

Abstract
Protein-truncating mutations in the dystrophin gene lead to the progressive muscle wasting disorder Duchenne muscular dystrophy, whereas in-frame deletions typically manifest as the milder allelic condition, Becker muscular dystrophy. Antisense oligomer-induced exon skipping can modify dystrophin gene expression so that a disease-associated dystrophin pre-mRNA is processed into a Becker muscular dystrophy-like mature transcript. Despite genomic deletions that may encompass hundreds of kilobases of the gene, some dystrophin mutations appear “leaky”, and low levels of high molecular weight, and presumably semi-functional, dystrophin are produced. A likely causative mechanism is endogenous exon skipping, and Duchenne individuals with higher baseline levels of dystrophin may respond more efficiently to the administration of splice-switching antisense oligomers. We optimized excision of exons 8 and 9 in normal human myoblasts, and evaluated several oligomers in cells from eight Duchenne muscular dystrophy patients with deletions in a known “leaky” region of the dystrophin gene. Inter-patient variation in response to antisense oligomer induced skipping in vitro appeared minimal. We describe oligomers targeting exon 8, that unequivocally increase dystrophin above baseline in vitro, and propose that patients with leaky mutations are ideally suited for participation in antisense oligomer mediated splice-switching clinical studies.
Keywords: antisense oligomers, Duchenne muscular dystrophy, exon skipping, personalized genetic therapy, splice-switching
Introduction
Dystrophin is an essential component of the oligomeric protein complex that protects the sarcolemma from the shearing forces of muscle contraction and relaxation. Diminished expression and/or production of a faulty dystrophin causes the X-linked recessive allelic conditions Duchenne (DMD) and Becker muscular dystrophy (BMD). DMD is the most common form of childhood muscle wasting, with patients losing ambulation before 12 years of age and, without appropriate healthcare and support, the condition is typically fatal in the second decade of life.1 In contrast, BMD presents with more variable phenotypes and may display a slower clinical progression, with varying levels of impairment2 and in some cases, only minimal symptoms such as cramps on exercise and elevation of serum creatine kinase.
Consequently, dystrophinopathies can be viewed as a spectrum, with DMD, the most severe phenotype being caused by the almost or complete failure of the protein. BMD typically describes those patients with milder symptoms and increased life expectancy resulting from internally deleted dystrophin isoforms retaining partial function. The wide range of dystrophinopathy phenotypes and the positive correlation between dystrophin levels and the age at which independent mobility is lost3 suggests that even a modest increase in expression of a functional dystrophin isoform could confer substantial clinical benefit.
The majority of DMD causing mutations are exon deletions that disrupt the reading frame, with two deletion prone regions of the gene identified at the 5′ end (minor hotspot) and involving exons 45–55 (major hotspot).4 The frame-shifting deletions lead to premature translational termination and destabilize the gene transcript through the induction of nonsense-mediated decay, whereas BMD-causing mutations are typically in-frame deletions that have limited impact on the critical binding domains. These observations led to the reading frame rule that correlates the genotype/phenotype relationship seen in DMD and BMD.5 Although more than 90% of mutations described in the dystrophin gene abide by this rule, there are exceptions where a protein-truncating mutation manifests as a mild phenotype, or an in-frame deletion has severe consequences (for review see).6 Some of these exceptions to the reading frame rule can be explained by the catastrophic effects of the mutation on crucial functional domains, whereas others arise from unanticipated consequences to the processing of the dystrophin mRNA. The in-frame, genomic deletion of exon 5 has been reported to cause DMD, rather than BMD as expected. Subsequent RNA studies revealed that the deletion of exon 5 led to the loss of exon 6 from the mature mRNA, resulting in a frame-shift that is consistent with a DMD phenotype, indicating cross-talk between exons during processing.7 Conversely, Ginjaar et al.8 reported a nonsense mutation in exon 29 that induced variable levels of skipping of this in-frame exon in three boys with DMD, leading to mild, moderate, and severe phenotypes within one family.
Individuals with deletions encompassing exons 3–7 can present with clinical severities ranging from BMD to DMD,9,10,11 and several mechanisms have been proposed that might moderate the predicted DMD phenotype in these cases; restoration of the reading frame by a second mutation, ribosomal frame-shifting, the use of alternative promoters, re-initiation of translation at an in-frame start codon in exon 89,12 and alternative splicing. We consider the latter possibility most likely, as transcripts capable of being translated into a BMD-like protein have been detected in dystrophic muscle containing another leaky mutation, the deletion of exon 45. Prior et al.13 identified naturally occurring transcripts missing exons 44 and 45 that could be translated into a functional dystrophin isoform. Induction of functional dystrophin after re-initiation of translation or the use of alternative promoters from this region of the dystrophin gene must be considered highly unlikely, since this would eliminate crucial actin binding domains.
Antisense oligomer (AO) mediated exon skipping during pre-mRNA processing to restore the reading frame or excise exons carrying stop codons is emerging as a promising therapy for DMD. This intervention can give rise to a mature mRNA that is translated into an internally truncated, but partially functional BMD-like dystrophin isoform. Proof-of-concept of this approach was reported after direct intra-muscular injections of splice-switching oligomers targeting exon 51 to restore the reading frame in a subset of DMD deletion patients, using either 2′-O-methyl (2OMe) modified bases on a phosphorothioate backbone,14 or a phosphorodiamidate morpholino oligomer.15 More recently, dystrophin expression was demonstrated in muscle after systemic delivery of both oligomers to patients with DMD with amenable mutations.16,17
Clinical evaluation of the morpholinooligomer targeting exon 51 (AVI-4658, now termed Eteplirsen) revealed that, although there were clear differences in dystrophin levels before and after oligomer administration, not all participants receiving the same dosage responded comparably. With this in mind, it is possible that variations in responses to exon skipping oligomers will be more pronounced when these are applied to mutations that naturally exhibited a degree of “leakiness” and result in variable disease severity.
Here we present in vitro data showing that leaky mutations in the minor deletion hotspot at the 5′ end of the dystrophin gene transcript are highly amenable to AO-induced exon 8 and 9 skipping, with only modest inter-patient variation. Several splice-switching AOs were evaluated, and similar trends in exon 8(+9) skipping efficiency by all AOs tested were observed in the different patient derived cells. We have previously shown that directing splice-switching oligomers to exon 8 resulted in human or canine dystrophin transcripts missing both exons 8 and 9.18,19 Indeed, in early canine exon skipping studies, RT-PCR carried out using primers targeting exons 1–9 failed to detect any splice switching, but when amplification conditions were modified to include exons 1–10, robust exon 8 and 9 skipping was detected.18 Although there appears to be cross-talk between dystrophin exons 8 and 9 during pre-mRNA processing, this communication appears one way, as splice-switching oligomers directed to exon 9 induce specific removal of the target exon alone. In summary, we have designed AOs that induce efficient exon 8 (+9) skipping, and propose that these compounds should be considered for clinical evaluation to address deletions of dystrophin exons 3–7 and 5–7.
Results
Preliminary AO screening
The splice motif predictor program, exonic splicing enhancer (ESE) finder20,21 was used to predict putative ESEs in exon 8 (Figure 1a). Using this information, splice-switching AOs were designed to induce skipping of exon 8, targeting the 3′ acceptor splice site, intra-exonic domains and the 5′ donor splice site (Figure 1b), and synthesized as 2OMe modified bases on a phosphorothioate backbone. These AOs were transfected into normal human myoblasts as cationic lipoplexes at 100, 200, and 400 nmol/l and subsequent RT-PCR analysis showed most oligomers induced pronounced excision of exons 8(+9) (Figure 1c). The loss of exon 9 does not disrupt the reading frame and is not considered contra-indicatory, and AOs targeting the 5′ end of the exon (i.e., splice acceptor and ESE motifs) induced the most efficient exon excision. These AOs also elicited substantial levels of exon 8(+9) skipping in myogenic cells from DMD patient 1 (DM1) with a deletion of exons 3–7, as shown in Figure 1d. Although, all oligomers induced efficient exon skipping at the concentrations tested, H8A(−6+24) appeared most effective in that minimal full-length dystrophin transcript product was observed. Different primer sets were used to amplify this region of the mRNA to minimize size differences between amplicons from normal and DMD gene transcripts (Table 4).
Figure 1.

Location of exonic splicing enhancers (ESEs) and antisense oligomer (AO) targets in dystrophin exon 8. (a) Locations of ESEs in exon 8 and 50 bases of flanking intronic sequence, predicted by ESE finder 3.0.20,21 Predicted splice factor binding sites, and relative scores are indicated on the y-axis. Red: SF2/ASF; blue: SC35; green: SRp40; yellow: SRp55. (b) Annealing sites of AOs targeted to dystrophin exon 8, relative to positions of predicted splice motifs shown in (a). Evaluation of selected AOs targeted to exon 8 at transfection concentrations indicated, in (c) normal human myoblasts and (d) myogenic cells derived from a patient with Duchenne muscular dystrophy missing dystrophin exons 3–7. The red bars represent additional oligomers, designed to target the exon 8 acceptor site, that were tested and found to induce some exon skipping.
Evaluation of 2OMe AOs in DMD myoblast (DM) cultures
DMD myoblasts from different patients, three carrying deletions of exons 3–7 and one carrying an exon 5–7 deletion were used to evaluate the four AOs previously shown to induce robust exon 8(+9) skipping in normal myogenic cells (Figure 2). Myoblasts from two of the patients harboring deletions of dystrophin exons 3–7, DM1 (Figure 2a) and DM3 (Figure 2b) appeared more amenable to induced exon skipping than DM4 patient cells, carrying a deletion of dystrophin exons 5–7. DM4 (Figure 2c) showed an overall lower response to all AOs but nevertheless maintained similar trends, with H8A(−6+24) inducing the most efficient exon removal, followed by H8A(+57+83). Pooled data from three transfections experiments in DM1, DM3, and DM4 cells is shown in Figure 2d. Preparations of DM2 patient cells were of variable myogenic quality, and generated less consistent data (data not shown).
Figure 2.

Evaluation of 2′-O-methyl (2OMe) antisense oligomers (AOs) targeting dystrophin exon 8 in Duchenne muscular dystrophy (DMD) myoblasts. (a–c) RT-PCR analysis of dystrophin transcripts from three DMD myogenic cell strains transfected with the four lead 2OMe AOs as lipoplexes, targeted to exon 8 over the concentration range 2.5–400 nmol/l. Gel images show full-length (FL) and induced products (refer to Table 4) from (a) DM1 (Δ3–7), (b) DM3 (Δ3–7), and (c) DM4 (Δ5–7). (d) Densitometry data showing relative exons 8 and 9 skipping as in (a–c) (n = 3), H8A(−6+18) shown in blue, H8A(−6+24) shown in red, H8A(+42+66) shown in green, and H8A(+57+83) shown in purple (mean + SEM).
Evaluation of 2OMe AOs in DMD fibroblast (DF) cultures
The same 2OMe AOs, as cationic lipoplexes (10–400 nmol/l), were transfected into Ad5.f50.AdApt.MyoD transformed fibroblasts from six unrelated DMD patients with deletions of exons 3–7 (Figure 3). In three separate experiments, patient fibroblasts DF2 and DF4 showed inconsistent exon skipping in response to all AOs tested (data not shown). DF1 showed a consistently poor but nevertheless dose-dependant response to all 2OMe oligomers over the range of concentrations tested (Figure 3a). DF3 (Figure 3b), DF5 (Figure 3c), and DF6 (Figure 3d), also responded in a dose-dependent manner to all AOs tested, with H8A(−6+24) inducing the highest levels of exon 8(+9) skipping. Pooled data from four transfections of DF1, DF3, DF5, and DF6 is shown in Figure 3e. Although the overall level of exon skipping varied, a clear dose-dependant response was evident and H8A(−6+24) was again one of the most consistent splice-switching oligomers.
Figure 3.

Evaluation of 2′-O-methyl (2OMe) antisense oligomers (AOs) targeting dystrophin exon 8 in MyoD transformed Duchenne muscular dystrophy (DMD) fibroblasts. (a–d) RT-PCR analysis from unrelated DMD patient fibroblast strains, all carrying a deletion of dystrophin exons 3–7, transfected with the four 2OMe AO lipoplexes, targeted to exons 8 at 10–400 nmol/l. Full-length (FL) and oligomer induced amplicons are indicated. (e) Densitometry was used to estimate relative exons 8 and 9 skipping induced by H8A(−6+18), shown in blue, H8A(−6+24), shown in red, H8A(+42+66), shown in green, and H8A(+57+83), shown in purple (n = 4) (mean + SEM).
Evaluation of phosphorodiamidate morpholino oligomers in DMD myogenic cells
Three sequences were selected for synthesis as phosphorodiamidate morpholino oligomers (PMOs) and were transfected by nucleofection into myogenic cells from patients with DMD (DM1–4), over the concentration range of 10–100 nmol/l (Figure 4a). Each oligomer induced a dose response in each of the myogenic cell strains, with DM1 again showing the most consistent response to the oligomers. H8A(−6+24) and H8A(−6+18) generated similar levels of exon skipping, whereas H8A(+57+83) was consistently less effective than the other AOs at all transfection concentrations. Pooled data (n = 4) from treatment of DM1–4 is shown in Figure 4b.
Figure 4.

Evaluation of phosphorodiamidate morpholino oligomers (PMOs), targeting dystrophin exon 8, in Duchenne muscular dystrophy (DMD) myoblasts. (a) RT-PCR products from four DMD myoblast strains (DM1 (Δ3–7), DM3 (Δ3–7), and DM4 (Δ5–7) myoblasts) transfected with three PMOs using nucleofection. Full-length and oligomer-induced products are shown. (b) Densitometry analysis (n = 4) showing relative exons 8 and 9 skipping induced by H8A(−6+18) (blue), H8A(−6+24) (red), H8A(+42+66) (green), and H8A(+57+83) (purple) (mean + SEM).
Dystrophin expression following PMO conjugated to the cell penetrating peptide K transfection
Myogenic cell strains DM1–4 were transfected with oligomers H8A(−6+18), H8A(−6+24), and H8A(+57+83), prepared as PMO conjugated to the cell penetrating peptide K (PPMOk) at a concentration of 2 µmol/l. Seven days after transfection, protein and RNA were extracted from the cells for western blot and RTPCR analysis. Extracts from normal myogenic cells indicate wild-type levels of protein, and untreated patient cells were included to assess base line dystrophin arising from “leaky” expression. The western blot membrane was probed with NCL-DYS1 and NCL-Hamlet1 to detect dystrophin and dysferlin, respectively (Figure 5a). The latter was included as a high molecular weight, muscle-specific loading control. In each of the untreated samples, dystrophin was detected, ranging from 3–7% of normal dystrophin levels (Figure 5b). Robust dystrophin expression was induced in DM1 and DM2 cells, reaching levels of 30–60% of those in normal myogenic cells, normalized to dysferlin expression. All patient cells showed increased dystrophin expression, with DM2 showing the greatest increase, from 4% in untreated to 43–62% in treated cells, representing a 10–14 fold increase (Figure 5c). DM1 also showed a marked increase in dystrophin levels from 7% in untreated cells, to 31–40% after oligomer application. DM3 and DM4 showed more modest increases in dystrophin, reaching 17–24% and 12–14% of normal dystrophin levels respectively, after oligomer treatment (Figure 5b,c).
Figure 5.

Dystrophin protein expression in PPMOk treated Duchenne muscular dystrophy (DMD) patient myoblasts. (a) Western blot showing dystrophin and dysferlin expression in PPMOk transfected (2 µmol/l), untreated (UT) DMD myogenic cells, and untreated normal human primary myogenic cells. Full-length dystrophin is 427 kDa, Δ5–9 dystrophin is ~402 kDa and Δ3–9 dystrophin is ~395 kDa. Dystrophin and dysferlin (230 kDa) were revealed by NCL-DYS1 and NCL-Hamlet1, respectively using the Western Breeze detection system. (b) Densitometry was used to estimate dystrophin expression relative to that in normal human myogenic cells, normalized to dysferlin. (c) Densitometry analysis showing relative dystrophin expression, determined by western blotting, as compared with untreated DMD myoblasts (fold increase) and normalized to dysferlin expression on the same blot. (d) RT-PCR analysis of exons 8 and 9 skipping levels. Full-length and oligomer-induced products are shown. (e) Densitometry analysis showing relative exons 8 and 9 skipping induced by H8A(−6+18) (blue), H8A(−6+24) (red), and H8A(+57+83) (purple). Data from untreated cells is shown in orange.
RT-PCR analysis showed that in each case, the target exons were removed efficiently by all of the oligomers tested, with perhaps H8A(+57+83) being marginally less effective as indicated by the residual full-length product (Figure 5d,e). H8A(−6+18) induced the highest protein levels in DM2 and DM3, and resulted inlevels of expression comparable to H8A(−6+24) in DM1 and DM4.
Discussion
Exon 51 was selected as the first target for proof-of-concept human exon skipping trials,14,15 as this strategy is potentially applicable to about 1 in 10 patients with DMD. Depending upon the specific database interrogated, the next most common dystrophin deletion subgroups should respond to skipping of exons 45, 44, or 53 (~1 in 13–20 boys), whereas skipping exon 8(+9) would only benefit ~1 in 30 individuals with DMD. Another consideration when selecting dystrophin deletions that would have the reading frame restored by exon 51 skipping was that these mutations are considered to be absolute, resulting in very low, or undetectable dystrophin. This was indeed the case for over half of the participants in the Muscular Dystrophy Exon Skipping Consortium trial,15 but dystrophin analysis of untreated muscle revealed that several boys had 1–2% of normal dystrophin levels, and one had an estimated 5% of control levels. In the absence of data from pretreatment biopsies, such amounts of dystrophin are likely to confound interpretation of the dystrophin induced by targeted exon skipping. However, rigorous dystrophin quantitation in the Morpholino studies,15,16 before and after treatment, revealed unequivocal but variable increases in dystrophin expression in muscle from some of the trial participants. Two participants responded to oligomer treatment by showing substantial increases in dystrophin expression (2–18% and 0.9–17%), whereas a third individual was found to have 7.7% dystrophin from a baseline of zero.16
Clinical application of exon 8 skipping should not be delayed because of the relatively small proportion of patients with DMD who are likely to benefit, but rather, exon 8 should be considered a priority, since deletions in this region of the dystrophin gene have shown a propensity for inconsistent correlation between the genotype and phenotype, and therefore may prove highly amenable to exon skipping therapy. Since exon 8 (and 9) skipping has been reported to occur naturally22,23 this region could be highly amenable to splice intervention. Furthermore, the presence of low levels of endogenous high molecular weight dystrophin, despite the frame-shifting deletions of up to 300 kB, should preclude an immune response to the oligomer-induced dystrophin.
Design of AOs to excise exon 8(+9)
The in silico prediction program ESE finder 3.020,21 was used to identify domains in dystrophin exon 8 that potentially influence pre-mRNA processing. In the first screen, AOs were designed to span the acceptor splice site, three intra-exonic domains predicted to contain ESEs implicated in exon recognition, H8A(+42+66), H8A(+96+120), and H8A(134+158), and the donor splice site. 2OMe AOs were used, since we have found these oligomers to be excellent as splice-switching research tools for AO design. These compounds can be synthesized in-house and readily transfect cells in vitro as cationic lipoplexes. Unlike many other dystrophin exons examined previously, where only one or two domains were found to be amenable targets for antisense oligomer-induced exon skipping19 exon 8 was unusual, in that the majority of nonoverlapping oligomers evaluated did induce some degree of targeted exon excision, indicating the involvement of several motifs in exon 8 recognition and inclusion in the mature mRNA.
It is tempting to speculate that the induction of exon 8(+9) skipping by several oligomers targeting different motifs may correlate to the leakiness of the flanking mutations. Perhaps a degree of mis-folding as a result of the deletion in the nascent pre-mRNA compromises dystrophin splicing so that exon 8(+9) is excluded from a proportion of the transcripts. Exclusion of exon 8(+9) from a dystrophin gene transcript missing exons 3–7 will restore the reading frame, allowing these transcripts to escape nonsense-mediated decay and accumulate to low levels, reflected by the detectable dystrophin.
Using the splice site analyser tool (ibis.tau.ac.il/ssat/SpliceSite), the acceptor and donor splice sites of exons 8 and 9 were unremarkable as compared with those of the flanking exons. However, dystrophin exon 9 skipping occurs frequently in the absence of any oligomer intervention, and has been reported to occur in mice, dogs, and humans.24 Exon 9 skipping alone was commonly detected in untreated DMD patient cells, and although this did not appear to be influenced by oligomers directed to exon 8, the propensity for spontaneous exon 9 excision may contribute to the high levels of dual exon skipping. Another feature that distinguishes exon 8 from many other dystrophin exons is the length of the preceding intron 7, in excess of 110 kb. Intron length does not necessarily reflect ease of excision from the mature mRNA, as introns 1 and 2 are in excess of 190 and 170 kb, respectively, and exon 2, (62 bp) is not efficiently dislodged from the dystrophin mRNA in normal myogenic cells (K. Greet, S.D. Wilton, unpublished data). In contrast, exon 3 (93 bp) was readily removed with oligomers targeting ESE motifs near the beginning of the exon, whereas targeting the exon 3 donor splice site failed to induce any skipping (unpublished data). Another “leaky” mutation is the deletion of exon 45, which is preceded by nearly 250 kb of noncoding intervening sequence. These massive introns contain microsatellite repeats, short interspersed elements, long interspresed elements, and transposable elements, and may contribute to the relatively high genetic instability of the dystrophin gene.25 Nevertheless, intron length alone is not the most important feature contributing to gene instability within the major deletion hotspot, as the small 6.6 kb intron 49 shows a higher density of deletion breakpoints than the 37-fold larger intron 44.
We concentrated on targeting splice-switching oligomers to two regions in exon 8, the acceptor splice site and a splice enhancer-rich intra-exonic domain in the first-third of the exon. Subtle changes in length and oligomer annealing site can influence AO efficiency26 hence, additional splice-switching oligomers were designed to target the annealing sites. H8A(−6+18) and H8A(+42+66), and AOs H8A(−6+24) and H8A(+57+83) proved to be effective. Although exon skipping was induced by oligomers annealing to the exon 8 donor splice site, we did not further pursue this target region. Our earlier studies targeting other human dystrophin exons found that the acceptor site and beginning of the exon are generally the better annealing sites for AO-induced exon excision.19,27
Oligomers H8A(−6+18), H8A(−6+24), H8A(+57+83), and H8A(+42+66) were selected for more detailed evaluation as 2OMe AOs after transfection into DMD patient myoblasts and MyoD transformed DMD patient fibroblasts. Again, H8A(−6+24) was generally the most effective splice-switching compound, although the other three oligomers were also very effective when evaluated using the 2OMe chemistry.
H8A(−6+18), H8A(−6+24), and H8A(+57+83) were synthesized as PMOs and PPMOk for further evaluation. Interestingly, when evaluated as a PMO in an in vitro titration study, H8A(+57+83) was not as efficient at splice switching as H8A(−6+18) and H8A(−6+24). However, when compared as PPMOks, all three oligomers induced similar levels of exon excision and protein production, suggesting a threshold effect at the single transfection concentration tested. This emphasizes the importance of dose response assays to identify the most efficient splice-switching oligomers.
The in vitro data presented here reveal the sequence H8A(−6+24) synthesized as the 2OMe AO chemistry as a lead candidate sequence for clinical application for amenable DMD mutations (e.g., Δ3–7, Δ4–7, Δ5–7, and Δ6–7). Although 2OMe AOs are chemically modified and have greater nuclease resistance than naturally occurring nucleic acids, degradation does occur. It could be surmised that although shorter 2OMe AO sequences (e.g., H8A(−6+18)) do induce splice switching, the longer the original oligomer (H8A(−6+24)), the more effective the degradation products. Differences in splice-switching efficiencies between H8A(−6+18) and H8A(−6+24) were less obvious when these sequences were evaluated as the more stable PMOs. In addition, the extra 6 bases in H8A(−6+24) do not appear to increase the potential for off-target effects, as both target sequences produced a single potential hit with 50 and 68% coverage, respectively, when submitted to a BLAST search of human genomic and transcript sequences (data not shown). However, if PMO oligomer production costs are considered, the shorter H8A(−6+18) may be more cost effective for clinical application.
Although shorter oligomers are more economical to produce and have less potential for cross-annealing to homologous sequences, it would be counter productive to use the shorter “inexpensive” oligomers if they are significantly less effective as splice switching agents. For example, H8A(−6+14), when transfected into normal cells as a 2OMe AO, induced only minimal exon skipping at concentrations of 300 nmol/l or above (data not shown). Nevertheless, it is sometimes difficult to distinguish relative skipping efficiencies of suboptimal compounds transfected into patient DMD cells, unless tested at low concentrations to allow discrimination of the more potent compounds. Exclusion of dystrophin exons 8 and 9 in cells from a patient missing exons 3–7, generates in-frame transcripts that escape nonsense-mediated decay, and the induced transcript would therefore have a longer half-life than the out-of-frame transcript. This could lead to overestimation of exon skipping efficiency, and hence, it is important to evaluate AOs in both normal and patient cells, and over a range of transfection concentrations.
Although myogenic cells from the patients with DMD are greatly preferred for oligomer evaluation in vitro, there are limitations to using muscle-derived cells from the patients with DMD, in particular, the invasive nature of muscle biopsies unless the patient is undergoing elective surgery. In addition, we have observed considerable variation in the proliferative and myogenic capability of cells obtained from dystrophic muscle, particularly when the tissue shows advanced dystrophic pathology. Although myogenic cells from different patients with DMD showed some variation in exon skipping efficiency in this study, both at the RNA and protein levels, exon 8(+9) was generally excluded in more than 50% of the transcript products after transfection at the higher concentrations. It is not possible to establish if this variation is because of the nature of the mutation (i.e., deletion breakpoints), the influence of genetic background on splicing, or limitations of in vitro cell culture, such as variations in myogenic capacity.
One alternative to using muscle-derived patient myogenic cells is the propagation of dermal fibroblasts and forced myogenesis.28 Although the dermal cells can be readily obtained with very minor discomfort to the patient, we have found that in vitro studies using fibroblasts tend to produce variable results. Although these cells are useful for RNA studies where typically >30% exon skipping was induced at the higher transfection concentrations, analyzing protein in these cells is more challenging, presumably reflecting relatively inefficient myogenic transformation.
When undertaking western blotting, we attempt to load equivalent amounts of normal, untreated and treated patient cell extracts to allow estimation of the relative amounts of base line and induced dystrophin. Western blot analysis showed that untreated myogenic cells from the DMD patients produced from 3 to 7% of the levels of dystrophin found in normal cells, consistent with reports of exon 3–7 (and 5–7) deletions as being “leaky”,9,10,11 and not strictly conforming to the genotype:phenotype correlations.5 Acknowledging the limitations of extrapolating in vitro data, these basal levels of dystrophin expression are in line with those expected in muscle from an individual with a severe BMD phenotype. Dystrophin levels of 3% or less than those in normal muscle are consistent with a DMD phenotype. Nicholson et al.29 reported that patients with DMD with low, but nevertheless detectable amounts of dystrophin became nonambulant some 18 months later than those who had no measurable dystrophin. Patients with dystrophin levels of 3–7 % of those in normal muscle could be expected to show severe BMD or the intermediate form of muscular dystrophy, consistent with the phenotypes reported for patients with mutations in this region of the dystrophin gene.
The variable basal levels of dystrophin expression in these patients could prove problematic when evaluating AOs in clinical studies, unless baseline dystrophin expression is established before treatment is initiated. Recent data on extended administration of Eteplirsen, an exon 51 splice-switching oligomer, indicates that the duration of treatment may be more important in determining the level of dystrophin expression than the actual dosage (personal communication). The dose escalating study reported by Cirak et al.16 extended over 12 weeks, and over that time the increases in dystrophin expression varied from 2–3 fold (i.e., 0.7–2.6% or 5–12.3%) to an impressive 9–18 fold (0 to 7–9%, 2 to 18%, and 0.9 to 17%).
It is readily acknowledged that there are enormous differences between in vivo and in vitro conditions, particularly when using an artificial system such as forced myogenesis of dermal fibroblasts. However, we were able to induce in excess of 10-fold increase in dystrophin expression, in line with some of the clinical data,16 and can only speculate if similar levels of dystrophin induction over a higher basal level of expression in these patients would result in significant functional improvements over time.
The in vitro study reported here cannot conclusively account for the origin of the basal dystrophin expression in these cells carrying deletions in the minor deletion hotspot. Although dystrophin was present in untreated cells, there was an unequivocal increase in dystrophin levels after the administration of oligomer. Therefore, analysis of basal dystrophin expression in DMD muscle, before oligomer treatment, should be considered and imperative in clinical studies. It remains to be determined if those DMD/severe BMD patients with higher basal dystrophin levels will be more responsive to oligomer mediated exon skipping. The detectable dystrophin protein in muscle from these patients, without treatment, reduces the possibility of an immune response to the induced dystrophin isoform. We propose H8A(−6+24) to be a strong candidate for the treatment of DMD patients with deletions of exons 3–7 and 5–7 and further suggest that the propensity for these patients to produce “leaky” dystrophin is a good reason to expedite exon 8 skipping clinical studies.
Materials and Methods
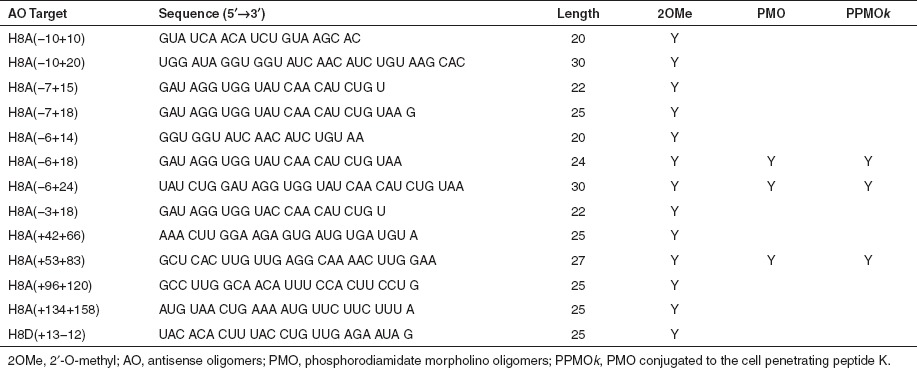
AO design and synthesis. Oligomers consisting of 2OMe modified bases on a phosphorothioate backbone were synthesized on an Expedite 8909 synthesizer (Applied Biosystems, Melbourne, Australia), as described by Adams et al.30 AO sequences are shown in Table 1, with nomenclature described by Mann et al.31 Oligomers, identified as efficient splice-switching agents, were then synthesized by Gene Tools (Philomath, OR) using the PMO chemistry, or AVI Biopharma (Bothell, WA) as a PPMOk.32,33
Table 1. AOs used in the study. Synthesized as 2OMe AO, PMO, and PPMOk.
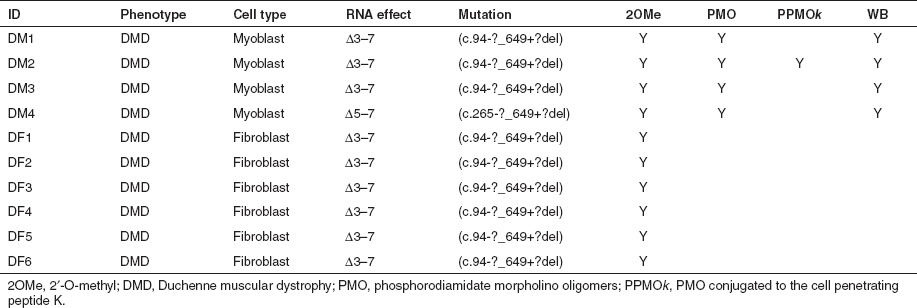
Cell culture and myogenic conversion of fibroblasts. Normal human myoblasts were prepared essentially as described by Rando et al.34 from de-identified muscle biopsies, obtained with informed consent, during elective surgery or following diagnostic muscle biopsies. Similarly donated after informed consent, de-identified fibroblasts were obtained from skin biopsies taken from the patients with DMD (Table 2). The use of human tissue has been approved by the University of Western Australia Human Ethics Committee (approval number RA/4/1/2295). Normal myoblasts were proliferated and differentiated as described previously by Harding et al.26 All cells were plated at 3 × 104 cells/well in 24 well plates that had been sequentially pre-treated for 1 hour with 50 µg/ml poly D-lysine (Sigma, Sydney, Australia) and 100 µg/ml Matrigel (BD Biosciences, Sydney, Australia).
Table 2. Summary table of DMD mutations and patient cells used in this study. DMD patient muscle-derived cells are identified as DM, and dermal fibroblasts converted to the myogenic lineage by forced myogenesis are shown as DF, western blot carried out (WB).
Following fine dissection of the skin biopsies, fibroblasts were cultured in Dulbecco's modified Eagle medium (Invitrogen, Melbourne, Australia) supplemented with 20% foetal calf serum (Serana, Bunbury, Australia), 1% GlutaMax-I (Gibco, Melbourne, Australia), 10 U/ml penicillin (Invitrogen), 10 mg/ml streptomycin (Invitrogen), and 250 ng/ml amphotericin B (Sigma). Fibroblasts were converted to myoblasts through forced myogenesis28 by transfection with a MyoD expressing adenovirus Ad5.f50.AdApt.MyoD (The Native Antigen Company, Oxford, UK), and then differentiated in low serum media. Briefly, patient fibroblasts were plated at 3 × 104 cells/well in 24 well plates that had been sequentially pre-treated for 1 hour with 50 µg/ml poly D-lysine (Sigma) and 100 µg/ml Matrigel (BD Biosciences). Twenty-four hours later, media was changed to DMEM supplemented with 5% horse serum and the MyoD adenovirus Ad5.f50.AdApt.MyoD at a multiplicity of infection of 200. Cells were allowed to differentiate for 48 hours after forced myogenesis with the MyoD adenovirus, before being transfected with oligomer.
Transfection of myogenic cells. 2OMeAOs were transfected as cationic lipoplexes with Lipofectamine 2000 (1:1 w/w) (Invitrogen) in Opti-MEM media (Gibco) as per the manufacturer's instructions.
PMOs were transfected by nucleofection using the Amaxa 4D-Nucleofector and the Cell Line P3-X kit (Lonza, Basel, Switzerland). Cells were trypsinized and counted as described above, and 8 × 104 cells per treatment group pelleted at 600 g and resuspended in 22 µl of solution P3. The PMO was added to the cuvette with 20 µl of the cell suspension, which was then placed in the Nucleofector and pulsed with program CM-132 according to the manufacturer's instructions. Pre-warmed Roswell Park Memorial Institute medium, (80 µl) (Invitrogen) was added to each cuvette immediately, and the cells were allowed to recover for 10 minutes at 37 °C. The 100 µl of cell suspension was then transferred to a fresh 1.5 ml microfuge tube containing 900 µl of differentiation medium.26 Each sample was then plated into 2 wells of a 24 well plate at 2.5–4 × 104 cells per well, allowing for some loss of cells during the nucleofection process and transfers. Nucleofected cells were maintained in differentiation media for 2–7 days after the treatment before extraction of RNA or protein.
PPMOk solutions were warmed for 5 minutes at 37 °C before being diluted in OptiMEM as indicated, and applied directly to the adherent cells at the specified concentrations. For each transfection procedure, untreated control samples were subjected to the same protocol, except that the splice-switching oligomer was excluded.
Previous studies have indicated that 48 hours after transfection is sufficient for dystrophin mRNA studies, but 7 days incubation allows time for dystrophin to accumulate to levels detectable by western blotting. PPMOk concentrations of 2 μmol/l were selected for induction of protein, as we have previously shown robust exon skipping over the transfection range of 300 nmol/l to 10 μmol/l.18
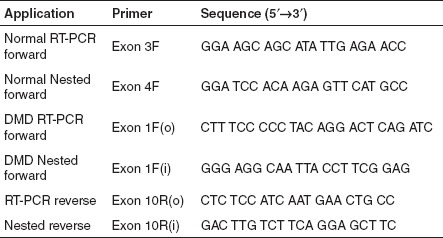
RNA extraction and nested RT-PCR. Total RNA was harvested 48 hours after transfection from duplicate wells using TRIzol (Invitrogen) according to the manufacturer's instructions, and resuspended in 40 µl of sterile water (Baxter Healthcare, Sydney, Australia). Amplification conditions were chosen that do not excessively enhance sensitivity of transcript detection, but allow some correlation between mRNA levels and induced dystrophin).35 Approximately 100 ng of total RNA was used as template for primary amplification using Superscript III One-step RT-PCR system with Platinum Taq (Invitrogen) to amplify exons 1–10 in samples from the patients with DMD, or 3–10 in samples from normal muscle. After 30 cycles (myoblasts) or 35 cycles (MyoD converted fibroblasts), a 1 µl aliquot was removed and subjected to nested PCR, amplifying exons 1–10 (DMD) or 4–10 (normal), for 30 cycles using AmpliTaq Gold (Applied Biosystems). Details of PCR primers (Geneworks, Adelaide, Australia) are shown in Table 3.
Table 3. RT-PCR primers used in the study. Inner and outer primers are represented by (i) and (o), respectively.
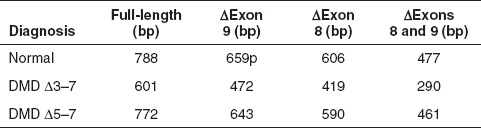
Gel analysis, imaging, and sequencing. PCR products were resolved on 2% agarose gels in TAE buffer and relative exon skipping efficiency estimated by densitometry of the full length and oligomer-induced PCR products on images captured by the Chemi-Smart 3000 system (Vilber Lourmat, MarnelaVallée, France), as described previously.30 The identities of induced transcripts were confirmed by band stab isolation,36 purification of templates using UltraClean spin columns (MoBio, Carlsbad, CA) and DNA sequencing using BigDye V3.1 terminator chemistry (Applied Biosystems) as per manufacturer's instructions. RT-PCR product sizes are summarized in Table 4. Sequencing was conducted at the Lotterywest State Biomedical Facility Genomics (Perth, Australia). Densitometric analysis was conducted using Bio1D-software (Scientific Software Group, Provo, UT).
Table 4. Expected sizes of exons 1–10 RT-PCR amplicons generated from normal and DMD patient RNA.
Western analysis of dystrophin expression. Western blotting was performed using a protocol derived from Cooper et al.37 and Nicholson et al.38 Seven days after transfection, cells were harvested by scraping, and resuspension in treatment buffer (50 µl/4.5 mg wet pellet weight) consisting of 125 mmol/l Tris-HCl pH 6.8, 15% sodium dodecyl sulfate, 10% glycerol, 0.5 mmol/l phenylmethylsulfonyl fluoride, 50 mmol/l dithiothreitol, bromophenol blue (0.004% w/v), and a protease inhibitor cocktail (15 µl/500 µl of treatment buffer) (Sigma). Samples were vortexed briefly, sonicated for 1 second, 4–8 times at a setting of 30/100 on an ultrasonic processor (Sonics, Newtown, CT) and heated at 95 °C for 5 minutes. Samples were then electrophoresed at 16 °C on a 3–10% Tris-Bis/Glycine sodium dodecyl sulfate gradient gel at pH 8.8, with a 3% stacking gel, pH 6.8. Fractionated proteins were transferred to a FluorotransW PVDF membrane (Pall, Melbourne, Australia) overnight at 18 °C at 290 mA, in a transfer buffer without methanol. Dystrophin was detected with NCL-DYS1 monoclonal anti-dystrophin (Novocastra, Newcastle upon Tyne, UK) applied at a dilution of 1:400, and dysferlin was detected using NCL-Hamlet1 at a dilution 1:1500 for 2 hours at room temperature. Detection was performed using a Western Breeze kit as per the manufacturer's instructions (Invitrogen). Enhanced chemiluminescence reactions were detected directly by the Chemi-Smart 3000 gel documentation system (Vilber Lourmat), using Chemi-Capt software for image acquisition and Bio-1D software (Scientific Software Group) for image analysis.
Acknowledgments
This work was supported by funding from the National Institutes of Health (5R01NS04414606), the Muscular Dystrophy Association, USA (1734027), Duchenne Ireland, the James and Matthew Foundation, Gavriel Meir Trust, Muscular Dystrophy Western Australia Inc., and the Medical and Health Research Infrastructure Fund of Western Australia. The authors also acknowledge support from the Department of Neuropathology, Royal Perth Hospital (Australia) and Department of Health, Government of Western Australia. FM is supported by the Great Ormond Street Hospital Children's Charity. The MRC Centre Biobank in London is also acknowledged.
REFERENCES
- Prior TW., and, Bridgeman SJ. Experience and strategy for the molecular testing of Duchenne muscular dystrophy. J Mol Diagn. 2005;7:317–326. doi: 10.1016/S1525-1578(10)60560-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emery AE. The muscular dystrophies. Lancet. 2002;359:687–695. doi: 10.1016/S0140-6736(02)07815-7. [DOI] [PubMed] [Google Scholar]
- Nicholson LV, Johnson MA, Bushby KM., and, Gardner-Medwin D. Functional significance of dystrophin positive fibres in Duchenne muscular dystrophy. Arch Dis Child. 1993;68:632–636. doi: 10.1136/adc.68.5.632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Dunnen JT, Grootscholten PM, Bakker E, Blonden LA, Ginjaar HB, Wapenaar MC.et al. (1989Topography of the Duchenne muscular dystrophy (DMD) gene: FIGE and cDNA analysis of 194 cases reveals 115 deletions and 13 duplications Am J Hum Genet 45835–847. [PMC free article] [PubMed] [Google Scholar]
- Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H., and, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics. 1988;2:90–95. doi: 10.1016/0888-7543(88)90113-9. [DOI] [PubMed] [Google Scholar]
- Aartsma-Rus A, Van Deutekom JC, Fokkema IF, Van Ommen GJ., and, Den Dunnen JT. Entries in the Leiden Duchenne muscular dystrophy mutation database: an overview of mutation types and paradoxical cases that confirm the reading-frame rule. Muscle Nerve. 2006;34:135–144. doi: 10.1002/mus.20586. [DOI] [PubMed] [Google Scholar]
- Muntoni F, Torelli S., and, Ferlini A. Dystrophin and mutations: one gene, several proteins, multiple phenotypes. Lancet Neurol. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Ginjaar IB, Kneppers AL, v d Meulen JD, Anderson LV, Bremmer-Bout M, van Deutekom JC.et al. (2000Dystrophin nonsense mutation induces different levels of exon 29 skipping and leads to variable phenotypes within one BMD family Eur J Hum Genet 8793–796. [DOI] [PubMed] [Google Scholar]
- Malhotra SB, Hart KA, Klamut HJ, Thomas NS, Bodrug SE, Burghes AH.et al. (1988Frame-shift deletions in patients with Duchenne and Becker muscular dystrophy Science 242755–759. [DOI] [PubMed] [Google Scholar]
- Winnard AV, Klein CJ, Coovert DD, Prior T, Papp A, Snyder P.et al. (1993Characterization of translational frame exception patients in Duchenne/Becker muscular dystrophy Hum Mol Genet 2737–744. [DOI] [PubMed] [Google Scholar]
- Chelly J, Gilgenkrantz H, Lambert M, Hamard G, Chafey P, Récan D.et al. (1990Effect of dystrophin gene deletions on mRNA levels and processing in Duchenne and Becker muscular dystrophies Cell 631239–1248. [DOI] [PubMed] [Google Scholar]
- Gangopadhyay SB, Sherratt TG, Heckmatt JZ, Dubowitz V, Miller G, Shokeir M.et al. (1992Dystrophin in frameshift deletion patients with Becker muscular dystrophy Am J Hum Genet 51562–570. [PMC free article] [PubMed] [Google Scholar]
- Prior TW, Bartolo C, Papp AC, Snyder PJ, Sedra MS, Burghes AH.et al. (1997Dystrophin expression in a Duchenne muscular dystrophy patient with a frame shift deletion Neurology 48486–488. [DOI] [PubMed] [Google Scholar]
- van Deutekom JC, Janson AA, Ginjaar IB, Frankhuizen WS, Aartsma-Rus A, Bremmer-Bout M.et al. (2007Local dystrophin restoration with antisense oligonucleotide PRO051 N Engl J Med 3572677–2686. [DOI] [PubMed] [Google Scholar]
- Kinali M, Arechavala-Gomeza V, Feng L, Cirak S, Hunt D, Adkin C.et al. (2009Local restoration of dystrophin expression with the morpholino oligomer AVI-4658 in Duchenne muscular dystrophy: a single-blind, placebo-controlled, dose-escalation, proof-of-concept study Lancet Neurol 8918–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirak S, Arechavala-Gomeza V, Guglieri M, Feng L, Torelli S, Anthony K.et al. (2011Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study Lancet 378595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goemans NM, Tulinius M, van den Akker JT, Burm BE, Ekhart PF, Heuvelmans N.et al. (2011Systemic administration of PRO051 in Duchenne's muscular dystrophy N Engl J Med 3641513–1522. [DOI] [PubMed] [Google Scholar]
- McClorey G, Moulton HM, Iversen PL, Fletcher S., and, Wilton SD. Antisense oligonucleotide-induced exon skipping restores dystrophin expression in vitro in a canine model of DMD. Gene Ther. 2006;13:1373–1381. doi: 10.1038/sj.gt.3302800. [DOI] [PubMed] [Google Scholar]
- Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C., and, Fletcher S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol Ther. 2007;15:1288–1296. doi: 10.1038/sj.mt.6300095. [DOI] [PubMed] [Google Scholar]
- Cartegni L, Wang J, Zhu Z, Zhang MQ., and, Krainer AR. ESEfinder: A web resource to identify exonic splicing enhancers. Nucleic Acids Res. 2003;31:3568–3571. doi: 10.1093/nar/gkg616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PJ, Zhang C, Wang J, Chew SL, Zhang MQ., and, Krainer AR. An increased specificity score matrix for the prediction of SF2/ASF-specific exonic splicing enhancers. Hum Mol Genet. 2006;15:2490–2508. doi: 10.1093/hmg/ddl171. [DOI] [PubMed] [Google Scholar]
- Chelly J, Kaplan JC, Maire P, Gautron S., and, Kahn A. Transcription of the dystrophin gene in human muscle and non-muscle tissue. Nature. 1988;333:858–860. doi: 10.1038/333858a0. [DOI] [PubMed] [Google Scholar]
- Surono A, Takeshima Y, Wibawa T, Ikezawa M, Nonaka I., and, Matsuo M. Circular dystrophin RNAs consisting of exons that were skipped by alternative splicing. Hum Mol Genet. 1999;8:493–500. doi: 10.1093/hmg/8.3.493. [DOI] [PubMed] [Google Scholar]
- Reiss J., and, Rininsland F. An explanation for the constitutive exon 9 cassette splicing of the DMD gene. Hum Mol Genet. 1994;3:295–298. doi: 10.1093/hmg/3.2.295. [DOI] [PubMed] [Google Scholar]
- McNaughton JC, Hughes G, Jones WA, Stockwell PA, Klamut HJ., and, Petersen GB. The evolution of an intron: analysis of a long, deletion-prone intron in the human dystrophin gene. Genomics. 1997;40:294–304. doi: 10.1006/geno.1996.4543. [DOI] [PubMed] [Google Scholar]
- Harding PL, Fall AM, Honeyman K, Fletcher S., and, Wilton SD. The influence of antisense oligonucleotide length on dystrophin exon skipping. Mol Ther. 2007;15:157–166. doi: 10.1038/sj.mt.6300006. [DOI] [PubMed] [Google Scholar]
- Arechavala-Gomeza V, Graham IR, Popplewell LJ, Adams AM, Aartsma-Rus A, Kinali M.et al. (2007Comparative analysis of antisense oligonucleotide sequences for targeted skipping of exon 51 during dystrophin pre-mRNA splicing in human muscle Hum Gene Ther 18798–810. [DOI] [PubMed] [Google Scholar]
- Lattanzi L, Salvatori G, Coletta M, Sonnino C, Cusella De Angelis MG, Gioglio L.et al. (1998High efficiency myogenic conversion of human fibroblasts by adenoviral vector-mediated MyoD gene transfer. An alternative strategy for ex vivo gene therapy of primary myopathies J Clin Invest 1012119–2128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson LV. The “rescue” of dystrophin synthesis in boys with Duchenne muscular dystrophy. Neuromuscul Disord. 1993;3:525–531. doi: 10.1016/0960-8966(93)90109-w. [DOI] [PubMed] [Google Scholar]
- Adams AM, Harding PL, Iversen PL, Coleman C, Fletcher S., and, Wilton SD. Antisense oligonucleotide induced exon skipping and the dystrophin gene transcript: cocktails and chemistries. BMC Mol Biol. 2007;8:57. doi: 10.1186/1471-2199-8-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aartsma-Rus A, Bremmer-Bout M, Janson AA, den Dunnen JT, van Ommen GJ., and, van Deutekom JC. Targeted exon skipping as a potential gene correction therapy for Duchenne muscular dystrophy. Neuromuscul Disord. 2002;12 Suppl 1:S71–S77. doi: 10.1016/s0960-8966(02)00086-x. [DOI] [PubMed] [Google Scholar]
- Jearawiriyapaisarn N, Moulton HM, Buckley B, Roberts J, Sazani P, Fucharoen S.et al. (2008Sustained dystrophin expression induced by peptide-conjugated morpholino oligomers in the muscles of mdx mice Mol Ther 161624–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abes R, Arzumanov AA, Moulton HM, Abes S, Ivanova GD, Iversen PL.et al. (2007Cell-penetrating-peptide-based delivery of oligonucleotides: an overview Biochem Soc Trans 35Pt 4775–779. [DOI] [PubMed] [Google Scholar]
- Rando TA., and, Blau HM. Primary mouse myoblast purification, characterization, and transplantation for cell-mediated gene therapy. J Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton SD., and, Fletcher S. Splice modification to restore functional dystrophin synthesis in Duchenne muscular dystrophy. Curr Pharm Des. 2010;16:988–1001. doi: 10.2174/138161210790883480. [DOI] [PubMed] [Google Scholar]
- Wilton SD, Lim L, Dye D., and, Laing N. Bandstab: a PCR-based alternative to cloning PCR products. BioTechniques. 1997;22:642–645. doi: 10.2144/97224bm14. [DOI] [PubMed] [Google Scholar]
- Cooper ST, Lo HP., and, North KN. Single section Western blot: improving the molecular diagnosis of the muscular dystrophies. Neurology. 2003;61:93–97. doi: 10.1212/01.wnl.0000069460.53438.38. [DOI] [PubMed] [Google Scholar]
- Nicholson LV, Davison K, Falkous G, Harwood C, O'Donnell E, Slater CR.et al. (1989Dystrophin in skeletal muscle. I. Western blot analysis using a monoclonal antibody J Neurol Sci 94125–136. [DOI] [PubMed] [Google Scholar]