result of inflammatory response to cancer and cigarette smoking results in increased protein bound drug and altered disposition.
Caution should be exercised in interpretation of studies which attempt to evaluate associations between tobacco smoking and drug pharmacokinetics. Crossover study designs (with and without tobacco exposure) are not ethically feasible in this setting, thus proper identification of comparable subject groups is essential. Retrospective use of medical records data on smoking history is discouraged due to inaccuracies in documentation and questionable validity (underestimation) of self-reporting.10 In addition, such studies may have insufficient power to observe clinically meaningful differences in systemic exposures of drugs with low therapeutic indices.
Alpha-1-Acid Glycoprotein
Alpha-1-acid glycoprotein (AAG; orosomucoid) is the 2nd most abundant serum protein and one of the major acute phase proteins in humans. The highly variable concentrations of serum AAG are mediated by a variety of factors including: systemic tissue injury, inflammation, infection, genetic variability, exposure to exogenous substances (e.g. tobacco smoke), glucocortoids, and several cytokines, most notably interleukin-1β, tumor necrosis factor-α, and interleukin-6.11 These changes in serum concentration have been correlated with increases in hepatic production.
The biological function of AAG remains unknown; however several physiologically significant activities, including a variety of immunomodulating effects, have been described.11 This protein also has potential to bind and/or carry numerous lipophilic drugs, containing one to seven binding sites per molecule. Although AAG's binding sites favor basic or neutral lipophilic drugs, it has also been shown to have the ability to bind acidic drugs such as phenobarbital.11 AAG may play a significant role in the pathophysiology and therapeutic response for a number of diseases, given the wide variance in its concentration associated with disease severity as well as its drug binding abilities.
Effect of Tobacco Smoking on Serum AAG
Serum concentrations of AAG are significantly increased in current smokers with respect to previous or never smokers.12 There is a direct correlation between the serum AAG concentration and cigarette smoking frequency, as shown in Figure 2.12 Serum AAG is significantly increased in men who have smoked for less than ten years (18%) and men who have smoked more than 10 years (14%) relative to men who have never smoked.13 Similarly, Tappia et. al. found serum AAG concentrations to be 39% higher in smokers compared to non-smokers.14 The inter-subject variability in serum AAG is greater in smokers. Induction of AAG by tobacco smoke also occurs in passive exposures. Children exposed to environmental tobacco smoke had significantly higher concentrations of serum AAG.15 Since the study used a definition of exposure as living with an adult who smoked, such could be considered either a second hand exposure (environmental smoke that is inhaled involuntarily or passively by someone who is not smoking) or a third hand exposure (small particles from cigarette smoke that are deposited on surfaces which remain after secondhand smoke is removed from area.)
Figure 2.
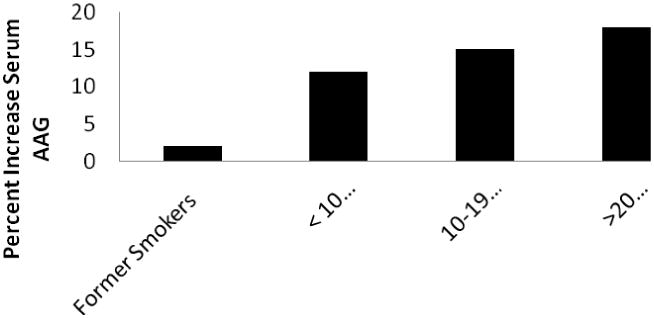
Percent increase in mean serum AAG concentrations in smokers (n = 4,586) compared to nonsmokers (n = 1489). Adapted from Lind, et al.12
Effect of Cancer on Serum AAG
The following types of cancers have been associated with elevations in serum AAG concentrations: breast, melanoma, colon, ovarian, esophageal, pancreatic, hepatic, prostate, kidney, rectal, laryngeal, stomach, leukemia, urinary, lung, urothelial, lymphoma, and uterine. [AU: As your original Table 1 was strictly a list, the information was incorporated into the text and the table was deleted.] Uslu et. al. found significantly higher serum concentrations of AAG in 35 male smokers with various stages of laryngeal cancer (10 stage II, 12 stage III, 13 stage IV) compared to 34 healthy smokers.16 Additionally, serum AAG was directly correlated to the stage of laryngeal cancer. Concentrations of AAG have been observed to be 2.5 times greater in breast and lung cancer patients, and 1.6 times greater in those with ovarian cancer relative to healthy controls.17 Budai, et al observed a four-fold increase in plasma AAG in melanoma, lymphoma, or ovarian cancer patients compared to healthy individuals.18
Anticancer Drug Pharmacokinetics & Pharmacodynamics Related to AAG
Many anticancer drugs are highly protein bound, at least in part to AAG. Examples include docetaxel, erlotinib, imatinib, paclitaxel, toremifene, vinblastine and vinorelbine.11 In addition, a number of investigational kinase inhibitors have high AAG binding capacity.
Since factors such as cigarette smoking, cancer type, and extent of disease influence the serum concentration of AAG, these factors may alter drug disposition and potentially clinical effect for anticancer drugs which are highly protein bound, however certain pharmacologic characteristics must be considered. It is anticipated that variation in serum AAG concentrations may have important pharmacokinetic implications if a drug is highly and mainly bound to AAG, has a small volume of distribution (e.g. plasma volume), and has a narrow therapeutic index (e.g. cytotoxic anticancer drugs).11
One of the best examples of the effect of serum AAG on the pharmacokinetics of anticancer drugs involves docetaxel. Docetaxel is > 98% bound to plasma proteins, in particular AAG. The effect of variability in serum AAG concentrations on docetaxel pharmacokinetics and pharmacodynamics is complex and important since both total and unbound docetaxel systemic exposures are directly correlated with the frequency of hematologic toxicity.19,20 Elevations in serum AAG are associated with reductions in the free fraction of docetaxel resulting in higher total drug exposure, presumably due to a reduction in the amount available for liver metabolism. Serum concentrations of AAG have been shown as important determinants of inter-patient variability in systemic docetaxel clearance accounting for approximately a 19% increase in patients with elevated AAG,21 although one study suggested that inter-patient variability in AAG concentrations does not explain variability in the plasma exposure to free drug.22 Patients with elevated plasma AAG concentrations prior to receiving docetaxel have been shown to experience lower toxicity rates and worse survival, consistent with a lower exposure to unbound drug. Incorporation of these data into models with other prognostic factors is still evolving.23 Association of high AAG concentrations with worse efficacy needs to be interpreted with caution, since higher baseline AAG may also be reflective of greater disease burden and thus provide another factor relating to poorer outcome.
Imatinib is metabolized primarily by CYP3A4 and potentially by CYP 1A2, 2D6, 2C9, and 2C19. A number of studies discovered relationships between imatinib plasma concentrations and therapeutic outcomes.24 High serum AAG concentrations have been shown to be significantly associated with lower imatinib clearance. Delbaldo et al. reported the inter-patient variability of imatinib clearance decreased from 38% to 29% when differences in serum AAG concentrations were taken into account.25 A pharmacokinetic model which utilizes AAG concentrations to estimate unbound plasma imatinib exposure has found associations between the latter with both toxicity and efficacy.26 Van Erp et al evaluated the pharmacokinetics, safety, and efficacy of imatinib using retrospective smoking history data in smokers (n=9) and non-smokers (n=25) with gastrointestinal stromal tumors or soft-tissue sarcoma. 27 Pharmacokinetic parameters were similar between the groups; however, this study was only marginally powered (69%) to observe a fairly large (50%) difference in apparent oral clearance. Smoking patients diagnosed with gastrointestinal stromal tumors who were treated with imatinib had shorter overall survival (P = 0.037), however the rate of anemia and fatigue was significantly higher in smokers relative to non-smokers (p 0.010 and p 0.011, respectively).
Drug Metabolism
Cytochrome P450
The cytochrome P450 monooxygenase system (CYP) is a multigene superfamily of heme-thiolate enzymes, which are important in the metabolism of foreign and endogenous compounds. Genetic variations, drug interactions, pathophysiological factors (e.g. tumor infiltration of the liver), or environmental factors can modify the activity of these enzymes leading to partial or complete inhibition as well as induction of the enzyme activity.
Cigarette smoking is one of the environmental factors that can influence the activity of multiple CYPs. Kushinsky and Louis reported in 1976 that injection of benzo[a]pyrene, which is formed as a result of cigarette smoking, caused a great increase in both liver weight and CYP450 content in rats.28 This induction may alter the efficacy of many drugs by decreasing their parent drug exposure and may also increase the risk of cancer by enhancing the metabolic activation of procarcinogens. CYP induction is mainly caused by polycyclic aromatic hydrocarbons (PAH), which are a product of the incomplete combustion of tobacco. PAH in tobacco smoke are believed to be responsible for the induction of CYPs 1A1, 1A2, 1B1 and 2E1. In contrast, tobacco smoke inhibits CYP2A6.
CYP1A1
CYP1A1 is an extrahepatic CYP450 found in the lung and placenta. Phenotyping the activity of CYP1A1 is generally evaluated ex vivo by measuring the activity of ethoxyresorufin-O-deethylase (EROD). Aryl hydrocarbons, found in cigarette smoke, such as bezo[a]pyrene, benzo-fluorene, tetrachlorodibenzo-p-dioxin, and fluoranthene regulate the induction of CYP1A1. The activity of CYP1A1 was 66% - 70% higher in men and women who smoke more than 10 cigarettes per day compared to non-smokers.29 CYP1A1 was induced up to 100- fold in lung tissue of Finnish active tobacco smokers who were operated on for lung tumors. The EORD activity ranged from 1-184 pmol/min/mg, and was the lowest among the long-term ex-smokers. There was a direct correlation between the number of cigarettes smoked per day and the EROD activity.30 Comparable results were obtained by Smith et al where mean EROD in microsomes from current smokers (12.11 ± 13.46 pmol/min/mg) were approximately 15-fold higher than those in microsomes from non-/former smokers (0.77 ± 1.74 pmol/min/mg;).31 A significant increase in the activity of EROD (1.44 – 1.77 fold) and in CYP1A1 mRNA (2.5 – 7.8 fold increase) was observed in blood lymphocytes isolated from patients suffering from tobacco associated lung cancer compared to controls.32
CYP1A2
CYP1A2 is almost exclusively expressed in the liver and is responsible for the metabolism of many clinically useful drugs. It accounts for approximately 13 - 15 % of the total hepatic CYP450. Drugs that are metabolized primarily by CYP1A2 exhibit a wide inter-individual variability due to differences in the enzyme's expression and activity resulting from genetic, environmental, and endogenous factors. Phenotyping the activity of CYP1A2 is possible ex vivo using probe substrates such as phenacetin, caffeine, theophylline or melatonin. Caffeine is the most widely used probe to assess CYP1A2 by comparing the ratio of its metabolites in urine. Polycyclic aromatic hydrocarbons in cigarette smoke are well known inducers of CYP1A2 activity and smoking has been associated with increased clearance of many CYP1A2 substrates. In a population based study (n=863), CYP1A2 activity was increased 1.66-fold in smokers consuming 11–20 cigarettes daily. These effects were significant in both Koreans (n = 150) and Swedes (n = 194).33
Induction of CYP1A2 by cigarette smoking may likely alter the pharmacokinetics of drugs that are primarily metabolized by this isoenzyme. Smoking schizophrenic patients have increased clearance of the CYP1A2 substrate clozapine which is associated with reduced efficacy.34 The induction of CYP1A2 by cigarette smoking has been shown to be reversible upon smoking cessation.35 Initial caffeine clearance significantly decreased by 36% in the first 4 days following smoking cessation. Thus, patients who are heavy smokers and receive a CYP1A2 substrate with a narrow therapeutic index may need immediate dose reductions following smoking cessation to avoid a sudden increase in systemic exposure and unwanted adverse events.
CYP1B1
Cytochrome P450 1B1 (CYP1B1) is a phase I enzyme involved in the activation of a broad spectrum of carcinogens such as polycyclic aromatic hydrocarbons. CYP1B1 is present in normal human bronchial epithelial cells and its activity is increased by cigarette smoking. It is responsible for the metabolism of 17 beta-estradiol to the 4-hydroxy-17 beta-estradiol.36 This highly active metabolite is genotoxic and may explain the increased risk of lung cancer among women compared to men. CYP1B1 mRNA expression has been shown to increase with higher tobacco consumption in smokers compared to non-smokers however induction is dependent on the CYP1B1 L432V gene polymorphism.37 Expression of CYP1B1 mRNA is increased in the bronchial mucosa of human tobacco smokers compared to non- smokers and short-term exposure to tobacco smoke condensate induces CYP1B1 in the tongue, esophagus, lung and colon of experimental mice.38 CYP1B1 gene expression was induced in bronchoalveolar lavage cells of smokers and CYP1B1 activity was 3-fold higher in bronchial biopsies of smokers compared to non-smokers.39
CYP2A6
CYP2A6 is an important Phase I enzyme, expressed predominantly in the liver, that metabolizes approximately 3% of therapeutic drugs. CYP2A6 expression and activity have demonstrated a wide (20- to >100-fold) inter-individual variation which is due primarily to genetic polymorphisms in the CYP2A6 gene. CYP2A6 activity is also modified by certain drugs and pathological and environmental factors. Nicotine C-oxidation leading to cotinine formation, a major nicotine metabolic pathway, is catalyzed by CYP2A6. Phenotyping CYP2A6 activity is performed by measuring the nicotine metabolite ratio (total trans-3-hydroxycotinine/total cotinine). Cigarette smoking significantly inhibits the metabolism of cotinine. Schoedel et al found that long-term, in vivo nicotine treatment significantly decreases in vitro nicotine metabolism by approximately 40% and the expression of a CYP2A6-like protein in African green monkey liver by approximately 60% with CYP1A2 activity altered from 0 to 7 fold. 40 Multiple studies have shown an inhibitory effect of cigarette smoking on CYP2A6 activity. For example, a significantly lower activity (up to 60%) of CYP2A6 was observed in smoking Serbians compared to non-smokers.41
CYP2E1
CYP2E1 is mainly expressed in the liver. It metabolizes low-molecular weight, lipophilic compounds such as acetone and therapeutic drugs such as acetaminophen. Phenotyping for CYP2E1 is typically determined by measuring the clearance of chlorzoxazone. Tobacco smoke induces CYP2E1 activity, protein, and mRNA expression in both the liver and the kidney of adult mice. Benowitz et al examined effects of cigarette smoking on the disposition kinetics of chlorzoxazone in a cross-over manner in 12 cigarette smokers in conditions where they either continued smoking or abstained from smoking.42 Cigarette smoking significantly induced metabolism of chlorzoxazone by 24% reflecting induction of CYP2E1.
Uridine 5′-Diphosphate-Glucuronosyltransferases (UGT) Enzymes
Uridine 5′-diphosphate-glucuronosyltransferase catalyzes glucuronidation, a major phase II metabolic reaction. The three human UGT families contain more than 20 known microsomal isoenzymes. Tobacco smoking exerts differential effects on UGT family isoenzymes and the literature contains conflicting results concerning the influence of smoking on UGT activity. UGT1A and UGT2B activity were significantly higher in placenta of mothers who smoked relative to non-smoking mothers.43 It was reported that UGT1A4 activity and UGT1A6 activity was higher in smokers relative to non-smokers whereas the activity of UGT1A1, and UGT2B7 was not affected by smoking.44 On the other hand, induction of UGT2B7 activity and not UGT1A4 by cigarette smoking was proposed to be responsible for the decreased exposure of lamotrigine in smokers. It is apparent that smoking induces UGTs, however, the extent of induction of individual UGT isoenzymes is not clear and needs further evaluation.
Examples of Tobacco-Associated Alterations in Anti-Cancer Drug Metabolism
Since cigarette smoking induces CYP1A1, 1A2, 1B1 and 2E1, and inhibits CYP2A6, the pharmacokinetics of the anticancer drugs metabolized by these enzymes could be different in smokers compared to non-smokers. Such pharmacokinetic changes could lead to differential response to treatment based on the type of interaction. Most of the marketed anticancer medications are primarily metabolized by CYP3A4, which is neither induced nor inhibited by smoking; therefore, there are a limited number of studies that explored the effect of smoking on the metabolism of anticancer medications, as discussed below.
Erlotinib is a kinase inhibitor utilized for the treatment of non-small cell lung cancer (NSCLC) and for metastatic pancreatic cancer in combination with gemcitibine. It is primarily metabolized by CYPs 3A4 and 1A2. Cigarette smoking induces CYP1A2 and may be responsible for the reduced systemic exposure of erlotinib observed in smokers. Hamilton, et al evaluated the systemic exposure of erlotinib in healthy smokers compared to non-smokers after receiving a single oral dose of 150 mg (n=16) or 300 mg (n=14).45 Following the 150 mg dose, the geometric mean ratio (smokers/nonsmokers) of AUC0-inf, C24, and Cmax were 36%, 12%, and 65%, respectively. Following the 300 mg dose, the geometric mean ratio (smokers/nonsmokers) of AUC0-inf and Cmax were 43% and 80%, respectively. A population pharmacokinetic analysis of erlotinib in patients with NSCLC receiving erlotinib as a single agent treatment showed that erlotinib clearance was 24% faster in current smokers relative to former smokers or never smokers.46 The pharmacokinetics of erlotinib were explored in NSCLC patients (n=22) who had a history of smoking at least 10 cigarettes per day for at least one year at the initiation of the study.47 Four dose levels were evaluated 200, 250, 300, and 350 mg given daily for 14 days. Erlotinib exposure following the 300 mg dose was similar to that previously observed following 150 mg dose in former or never smokers. The maximum tolerated dose of erlotinib in NSCLC patients who smoke was 300 mg. This reduction in erlotinib exposure in smokers may lead to treatment resistance and warrant the need for increasing the dose of erlotinib in smokers, since it was shown that current smokers had a toxicity profile following 300 mg comparable to that observed in non-smokers following a 150 mg dose. In addition, smoking history was shown to be one of the predictors of erlotinib outcome.48 The prescribing information for erlotinib has been recently changed to reflect these data, however cautious monitoring of adverse effects is advised and readjustment of the dose if the patient discontinues smoking will be necessary.
Tegafur is a chemotherapeutic 5-fluorouracil prodrug that is bioactivated to 5-fluorouracil by CYPs 1A2, 2A6, and 2C8. Potential effects of tobacco smoking on tegafur pharmacokinetics are difficult to predict since smoking induces CYP1A2 but inhibits CYP2A6. Chuh et al evaluated the pharmacokinetics of tegafur in East Asian (n=18) and Caucasian (n=19) patients undergoing therapy for refractory solid malignancies.49 Tegafur clearance significantly increased with increasing cotinine: nicotine ratio (r2 = 0.35, P = 0.002). Although the authors emphasize the role of CYP2A6 in the bioactivation of tegafur, the increased clearance of tegafur with increased nicotine:cotinine ratio highlight the induction of CYP1A2 and its role in tegafur clearance.
Irinotecan is a topoisomerase I inhibitor indicated as a component of the first-line therapy of colon and rectal cancer. Irinotecan is a prodrug which is converted to the active metabolite, SN-38, via carboxylesterases. SN-38 is subsequently conjugated, predominantly by UGT1A1. Iriontecan is also converted to the relatively inactive metabolites 7-ethyl-10-[4-N-(5-aminopentanoic acid)-1-piperidino]carbonyloxycamptothecin (APC), and 7-ethyl-10-(4-amino-1-piperidino) carbonyloxycamptothecin (NPC)], via CYP3A4/5. Smoking induces many UGT isoenzymes and is expected to reduce the exposure to the SN-38 metabolite, hence decreasing the adverse effects of irinotecan, primarily neutropenia. Since smoking does not induce CYP3A4,50 it is not expected to affect the rate of formation of the inactive metabolites APC and NPC. These projections were confirmed by van der Bol et al.51 In a study of 49 smokers and 141 non-smokers treated with irinotecan, the exposure to irinotecan and SN-38 were significantly lower (15% and 40%, respectively) in smokers relative to non-smokers. The odds ratio of grade 3 to 4 neutropenia was significantly lower in smokers relative to non-smokers (OR=0.1, P = 0.006). There was no significance difference in CYP3A4 phenotype between smokers and non-smokers. As a result, personalization of irinotecan therapy by increasing irinotecan doses in smokers has been proposed.52
Nicotine and Cancer Biology
Over 1 million cancer related deaths are attributable worldwide to tobacco use. A direct link between tobacco smoking and at least 19 different cancer types has been firmly drawn from human epidemiological studies.53 The biologic effects of the multiple mutagens/carcinogens found in tobacco smoke have been well documented in both animals and humans, however less studied are the properties of nicotine itself on tumor biology. The following sections review the emerging data related to the mechanisms by which nicotine may augment cancer growth from a cellular to a whole tumor basis.
Nicotine Receptors on Cancer Cells
The presence of nicotinic acetylcholine receptors (nAChRs) in non-neuronal cells suggests a function for this receptor class outside of synaptic signaling. These receptors are found in a wide variety of epithelial tissue types. Cancers that express the main nAChR receptor subtype responsible for driving tumor progression in most tumors of non-neuronal origin (alpha7-nAChR) are frequently found along the upper aerodigestive tract and bronchial tree, where direct exposure to nicotine and other carcinogens is frequent in tobacco consuming individuals.54 These include head and neck squamous cell carcinoma (HNSCC), esophageal carcinoma, small cell and non-small cell lung cancer (SCLC/NSCLC).55,56 Although other nAChR subtypes have been found in these cancers, the preponderance and induction of alpha7 in human cancers has resulted in proposed targeting strategies of this specific receptor subtype in bronchial-based cancers. Breast and bladder carcinomas also express alpha7-nAChRs.
Signaling Effects and Tumor Proliferation
The lack of intrinsic excitability in tumor cells expressing nAChRs suggests that alternative signaling pathways to traditional depolarization are employed. Treatment of SCLC cells with nicotine stimulates calcium influx responsible for second messenger signaling to promote DNA synthesis. In addition, nicotine stimulates (via norepinephrine) beta-adrenergic receptor signaling in several different animal tumor models, resulting in elevated tumor growth and neovascularization (angiogenesis).57 These reports demonstrate direct and indirect roles for nicotine in stimulating second messenger signaling.
Additional studies suggest that nAChRs also act in a manner analogous to conventional transmembrane growth factor receptors, where nAChR activation triggers signaling cascades that are commonly activated in tumor cells by extracellular signals. Supporting this idea is activation of alpha7-nAChR by the endogenous ligand secreted mammalian Ly-6/urokinase plasminogen activator receptor-related protein-1 (SLURP1), as well as binding of the related ligand SLURP-2 to alpha3-nAChRs.58 SLURP1 directly binds alpha7-nAChR to competitively impair nicotine binding, indicating that alpha7-nAChR activity can be triggered by paracrine signals outside of nicotine to modulate intracellular signaling cascades.
Pharmacological blockade of alpha7-nAChR impairs the growth of several tumor cell types, indicating that alpha7 is the main AChR subtype responsible for mediating mitogenic signaling. Nicotine binding to the alpha7 and other nAChRs triggers several specific signaling pathways that influence tumor growth, survival and invasion. Some of the more prominent signaling events engaged by nAChR activation include elevated activation of the serine kinase Raf-MEK-ERK pathway and protein kinase C (PKC) through alpha7-nAChR in SCLC cells exposed to nicotine, resulting in phosphorylation of the transcription factor Myc and stimulation of gene expression.59 Nicotine induced Raf activation through alpha7-nAChR in several NSCLC cell lines also stimulates cyclin D and cyclin E dependent kinase production, resulting in cell cycle progression.60
Nicotine also stimulates phosphorylation and inactivation of retinoblastoma protein (Rb), releasing inhibitory binding to the E2F1 transcription factor to promote cell cycle progression. The growth promoting transcription factor NF-kappaB is also promoted by nicotine through the PI3-kinase/Akt pathway in NSCLC cell lines.61
Collective activation and/or enhanced signaling of these pathways by nicotine could contribute to increased tumor proliferation in cancer patients with continued tobacco exposure, perhaps compromising clinical treatment regimes. In support of this, most studies on cells in culture are treated with nicotine concentrations of 1 micromolar (162 ng/mL), which is similar to the concentration found in the plasma of chronic smokers (approximately 45 ng/mL).62
Nicotine's Effects on Tumor Biology
Apoptosis
Escape from programmed cell death (apoptosis) is a hallmark of cancer that contributes to tumor progression. Impairment of apoptotic signaling has been linked to nicotine based nAChR activation in many cancer types through the utilization of multiple signaling pathways that circumvent apoptosis by inactivating phosphorylation events on members of the Bcl-2 protein family. Nicotine stimulation in lung cancer cells results in phosphorylation of the apoptosis-promoting protein Bax by Akt as well as NF-kappaB activation to prevent cell death.63 Nicotine stimulates phosphorylation of the pro-apoptotic protein BAD on three sites in lung cancer cells, preventing activation of mitochondrial-based cell death by becoming sequestered in the cytoplasm.64 The redundancy of nicotine-based inactivation of apoptosis by targeting different Bcl-2 family proteins creates an effective means to sustain tumor cell survival as well as promoting chemotherapeutic resistance in cancer patients with continued tobacco exposure.65
In addition to impairing mitochondrial-targeting Bcl proteins, nAChR activation also promotes cell survival through induction and activation of members of the Inhibitor of Apoptosis (IAP) protein family, which function by binding and preventing activation of the caspase family of pro-apoptotic proteases. Nicotine treatment of NSCLC cells induces expression of the IAP protein survivin, enhancing protection against cisplatin-mediated apoptosis.66 Nicotine treatment also stimulates increases in the related protein XIAP. The antagonistic role of nicotine-based AChR signaling in canonical pro- and anti-apoptotic pathways presents a collective challenge for management of tobacco-related malignancies by radiation and chemotherapy.
Angiogenesis
Angiogenesis refers to the formation of new blood vessels associated with developing solid tumors. Neovasculature resultant from angiogenic mobilization and proliferation of endothelial cells is required to form the vascular network needed to provide essential nutritive delivery and waste removal from the growing tumor mass. Angiogenic vasculature also provides routes for invasive escape of tumor cells, promoting metastatic spread.
Enhanced vascular endothelial proliferation was the first reported nicotine-mediated effect on cells at concentrations down to approximately 16 pg/mL.67 Subsequent reports have demonstrated that second hand tobacco smoke and direct nicotine-mediated activation of alpha7-nAChR stimulates endothelial proliferation resulting in tumor angiogenesis and vascular network formation in animals.68 This is consistent with observations in cancer patients exposed to tobacco that have increased tumor size and vascular content compared to non-exposed individuals.69 Many of the signaling pathways responsible for alpha7-nAChR mediated tumor angiogensis are the same as those utilized for tumor growth, including the PI3-kinase/Akt, ERK and NF-kappaB (see above).
Angiogenesis can also be promoted by mobilization of endothelial progenitor cells from the marrow to sites of tumor formation. Nicotine treatment of tumor-bearing mice results in enhanced tumor growth due to epithelial progenitor cell recruitment and subsequent vessel formation, where nicotine can act directly on these cells to enhance proliferation and motility.70
Tumor Invasion and Metastasis
Epithelial to mesenchymal transition (EMT) is a key event in tumor progression that results in an altered tumor cell phenotype predisposed for invasion and extracapsular metastatic spread. It is accompanied by a series of epigenetic changes that downregulate key epithelial genes while simultaneously upregulating genes associated with the less differentiated mesenchyme. Sustained nicotine treatment of lung and breast cancer cells results in reduction of the epithelial marker proteins beta-catenin and E-cadherin, while upregulating expression of the mesenchymal proteins fibronectin and vimentin.71 Tumor cells that attain the mesenchymal phenotype are more invasive and motile, possessing the capability to degrade basement membrane extracellular matrix components through the cooperative activity of multiple different matrix metalloproteinases. These results are supported by observations in breast cancer patients and animal models where cigarette smoke exposure enhances tumor cell metastasis to the lungs and lymph nodes.72,73 Nicotine drives metastasis in pancreatic ductal carcinoma through induction of osteopontin, a pro inflammatory molecule present in individuals with chronic tobacco exposure.74 These studies suggest that nicotine promotes metastatic tumor progression by inducing EMT and augmenting the invasive properties of mesenchymal tumor cells.
Nicotine in Murine Lung Cancer Models
Two recently published studies have attempted to evaluate the effects of exogenous nicotine administration on tumor growth using murine models of tobacco induced lung cancer. Low dose nicotine administration, intended to mimic replacement therapy used in human cessation treatments, did not appear to affect the incidence, multiplicity or progression of tobacco induced lung cancers. 75 While these data are important in the context of longer term use of nicotine for smoking cessation the studies did not evaluate potential effects on response to treatment and are inconsistent with other work.60 The translatability of artificial animal models to human situations should also be interpreted with caution.
Conclusion
It is unlikely that we can anticipate 100 percent smoking cessation rates in most clinical oncology settings due both to human nature and inadequate access to smoking cessation services. Even in the resource-rich environment of NCI designated cancer centers, a recent survey has shown that up to 40 percent of these sites do not provide tobacco use treatment. Most drugs do not have formal assessment of efficacy/toxicity in smokers compared to nonsmokers, however the examples provided and mechanisms reviewed in this article may allow clinicians to suspect such interactions based on a drug's clinical pharmacology and monitor accordingly to individualize dosing.
There are substantial data suggesting inter-individual variation in the pharmacokinetics of anticancer drugs are associated with clinically meaningful variability in toxicity and efficacy. Furthermore, the importance of pharmacokinetics in drug dosing is exemplified by the contemporary practice of determining the phase II (optimal biologic) dose for newer, less toxic, molecularly targeted anticancer drugs using preclinically defined target plasma exposures. Studies of selected drugs have shown that the degree of pharmacokinetic perturbation imparted by smoking is of the same magnitude of other, clinically important drug-drug interactions and sufficient to affect dosing recommendations.Globally, tobacco smoking remains an immense issue to consider, particularly as new drug development continues to expand internationally. Studies that investigate the effects of smoking on the clinical pharmacology of drugs are almost always conducted post marketing (e.g. erlotinib), thus the clinical community should remain vigilant for situations where, based on a drug's pharmacology, such formal studies can be rationalized.
The potential undesirable pharmacologic effects of nicotine on tumor growth and development are only beginning to be explored and may partially explain the worse prognosis for patients who continue to smoke during treatment. If such findings are substantiated, additional translational studies should evaluate any detrimental biologic effects of nicotine delivered by cessation products to cancer patients, as typical blood concentrations from such are within a log of those used in cell culture to stimulate cancer growth.
Acknowledgments
Disclaimer: The opinions expressed in this article are those of the authors and do not necessarily represent an official position of the U.S. Food and Drug Administration.
Dr Weed is supported by NIH grant P20 RR016440-10. Dr Petros is supported by the Mylan Chair of Pharmacology at West Virginia University.
References
- 1.U.S. Centers for Disease Control & Prevention. State-Specific Prevalence and Trends in Adult Cigarette Smoking --- United States, 1998—2007. MMWR. 2009 Mar 13;58:221–226. [PubMed] [Google Scholar]
- 2.Garces YI, Yang P, Parkinson J, et al. The relationship between cigarette smoking and quality of life after lung cancer diagnosis. Chest. 2004;126:1733–41. doi: 10.1378/chest.126.6.1733. [DOI] [PubMed] [Google Scholar]
- 3.Burke L, Miller LA, Saad A, Abraham J. Smoking behaviors among cancer survivors: An observational clinical study. J Oncol Practice. 2009;5:6–9. doi: 10.1200/JOP.0912001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gritz E, Fingeret MC, Vidrine DJ, et al. Successes and failures of the teachable moment – Smoking cessation in cancer patients. Cancer. 2006;106:17–27. doi: 10.1002/cncr.21598. [DOI] [PubMed] [Google Scholar]
- 5.Morgan G, Schnoll RA, Alfano CM, et al. National Cancer Institute Conference on Treating Tobacco Dependence at Cancer Centers. J Oncol Pract. 2011;7:178–82. doi: 10.1200/JOP.2010.000175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ebbert JO, Williams BA, Sun Z, et al. Duration of smoking abstinence as a predictor for non-small-cell lung cancer survival in women. Lung Cancer. 2005;47:165–72. doi: 10.1016/j.lungcan.2004.07.045. [DOI] [PubMed] [Google Scholar]
- 7.Fortin A, Wang CS, Vigneault E. Influence of smoking and alcohol drinking behaviors on treatment outcomes of patients with squamous cell carcinomas of the head and neck. Int J Radiat Oncol Biol Phys. 2009;74:1062–9. doi: 10.1016/j.ijrobp.2008.09.021. [DOI] [PubMed] [Google Scholar]
- 8.Petros WP, Evans WE. Anticancer agents. In: Burton ME, Shaw LM, Schentag JJ, Evans WE, editors. Applied pharmacokinetics & Pharmacodynamics – Principles of therapeutic drug monitoring. Philadelphia: Lippincott Williams & Wilkins; 2006. pp. 617–636. [Google Scholar]
- 9.Benowitz NL. Nicotine addiction. N Engl J Med. 2010;362:2295–303. doi: 10.1056/NEJMra0809890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wells AJ, English PB, Posner SF, Wagenknecht LE, Perez-Stable EJ. Misclassification rates for current smokers misclassified as nonsmokers. Am J Public Health. 1998;88:1503–9. doi: 10.2105/ajph.88.10.1503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Israili ZH, Dayton PG. Human alpha-1-glycoprotein and its interactions with drugs. Drug Metab Rev. 2001;33:161–235. doi: 10.1081/dmr-100104402. [DOI] [PubMed] [Google Scholar]
- 12.Lind P, Engström G, Stavenow L, Janzon L, Lindgärde F, Hedblad B. Risk of myocardial infarction and stroke in smokers is related to plasma levels of inflammation-sensitive proteins. Arterioscler Thromb Vasc Biol. 2004;24:577–82. doi: 10.1161/01.ATV.0000116863.37311.82. [DOI] [PubMed] [Google Scholar]
- 13.Moszczyński P, Zabiński Z, Moszczyński P, Jr, Rutowski J, Słowiński S, Tabarowski Z. Immunological findings in cigarette smokers. Toxicol Lett. 2001;118:121–7. doi: 10.1016/s0378-4274(00)00270-8. [DOI] [PubMed] [Google Scholar]
- 14.Tappia PS, Troughton KL, Langley-Evans SC, Grimble RF. Cigarette smoking influences cytokine production and antioxidant defenses. Clin Sci (Lond) 1995;88:485–89. doi: 10.1042/cs0880485. [DOI] [PubMed] [Google Scholar]
- 15.Shima M, Adachi M. Effects of environmental tobacco smoke on serum levels of acute phase proteins in schoolchildren. Prev Med. 1996;25:617–24. doi: 10.1006/pmed.1996.0097. [DOI] [PubMed] [Google Scholar]
- 16.Uslu C, Taysi S, Ackay F, Sutbeyaz MY, Bakan N. Serum free and bound sialic acid and alpha-1-acid glycoprotein in patients with laryngeal cancer. Ann Clin Lab Sci. 2003;33:156–9. [PubMed] [Google Scholar]
- 17.Duché J, Urien S, Simon N, Malaurie E, Monnet I, Barré Expression of the genetic variants of human alpha-1-acid glycoprotein in cancer. Clin Biochem. 2000;33:197–202. doi: 10.1016/s0009-9120(00)00048-5. [DOI] [PubMed] [Google Scholar]
- 18.Budai L, Ozohanics O, Ludányi K, Drahos L, Kremmer T, Krenyacz J, Vékey K. Investigation of genetic variants of alpha-1 acid glycoprotein by ultra-performance liquid chromatography-mass spectrometry. Anal Bioanal Chem. 2009;393:991–8. doi: 10.1007/s00216-008-2518-6. [DOI] [PubMed] [Google Scholar]
- 19.Baker SD, Sparreboom A, Verweij J. Clinical pharmacokinetics of docetaxel. Clin Pharmacokinet. 2006;45:235–252. doi: 10.2165/00003088-200645030-00002. [DOI] [PubMed] [Google Scholar]
- 20.Minami H, Kawada K, Sasaki Y, et al. Pharmacokinetics and pharmacodynamics of protein-unbound docetaxel in cancer patients. Cancer Sci. 2006;97:235–241. doi: 10.1111/j.1349-7006.2006.00166.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bruno R, Viivier N, Vergniol JC, et al. A population pharmacokinetic model for docetaxel (Taxotere), model building and validation. J Pharmacokin Biopharm. 1996;24:153–172. doi: 10.1007/BF02353487. [DOI] [PubMed] [Google Scholar]
- 22.Baker SD, Li J, ten Tije AJ, et al. Relationship of systemic exposure to unbound docetaxel and neutropenia. Clin Pharmacol Ther. 2005;77:43–53. doi: 10.1016/j.clpt.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 23.Puisset F, Alexandre J, Treluyer JM, et al. Clinical Pharmacodyanmic factors in docetaxel toxicity. Br J Cancer. 2007;97:290–296. doi: 10.1038/sj.bjc.6603872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.von Mehren M, Widmer N. Correlations between imatinib pharmacokinetics, pharmacodynamics, adherence, and clinical response in advanced metastatic gastrointestinal stromal tumor (GIST): An emerging role for drug blood level testing? Cancer Treat Rev. 2011;37:291–299. doi: 10.1016/j.ctrv.2010.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Delbaldo C, et al. Pharmacokinetic-pharmacodynamic relationships of imatinib and its main metabolite in patients with advanced gastrointestinal stromal tumors. Clin Cancer Res. 2006;12:6073–6078. doi: 10.1158/1078-0432.CCR-05-2596. [DOI] [PubMed] [Google Scholar]
- 26.Widmer N, Decosterd LA, Leyvraz S, et al. Relationship of imatinib-free plasma levels and target genotype with efficacy and tolerability. Br J Cancer. 2008;98:1633–1640. doi: 10.1038/sj.bjc.6604355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.van Erp N, Gelderblom H, van Glabbeke M, et al. Effect of cigarette smoking on imatinib in patients in the soft tissue and bone sarcoma group of the EORTC. Clin Cancer Res. 2008;14:8308–8313. doi: 10.1158/1078-0432.CCR-08-1303. [DOI] [PubMed] [Google Scholar]
- 28.Kushinsky R, Louis CJ. The effect of cigarette smoke on aryl hydrocarbon hydroxylase activity and cytochrome P450 content in rat liver and lung microsomes. Oncology. 1976;33:197–200. doi: 10.1159/000225143. [DOI] [PubMed] [Google Scholar]
- 29.Vistisen K, Loft S, Poulsen HE. Cytochrome P450 IA2 activity in man measured by caffeine metabolism: effect of smoking, broccoli and exercise. Adv Exp Med Biol. 1991;283:407–11. doi: 10.1007/978-1-4684-5877-0_55. [DOI] [PubMed] [Google Scholar]
- 30.Anttila S, Tuominen P, Hirvonen A, et al. CYP1A1 levels in lung tissue of tobacco smokers and polymorphisms of CYP1A1 and aromatic hydrocarbon receptor. Pharmacogenetics. 2001 Aug;11:501–9. doi: 10.1097/00008571-200108000-00005. [DOI] [PubMed] [Google Scholar]
- 31.Smith GB, Harper PA, Wong JM, et al. Human lung microsomal cytochrome P4501A1 (CYP1A1) activities: impact of smoking status and CYP1A1, aryl hydrocarbon receptor, and glutathione S-transferase M1 genetic polymorphisms. Cancer Epidemiol Biomarkers Prev. 2001 Aug;10:839–53. [PubMed] [Google Scholar]
- 32.Shah PP, Saurabh K, Pant MC, et al. Evidence for increased cytochrome P450 1A1 expression in blood lymphocytes of lung cancer patients. Mutat Res. 2009 Nov 2;670:74–8. doi: 10.1016/j.mrfmmm.2009.07.006. [DOI] [PubMed] [Google Scholar]
- 33.Zhou SF, Wang B, Yang LP, Liu JP. Structure, function, regulation and polymorphism and the clinical significance of human cytochrome P450 1A2. Drug Metab Rev. 2010 May;42:268–354. doi: 10.3109/03602530903286476. [DOI] [PubMed] [Google Scholar]
- 34.Mauri MC, Volonteri LS, Colasanti A, Fiorentini A, De Gaspari IF, Bareggi SR. Clinical pharmacokinetics of atypical antipsychotics: a critical review of the relationship between plasma concentrations and clinical response. Clin Pharmacokinet. 2007;46:359–88. doi: 10.2165/00003088-200746050-00001. [DOI] [PubMed] [Google Scholar]
- 35.Faber MS, Fuhr U. Time response of cytochrome P450 1A2 activity on cessation of heavy smoking. Clin Pharmacol Ther. 2004 Aug;76:178–84. doi: 10.1016/j.clpt.2004.04.003. [DOI] [PubMed] [Google Scholar]
- 36.Bloom J, Hinrichs AL, Wang JC, et al. The contribution of common CYP2A6 alleles to variation in nicotine metabolism among European-Americans. Pharmacogenet Genomics. 2011 Jul;21:403–16. doi: 10.1097/FPC.0b013e328346e8c0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Helmig S, Seelinger JU, Philipp-Gehlhaar M, et al. Cyp1B1 mRNA expression in correlation to cotinine levels with respect to the Cyp1B1 L432V gene polymorphism. Eur J Epidemiol. 2010 Dec;25:867–73. doi: 10.1007/s10654-010-9505-x. [DOI] [PubMed] [Google Scholar]
- 38.Port JL, Yamaguchi K, Du B, et al. Tobacco smoke induces CYP1B1 in the aerodigestive tract. Carcinogenesis. 2004;25:2275–81. doi: 10.1093/carcin/bgh243. [DOI] [PubMed] [Google Scholar]
- 39.Thum T, Erpenbeck VJ, Moeller J, et al. Expression of xenobiotic metabolizing enzymes in different lung compartments of smokers and nonsmokers. Environ Health Perspect. 2006 Nov;114:1655–61. doi: 10.1289/ehp.8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoedel KA, Sellers EM, Palmour R, Tyndale RF. Down-regulation of hepatic nicotine metabolism and a CYP2A6-like enzyme in African green monkeys after long-term nicotine administration. Mol Pharmacol. 2003 Jan;63:96–104. doi: 10.1124/mol.63.1.96. [DOI] [PubMed] [Google Scholar]
- 41.Hukkanen J, Jacob Iii P, Peng M, et al. Effects of nicotine on cytochrome P450 2A6 and 2E1 activities. Br J Clin Pharmacol. 2010 Feb;69:152–9. doi: 10.1111/j.1365-2125.2009.03568.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Benowitz NL, Peng M, Jacob P., 3rd Effects of cigarette smoking and carbon monoxide on chlorzoxazone and caffeine metabolism. Clin Pharmacol Ther. 2003 Nov;74:468–74. doi: 10.1016/j.clpt.2003.07.001. [DOI] [PubMed] [Google Scholar]
- 43.Collier AC, Tingle MD, Paxton JW, et al. Metabolizing enzyme localization and activities in the first trimester human placenta: the effect of maternal and gestational age, smoking and alcohol consumption. Hum Reprod. 2002 Oct;17:2564–72. doi: 10.1093/humrep/17.10.2564. [DOI] [PubMed] [Google Scholar]
- 44.Court MH. Interindividual variability in hepatic drug glucuronidation: studies into the role of age, sex, enzyme inducers, and genetic polymorphism using the human liver bank as a model system. Drug Metab Rev. 2010 Feb;42:209–24. doi: 10.3109/03602530903209288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hamilton M, Wolf JL, Rusk J, et al. Effects of smoking on the pharmacokinetics of erlotinib. Clin Cancer Res. 2006 Apr 1;12:2166–71. doi: 10.1158/1078-0432.CCR-05-2235. [DOI] [PubMed] [Google Scholar]
- 46.Lu JF, Eppler SM, Wolf J, et al. Clinical pharmacokinetics of erlotinib in patients with solid tumors and exposure-safety relationship in patients with non-small cell lung cancer. Clin Pharmacol Ther. 2006 Aug;80:136–45. doi: 10.1016/j.clpt.2006.04.007. [DOI] [PubMed] [Google Scholar]
- 47.Hughes AN, O'Brien ME, Petty WJ, et al. Overcoming CYP1A1/1A2 mediated induction of metabolism by escalating erlotinib dose in current smokers. J Clin Oncol. 2009 Mar 10;27:1220, 6. doi: 10.1200/JCO.2008.19.3995. Epub 2009 Jan 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lilenbaum R, Axelrod R, Thomas S, et al. Randomized phase II trial of erlotinib or standard chemotherapy in patients with advanced non-small-cell lung cancer and a performance status of 2. J Clin Oncol. 2008 Feb 20;26:863–9. doi: 10.1200/JCO.2007.13.2720. [DOI] [PubMed] [Google Scholar]
- 49.Chuah B, Goh BC, Lee SC, et al. Comparison of the pharmacokinetics and pharmacodynamics of S-1 between Caucasian and East Asian patients. Cancer Sci. 2011 Feb;102:478–83. doi: 10.1111/j.1349-7006.2010.01793.x. [DOI] [PubMed] [Google Scholar]
- 50.Lin JH, Lu AY. Interindividual variability in inhibition and induction of cytochrome P450 enzymes. Annu Rev Pharmacol Toxicol. 2001;41:535–67. doi: 10.1146/annurev.pharmtox.41.1.535. [DOI] [PubMed] [Google Scholar]
- 51.van der Bol JM, Mathijssen RH, Loos WJ, et al. Cigarette smoking and irinotecan treatment: pharmacokinetic interaction and effects on neutropenia. J Clin Oncol. 2007 Jul 1;25:2719–26. doi: 10.1200/JCO.2006.09.6115. [DOI] [PubMed] [Google Scholar]
- 52.Benowitz NL. Cigarette smoking and the personalization of irinotecan therapy. J Clin Oncol. 2007 Jul 1;25:2646–7. doi: 10.1200/JCO.2007.10.7359. Epub 2007 Jun 11. [DOI] [PubMed] [Google Scholar]
- 53.Hect SS. Tobacco. In: DeVita VT Jr, Lawrence TS, Rosenberg SA, editors. Cancer – Principles and Practice of Oncology. Vol. 2011. Philadelphia: Wolters Kluwer/Lippincott Williams & Wilkins; pp. 150–160. [Google Scholar]
- 54.Catassi A, Servent D, Paleari L, Cesario A, Russo P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res. 2008;659:221–31. doi: 10.1016/j.mrrev.2008.04.002. Epub 2008 Apr 11. [DOI] [PubMed] [Google Scholar]
- 55.Carracedo DG, Rodrigo JP, Nieto CS, Gonzalez MV. Epithelial cell nicotinic acetylcholine receptor expression in head and neck squamous cell carcinoma pathogenesis. Anticancer Res. 2007;27:835–9. [PubMed] [Google Scholar]
- 56.Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci U S A. 1990;87:3294–8. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Al-Wadei HA, Plummer HK, 3rd, Schuller HM. Nicotine stimulates pancreatic cancer xenografts by systemic increase in stress neurotransmitters and suppression of the inhibitory neurotransmitter gamma-aminobutyric acid. Carcinogenesis. 2009;30:506–11. doi: 10.1093/carcin/bgp010. Epub 2009 Jan 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Grando SA. Basic and clinical aspects of non-neuronal acetylcholine: biological and clinical significance of non-canonical ligands of epithelial nicotinic acetylcholine receptors. J Pharmacol Sci. 2008;106:174–9. doi: 10.1254/jphs.fm0070087. Epub 2008 Feb 16. [DOI] [PubMed] [Google Scholar]
- 59.Jull BA, Plummer HK, 3rd, Schuller HM. Nicotinic receptor-mediated activation by the tobacco-specific nitrosamine NNK of a Raf-1/MAP kinase pathway, resulting in phosphorylation of c-myc in human small cell lung carcinoma cells and pulmonary neuroendocrine cells. J Cancer Res Clin Oncol. 2001;127:707–17. doi: 10.1007/s004320100289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Davis R, Rizwani W, Banerjee S, et al. Nicotine promotes tumor growth and metastasis in mouse models of lung cancer. PLoS One. 2009;4:e7524. doi: 10.1371/journal.pone.0007524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsurutani J, Castillo SS, Brognard J, et al. Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis. 2005;26:1182–95. doi: 10.1093/carcin/bgi072. Epub 2005 Mar 24. [DOI] [PubMed] [Google Scholar]
- 62.Dasgupta P, Rastogi S, Pillai S, et al. Nicotine induces cell proliferation by beta-arrestin-mediated activation of Src and Rb-Raf-1 pathways. J Clin Invest. 2006;116:2208–2217. doi: 10.1172/JCI28164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Tsurutani J, Castillo SS, Brognard J, et al. Tobacco components stimulate Akt-dependent proliferation and NFkappaB-dependent survival in lung cancer cells. Carcinogenesis. 2005;26:1182–95. doi: 10.1093/carcin/bgi072. Epub 2005 Mar 24. [DOI] [PubMed] [Google Scholar]
- 64.Jin Z, Gao F, Flagg T, Deng X. Nicotine induces multi-site phosphorylation of Bad in association with suppression of apoptosis. J Biol Chem. 2004;279:23837–44. doi: 10.1074/jbc.M402566200. Epub 2004 Mar 22. [DOI] [PubMed] [Google Scholar]
- 65.Catassi A, Servent D, Paleari L, Cesario A, Russo P. Multiple roles of nicotine on cell proliferation and inhibition of apoptosis: implications on lung carcinogenesis. Mutat Res. 2008;659:221–31. doi: 10.1016/j.mrrev.2008.04.002. Epub 2008 Apr 11. [DOI] [PubMed] [Google Scholar]
- 66.Dasgupta P, Kinkade R, Joshi B, Decook C, Haura E, Chellappan S. Nicotine inhibits apoptosis induced by chemotherapeutic drugs by up-regulating XIAP and survivin. Proc Natl Acad Sci U S A. 2006;103:6332–7. doi: 10.1073/pnas.0509313103. Epub 2006 Apr 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Villablanca AC. Nicotine stimulates DNA synthesis and proliferation in vascular endothelial cells in vitro. J Appl Physiol. 1998;84:2089–98. doi: 10.1152/jappl.1998.84.6.2089. [DOI] [PubMed] [Google Scholar]
- 68.Zhu BQ, Heeschen C, Sievers RE, et al. Second hand smoke stimulates tumor angiogenesis and growth. Cancer Cell. 2003;4:191–6. doi: 10.1016/s1535-6108(03)00219-8. [DOI] [PubMed] [Google Scholar]
- 69.Singh S, Pillai S, Chellappan S. Nicotinic acetylcholine receptor signaling in tumor growth and metastasis. J Oncol. 2011;2011:456743. doi: 10.1155/2011/456743. Epub 2011 Mar 30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Heeschen C, Chang E, Aicher A, Cooke JP. Endothelial progenitor cells participate in nicotine-mediated angiogenesis. J Am Coll Cardiol. 2006;48:2553–60. doi: 10.1016/j.jacc.2006.07.066. Epub 2006 Nov 28. [DOI] [PubMed] [Google Scholar]
- 71.Dasgupta P, Rizwani W, Pillai S, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. doi: 10.1002/ijc.23894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Murin S, Inciardi J. Cigarette smoking and the risk of pulmonary metastasis from breast cancer. Chest. 2001;119:1635–40. doi: 10.1378/chest.119.6.1635. [DOI] [PubMed] [Google Scholar]
- 73.Murin S, Pinkerton KE, Hubbard NE, Erickson K. The effect of cigarette smoke exposure on pulmonary metastatic disease in a murine model of metastatic breast cancer. Chest. 2004;125:1467–71. doi: 10.1378/chest.125.4.1467. [DOI] [PubMed] [Google Scholar]
- 74.Lazar M, Sullivan J, Chipitsyna G, et al. Induction of monocyte chemoattractant protein-1 by nicotine in pancreatic ductal adenocarcinoma cells: role of osteopontin. Surgery. 2010;148:298–309. doi: 10.1016/j.surg.2010.05.002. Epub 2010 Jun 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Shields PG. Long-term nicotine replacement therapy: cancer risk in context. Cancer Prev Res. 2011;4:1719–23. doi: 10.1158/1940-6207.CAPR-11-0453. [DOI] [PubMed] [Google Scholar]