

Abstract
Stachyose synthase (STS) (EC 2.4.1.67) was purified to homogeneity from mature seeds of adzuki bean (Vigna angularis). Electrophoresis under denaturing conditions revealed a single polypeptide of 90 kD. Size-exclusion chromatography of the purified enzyme yielded two activity peaks with apparent molecular masses of 110 and 283 kD. By isoelectric focusing and chromatofocusing the protein was separated into several active forms with isoelectric point values between pH 4.7 and 5.0. Purified STS catalyzed the transfer of the galactosyl group from galactinol to raffinose and myo-inositol. Additionally, the enzyme catalyzed the galactinol-dependent synthesis of galactosylononitol from d-ononitol. The synthesis of a galactosylcyclitol by STS is a new oberservation. Mutual competitive inhibition was observed when the enzyme was incubated with both substrates (raffinose and ononitol) simultaneously. Galactosylononitol could also substitute for galactinol in the synthesis of stachyose from raffinose. Although galactosylononitol was the less-efficient donor, the Michaelis constant value for raffinose was lower in the presence of galactosylononitol (13.2 mm) compared with that obtained in the presence of galactinol (38.6 mm). Our results indicate that STS catalyzes the biosynthesis of galactosylononitol, but may also mediate a redistribution of galactosyl residues from galactosylononitol to stachyose.
RFO (e.g. raffinose, stachyose, and verbascose) are among the most widespread soluble α-galactosides that are accumulated during the development and maturation of seeds (Dey, 1985). In plants containing cyclitols, such as the methylated inositols ononitol (1d-4-O-methyl-myo-inositol) and pinitol (1d-3-O-methyl-chiro-inositol), several nonreducing galactosylcylitols occur in addition to RFO. Galactosylcyclitols are frequently found in similar or even higher amounts than RFO in seeds of many important grain legumes, such as lentil, chickpea, and soybean (Quemener and Brillouet, 1983; Horbowicz and Obendorf, 1994). Both families of soluble α-galactosides share some common functions. They are generally regarded as being reserve carbohydrates for the germinating seedling and have been proposed to participate in the acquisition of desiccation tolerance and in the viability of seeds (Horbowicz and Obendorf, 1994). The occurrence of galactosides of methylated inositols is restricted to seeds, whereas RFO are also found in storage tubers and leaves, and serve as transport carbohydrates in the phloem (Kandler and Hopf, 1980).
RFO are synthesized by a set of distinct galactosyltransferases, which sequentially add Gal units to Suc, yielding raffinose (raffinose synthase, EC 2.4.1.82), stachyose (STS, EC 2.4.1.67), and higher homologs (Kandler and Hopf, 1980, 1984). The galactosyl donor for these reactions is galactinol (O-α-d-galactopyranosyl-[1→1]-l-myo-inositol), which in turn is synthesized from myo-inositol and UDP-Gal by the enzyme galactinol synthase (EC 2.4.1.123). This enzyme was characterized from cotyledons of bean (Liu et al., 1995) and other plant sources (Handley and Pharr, 1982; Webb, 1982; Smith et al., 1991). Considerably less is known about raffinose synthase, which has only been examined as a partially purified preparation from broad bean seeds (Lehle and Tanner, 1973). STS was originally described in seeds of bean (Tanner and Kandler, 1968) but has only been purified to homogeneity from leaves of melon (Holthaus and Schmitz, 1991).
Although the structure and distribution of many galactosylcyclitols are well documented (for review, see Obendorf, 1997), very little information is available on the biochemistry of these galactosides. We have chosen ononitol (1d-4-O-methyl-myo-inositol) and galactosylononitol (O-α-d-galactopyranosyl-[1→3]-4-O-methyl-d-myo-inositol) as a model system for the characterization of galactosylcyclitol biosynthetic pathways (Richter et al., 1997). In an initial study we identified a galactosyltransferase activity in extracts of seeds of adzuki bean (Vigna angularis) that utilizes galactinol in the biosynthesis of galactosylononitol (Peterbauer and Richter, 1997). By analogy with similar galactosyltransferases, we have termed this activity GOS (Fig. 1). GOS and STS activity copurified with a constant activity ratio during several chromatographic steps, suggesting that galactosylononitol synthesis may be catalyzed by STS. The present study therefore was aimed at clarifying whether STS participates in galactosylononitol metabolism. We report on the purification and characterization of the enzyme from seeds of V. angularis, and demonstrate that STS is a multisubstrate enzyme that is able to synthesize galactosylononitol (GOS activity) in addition to stachyose (STS activity).
Figure 1.

Reaction scheme of GOS activity. The galactosyl moiety of galactinol is transfer red to d-ononitol, yielding galactosylononitol and myo-inositol. The reaction is reversible.
MATERIALS AND METHODS
Plant Material and Chemicals
Seeds from adzuki bean (Vigna angularis [Willd.] Ohwi & Ohashi) were obtained from a local market. Galactinol was purified from leaves of sage (Salvia officinalis) as previously described (Kuo, 1992). Ononitol and galactosylononitol were isolated from seeds of V. angularis as previously described (Richter et al., 1997). Further substrates (d-pinitol, sequoyitol, d- and l-bornesitol, l-quebrachitol, 1-O-methyl-scyllo-inositol, d- and l-chiro-inositol, d-1-O-methyl-muco-inositol, and muco-inositol) were isolated and purified as previously described (Wanek and Richter, 1995). All other chemicals were obtained from commercial sources and were of the highest purity available.
Enzyme Purification
Preparation of Extract
Seeds were frozen in liquid N2 and ground to a fine powder in a sample mill (IKA A10, Janke and Kunkel, Germany). Approximately 125 g of the powder was suspended in 480 mL of ice-cold extraction buffer (100 mm Hepes-NaOH, pH 7.0, 1 mm DTT, 1 mm EGTA, and 20 mm MgCl2) that contained 5 g of polyvinylpolypyrrolidone, and was further homogenized with a Polytron tissue homogenizer. The suspension was filtered through fine-mesh nylon (42 μm) and centrifuged at 26,000g at 4°C for 30 min. The supernatant was used for further protein purification.
Protamine Sulfate and Ammonium Sulfate Precipitation
All manipulations were carried out at 4°C. A 10% (w/v) protamine sulfate solution (in extraction buffer) was slowly added to the crude extract to a final concentration of 2 g L−1. Precipitated protein was removed by centrifugation (30 min at 26,000g) and the supernatant was fractionated with solid ammonium sulfate. Proteins precipitating between 35 and 55% saturation were collected by centrifugation for 20 min at 26,000g.
HIC
The pellet of the ammonium sulfate fractionation was dissolved in HIC buffer (50 mm Hepes-NaOH, pH 7.0, 1 mm DTT, and 1 m ammonium sulfate) and loaded onto a 70-mL column of Phenyl Sepharose HP (Pharmacia) preequilibrated in the same buffer at 20°C. Bound protein was eluted by applying a linear gradient of 1.0 to 0.0 m ammonium sulfate in HIC buffer (700 mL) at a constant flow rate of 5 mL min−1. Fifteen-milliliter fractions were collected and assayed for STS activity. Active fractions were pooled and concentrated by repeated ultrafiltration (Ultrafree-15, Biomax 10K, Millipore) in PAGE sample buffer (125 mm Tris-HCl, pH 6.8, containing 1 mm DTT, 10% [v/v] glycerol, and 0.0015% [w/v] bromphenol blue) at 4°C.
Preparative Native PAGE
Preparative PAGE was performed under nondenaturating conditions on a Prep Cell (model 491, Bio-Rad). A 7% acrylamide resolving gel (7 cm high) and a 3.75% acrylamide stacking gel (2 cm high) were cast in a 37-mm i.d. gel tube in 123 mm bis-Tris-HCl (pH 6.61). The upper running buffer consisted of 44 mm Tes and 113 mm bis-Tris (pH 7.25). The lower running buffer (63 mm bis-Tris-HCl, pH 5.9) was cooled by circulation through a water bath (4°C). The sample of the HIC step (3 mL) was electrophoresed at 12 W of constant power. Proteins were eluted with bis-Tris-HCl (113 mm, pH 7.0, 1 mm DTT, and 10% [v/v] glycerol) at a flow rate of 0.8 mL min−1, and 4-mL fractions were collected. Every second fraction was assayed for STS activity.
AEC
Active PAGE fractions were pooled, desalted, and concentrated at 4°C by repeated ultrafiltration in AEC sample buffer (20 mm bis-Tris-HCl, pH 7.0, 1 mm DTT and 10% [v/v] glycerol) as described above. The sample was applied at 2 mL min−1 to an anion-exchange column (6-mL bed volume, Resource Q, Pharmacia,) preequilibrated in AEC sample buffer. Bound protein was eluted with 90 mL of a linear gradient of 0.0 to 0.3 m NaCl in AEC sample buffer. Fractions (1.5 mL each) that contained STS activity were pooled, concentrated at 4°C by ultrafiltration (Centricon-10, Amicon, Beverly, MA), and stored in liquid N2.
Enzyme and Protein Assay
STS activity was routinely determined in reaction mixtures that contained 50 mm Hepes-NaOH (pH 7.0), 1 mm DTT, 10 mm galactinol, 50 mm raffinose, and 3 to 30 pkat of enzyme activity in a final volume of 60 μL. Assays were incubated at 30°C for 30 min and terminated by boiling for 5 min. After centrifugation, the supernatant was deionized by the use of ion-exchange resins (Dowex 50–100 mesh: 50WX8, H+-form; 1X8, formate form; Sigma). The formation of stachyose was monitored by HPLC with pulsed amperometric detection (DX 500, Dionex, Sunnyvale, CA) on a Carbopac PA10 column (250 × 4 mm, Dionex) with 100 mm NaOH as the eluent at a flow rate of 1 mL min−1 (30°C).
For determination of GOS activity, enzyme preparations were incubated with 20 mm d-ononitol and 10 mm galactinol. Reaction products were separated by HPLC with pulsed amperometric detection on a Carbopac MA1 column (250 × 4 mm, Dionex) with 100 mm NaOH at a flow rate of 0.4 mL min−1 at 20°C. The identity of reaction products was checked by GC-MS (see below).
Protein concentrations were estimated with BSA as a standard, using the Bradford dye-binding procedure (protein assay, Bio-Rad).
Enzyme Characterization
Kinetic Analysis and Substrate Specificity
Km and Vmax values were obtained from slope and intercept replots of initial rate data as described by Rudolph and Fromm (1979). The concentrations of substrates were varied as follows: raffinose from 5.5 to 50 mm at (a) 1.1 to 10 mm galactinol or (b) 2.2 to 20 mm galactosylononitol, and d-ononitol from 2.2 to 20 mm at 1.1 to 10 mm galactinol. Kinetic values estimated at only one concentration of the fixed substrate are referred to as Km(app) and Vmax(app) values, respectively. Inhibition constants were calculated from replots of the slopes from primary double-reciprocal plots against the inhibitor concentration.
The reaction products of substrate-specificity assays were analyzed by GC-MS (Richter, 1992). Deionized assays were taken to dryness and the reaction products were converted to trimethylsilyl derivatives by treatment with pyridine: N,O-bis-(trimethylsilyl)-trifluoroacetimide:trimethyl-chlorosilane (40:10:1; v/v) at 75°C for 60 min. The trimethylsilyl derivatives were separated on a fused silica column (DB5-ms, 30 m in length, 0.25 mm i.d., and 0.1-μm film thickness; J & W Scientific, Folsom, CA) with He as the carrier gas at 140 kPa column-head pressure. The oven temperature was programmed from 110°C (1 min) to 320°C at 8°C min−1. Mass spectral data were obtained with an ion-trap mass spectrometer (Saturn 3, Varian, Sugarland, TX) at a source temperature of 260°C in the electron-impact-ionization mode.
SDS-PAGE and Size-Exclusion Chromatography
Discontinuous SDS-PAGE was carried out under reducing conditions using 8 to 18% precast gradient gels (Excel Gel SDS, Pharmacia), according to the manufacturer′s recommendations, and calibrated with SDS molecular-mass standards in the range of 14.4 to 97.4 kD (Bio-Rad). Proteins were visualized by silver staining. The molecular mass of native STS was estimated by using size-exclusion chromatography. A 0.1-mL sample of purified enzyme was applied at 0.5 mL min−1 to a Superdex 200 HR 10/30 column (Pharmacia) pre-equilibrated in 20 mm NaPi (pH 7.0), 150 mm NaCl, and 0.5 mm DTT. Fractions of 0.25 mL were collected and assayed for STS and GOS activity. The column was calibrated with reference proteins of known molecular masses (18–300 kD, Combithek, Boehringer Mannheim).
IEF and Chromatofocusing
IEF was performed on Ampholine gels (pH 3.5–9.5, Pharmacia), calibrated with standard proteins (IEF-mix, pI 3.6–9.3, Sigma). Proteins were visualized by silver staining. For chromatofocusing, a sample of purified enzyme was diluted with 25 mm bis-Tris-HCl (pH 6.3) containing 0.5 mm DTT, concentrated by ultrafiltration (Centricon-10, Amicon), and applied at 1 mL min−1 to a Mono P HR 5/5 column (Pharmacia) pre-equilibrated in the same buffer. The column was eluted with a pH gradient (6.3–4.0) formed by 10% (v/v) Polybuffer 74 (Pharmacia) containing 0.5 mm DTT. Fractions of 0.25 mL were collected, and an equal volume of 0.5 m NaPi (pH 7.0, 1 mm DTT) was added. Fractions were desalted and concentrated by repeated ultrafiltration (Centricon-10, Amicon) in 20 mm NaPi (pH 7.0) and 1 mm DTT at 10°C, and assayed for STS and GOS activity.
N-Terminal Amino Acid Sequencing
A sample of STS was subjected to SDS-PAGE as described above and electroblotted onto a PVDF membrane (Bio-Rad) with 10 mm 3-(cyclohexylamino)-1-propane-sulfonic acid (pH 11.0) containing 10% (v/v) methanol. The membrane was stained with Coomassie blue R-250, the band at 90 kD was cut out, and the N-terminal amino acid sequence was analyzed at the Institute of Biochemistry (University of Vienna) using a sequencer (model 476A, Applied Biosystems).
RESULTS
Purification of STS
STS was purified 244-fold from mature seeds of V. angularis. The results of a typical purification procedure are summarized in Table I. An initial protamine sulfate and ammonium sulfate fractionation removed lipid contaminants and 38% of the protein from the crude extract. HIC on Phenyl Sepharose HP (Fig. 2A) provided a significant purification and substantially reduced the total amount of protein. The key step in the purification procedure was native preparative PAGE at neutral pH (Fig. 2B). By using this technique STS was resolved highly purified and with an excellent yield of activity (Fig. 2B). Remaining contaminants were removed by AEC on a Resource Q column (Fig. 2C), yielding a 244-fold purification with a recovery of 7.9% (Table I). The final preparation had a specific STS activity of 11.2 nkat mg−1 protein and was apparently homogenous by SDS-PAGE (Fig. 3A). N-terminal sequencing of the purified protein revealed a single sequence of 24 amino acids (NDPVNATLGLEPsEKVFDLLDGKL).
Table I.
Purification of STS from mature seeds of V. angularis
| Purification Step | Total Protein | Total Activity | Specific Activity | Purification | Recovery |
|---|---|---|---|---|---|
| mg | nkat | nkat mg−1 | fold | % | |
| Crude extract | 5780 | 265.9 | 0.046 | 1 | 100.0 |
| Protamine/ammonium sulfate | 2173 | 214.6 | 0.099 | 2 | 80.7 |
| Phenyl Sepharose HP | 51.1 | 89.7 | 1.76 | 38 | 33.7 |
| Native PAGE | 5.23 | 54.1 | 10.3 | 225 | 20.4 |
| Resource Q | 1.87 | 21.0 | 11.2 | 244 | 7.9 |
Enzyme activity was assayed with 10 mm galactinol and 50 mm raffinose.
Figure 2.

Purification of STS from seeds of V. angularis. A, HIC of proteins on Phenyl Sepharose HP after protamine sulfate treatment and ammonium sulfate fractionation (dashed line, 1.0–0.0 m ammonium sulfate). B, Native preparative PAGE of pooled fractions from the HIC on a Bio-Rad PrepCell 491. C, Final purification by AEC on Resource Q (dashed line, 0.0–0.3 m NaCl).
Figure 3.

Analysis of STS at various stages of purification. A, SDS-PAGE. Lane 1, Crude protein extract; lane 2, 35 to 55% ammonium sulfate precipitate after protamine sulfate treatment; lane 3, pooled fractions after chromatography on Phenyl Sepharose HP; lane 4, molecular mass markers; lane 5, pooled fractions after native PAGE; and lane 6, pooled fractions after chromatography on Resource Q. B, IEF of purified STS. Lanes were loaded with 0.1 to 1.0 μg of protein and stained with silver nitrate.
Physicochemical Properties and Effectors
Purified STS was subjected to size-exclusion chromatography on a Superdex 200 HR column for determination of the native molecular mass. A minor and a major peak of 283 and 110 kD, respectively, were observed (Fig. 4A). The 110-kD protein was isolated and reinjected onto the size-exclusion column. Again a 283-kD peak was observed in addition to the 110-kD peak, indicating that the enzyme forms aggregates under the conditions used. On SDS-PAGE gels both the 110- and 283-kD forms exhibited a single band corresponding to a molecular mass of 90.1 kD (Fig. 3A).
Figure 4.

Size-exclusion chromatography (A) and chromatofocusing (B) of purified STS. STS (•) and GOS (○) activities were determined by HPLC as described in Methods. A, Elution profile from a Superdex 200 HR 10/30 column. Inset, Native molecular mass estimation. The apparent molecular masses of STS (110 and 283 kD) are indicated by arrows. B, Elution profile from a Mono P HR 5/5 column. The pH gradient (□) was formed with Polybuffer 74.
Purified STS was subjected to IEF under native conditions, yielding several bands in the range of pI 4.7 to 5.0 (Fig. 3B). By chromatofocusing on a Mono P column, several unresolved active peaks were observed in a similar pH range (Fig. 4B), possibly due to aggregate formation. Since the enzyme was unstable at low pH values, no attempts were made to get further insight into the observed microheterogeneity.
STS exhibited a temperature optimum at around 35°C and a one-half-maximum activity at approximately 23°C (Fig. 5A). The pH-dependent activity curve showed a maximum between pH 6.5 and 7.0 in McIlvaine buffer (NaPi-citrate) (Fig. 5B). STS activity did not require cations, but was strongly inhibited by Mn2+, Zn2+, Cu2+, and Fe2+ (Table II). The presence of DTT slightly increased the activity, although 20 μm PCMBS had only a minimal effect on the enzyme (Table II). The purified enzyme was stable for at least 4 months when stored in liquid N2. When stored at −20°C, 85% of activity was lost within 4 months.
Figure 5.

Effect of temperature (A) and pH (B) on STS (•) and GOS (○) activity. A, Temperature-dependent activity profiles assayed in 50 mm Hepes-NaOH (pH 7.0) and 1 mm DTT. Data were adjusted relative to the maximum activity measured. B, pH-dependent activity profiles assayed in McIlvaine buffer and 1 mm DTT at 30°C. Data were adjusted relative to the maximum activity measured.
Table II.
Influence of cations and other agents on STS activity from V. angularis
| Compound | Concentration | Relative Rates |
|---|---|---|
| mm | % | |
| None | – | 100.0 |
| DTT | 2 | 122.2 |
| Na2EDTA | 5 | 110.7 |
| MgCl2 | 5 | 111.0 |
| KCl | 5 | 105.0 |
| NaCl | 5 | 103.6 |
| Fe(II)SO4 | 5 | 16.5 |
| MnCl2 | 5 | 0.5 |
| ZnCl2 | 5 | 0.2 |
| CuSO4 | 5 | 0.2 |
| PCMBSa | 0.02 | 96.5 |
The enzyme activity was assayed with 10 mm galactinol, 50 mm raffinose, and the indicated additions. Activity of the control (no addition) was 25 pkat per assay.
PCMBS, p-Chloromercuribenzenesulfonic acid.
Reactions and Kinetics
The purified enzyme catalyzed several galactosyl transfer reactions (Table III). For the genuine STS activity,
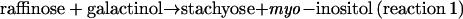 |
Km values of 15.8 and 38.6 mm were estimated at nonsaturating substrate levels for galactinol and raffinose, respectively, with a Vmax of 77.5 nkat mg−1 protein (Table IV). Apparent affinities (Km[app] values) for both substrates were markedly affected by the concentration of the respective fixed cosubstrate. In the presence of the lowest concentration of galactinol used (1.1 mm), a Km(app) value of 2.4 mm was found for raffinose, whereas a Km(app) of 1.8 mm was found for galactinol in the presence of 5.5 mm raffinose. myo-Inositol acted as a competitive inhibitor relative to raffinose with a Ki of 4.6 mm (data not shown). As already described for STS from other plant sources (Tanner and Kandler, 1968; Gaudreault and Webb, 1981), the enzyme from V. angularis also catalyzed the following exchange reaction:
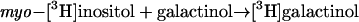 |
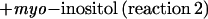 |
When assayed with 10 mm galactinol, a Km(app) of 5.2 mm was estimated for myo-[3H]inositol. However, the enzyme was not specific for galactinol and myo-inositol, but also catalyzed the following reaction, in a manner similar to that of reaction 1:
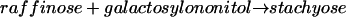 |
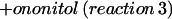 |
Thus, the enzyme accepted galactosylononitol instead of galactinol as a galactosyl donor, indicating an alternative biosynthetic pathway for stachyose. A fairly high Km value was obtained for galactosylononitol (31.3 mm) with a Vmax of 46.2 nkat mg−1, whereas the Km value for raffinose (13.2 mm) was considerably lower in the presence of galactosylononitol instead of galactinol (Table IV). In addition, the purified enzyme was capable of catalyzing the synthesis of galactosylononitol from galactinol and ononitol (GOS activity, Fig. 1):
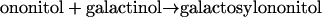 |
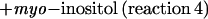 |
This reaction closely resembles (exchange) reaction 2, with ononitol substituting for myo-[3H]inositol. For the GOS reaction, Km values were estimated to be 6.3 and 18.1 mm for galactinol and ononitol, respectively (Table IV). The reaction was readily reversible. A rate of 21.6 nkat mg−1 was observed in the presence of 10 mm galactosylononitol and 20 mm myo-inositol.
Table III.
Substrate specificity of STS
| Galactosyl Acceptor | Concentration | Activity |
|---|---|---|
| mm | % | |
| Raffinose | 50 | 100.0a |
| Raffinose (assayed with 10 mm galactosylononitol) | 50 | 37.8 |
| Stachyose | 50 | n.d.b |
| Suc | 50 | n.d. |
| myo-Inositolc | 20 | 224.8 |
| 1d-4-O-Methyl-myo-inositol (d-ononitol) | 20 | 103.5 |
| 5-O-Methyl-myo-inositol (sequoyitol) | 20 | 126.9 |
| 1d-1-O-Methyl-myo-inositol (d-bornesitol) | 20 | n.d. |
| 1l-1-O-Methyl-myo-inositol (l-bornesitol) | 20 | n.d. |
| scyllo-Inositol | 20 | 16.9 |
| 1-O-Methyl-scyllo-inositol | 20 | 4.0 |
| d-chiro-Inositol | 20 | n.d. |
| 1d-3-O-Methyl-chiro-inositol (d-pinitol)d | 20 | 0.9 |
| l-chiro-Inositol | 20 | n.d. |
| 1l-2-O-Methyl-chiro-inositol (l-quebrachitol) | 20 | n.d. |
| muco-Inositol | 20 | n.d. |
| 1d-1-O-Methyl-muco-inositol | 20 | n.d. |
| epi-Inositol | 20 | 23.2 |
Reactions were assayed with 10 mm galactinol unless otherwise indicated.
Corresponds to an activity of 56 pkat per assay.
n.d., Not detected.
Assayed with 20 mm myo-[3H]inositol (10 MBq mmol−1). Reaction products were separated by HPLC on a Carbopac MA1 and monitored with a radiodetector.
The product of this reaction was identified as galactopinitol A.
Table IV.
Kinetic parameters of STS in galactosyl transfer reactions
| Substrates (Cosubstratesa) | Km | Vmax/Km |
|---|---|---|
| mm | ||
| Donors | ||
| Galactinol (raffinose) | 15.8 | 4.9 |
| Galactinol (ononitol) | 6.3 | 10.5 |
| Galactosylononitol (raffinose) | 31.3 | 1.5 |
| Acceptors | ||
| Raffinose (galactinol) | 38.6 | 2.0 |
| Raffinose (galactosylononitol) | 13.2 | 3.5 |
| Ononitol (galactinol) | 18.1 | 3.7 |
| myo-Inositol (galactinol) | 5.2b | 5.9 |
The respective cosubstrate is shown in parentheses.
Value corresponds to an apparent Km(app) determined at 10 mm galactinol.
STS and GOS activities exhibited identical temperatures and pH optima (Fig. 5, A and B) and were catalyzed by all forms of the enzyme separated by size-exclusion chromatography (Fig. 4A) and chromatofocusing (Fig. 4B), although STS activity appeared to be less stable than GOS activity at low pH values. Simultaneous incubation of the enzyme with galactinol and both acceptors (ononitol and raffinose) resulted in mutual inhibition (Fig. 6). Ononitol acted as a competitive inhibitor toward raffinose, whereas raffinose acted as a competitive inhibitor with respect to ononitol. Apparent Ki values of 18.7 and 17.9 mm for ononitol and raffinose, respectively, were estimated from replots of slopes (Fig. 6).
Figure 6.

Mutual inhibition of STS and GOS activity. The reaction mixtures contained 10 mm galactinol, 0 to 50 mm raffinose, and 0 to 20 mm ononitol. Assays were analyzed for both stachyose and galactosylononitol. Data were plotted as follows: A, Formation of stachyose at different concentrations of ononitol with raffinose as the variable substrate. Ononitol concentrations were: 1, 0 mm; 2, 2.9 mm; 3, 4 mm; 4, 6.7 mm; and 5, 20 mm. B, Formation of galactosylononitol at different concentrations of raffinose with ononitol as the variable substrate. The raffinose concentrations were: 1, 0 mm; 2, 7.1 mm; 3, 10 mm; 4, 16.7 mm; and 5, 50 mm.
Several isomeric inositols and inositol O-methyl-ethers were tested as possible galactosyl acceptors (Table III). Under the conditions used, myo-inositol was the most effective acceptor (224% compared with raffinose). Methylation or epimerization of the hydroxyl groups at carbons C-2, C-4, C-5, and C-6 of the myo-inositol ring yielded the derivatives d-ononitol, sequoyitol, scyllo-inositol, O-methyl-scyllo-inositol, and epi-inositol, respectively, which were utilized at rates between 4 and 127%. Those derivatives of myo-inositol, that are modified at C-1 or C-3 (d- and l-bornesitol, d- and l-chiro-inositol, l-quebrachitol, muco-inositol, and d-1-O-methyl-muco-inositol) were found to be inactive in the system used. d-Pinitol, a naturally occurring cyclitol in many legumes, was only accepted at a very low rate. All attempts to demonstrate a galactosyl transfer from galactinol to Suc or to stachyose were not successful (Table III).
DISCUSSION
Although STS has previously been characterized in enzyme preparations from several different plant species and has been purified to homogeneity from leaves of melon (Holthaus and Schmitz, 1991), this is the first report to our knowledge on the purification of the enzyme to homogeneity from a seed source. STS from V. angularis exhibited a broad pH optimum between pH 6.5 and 7.0 and a temperature optimum at around 35°C, similar to that of the partially purified enzyme from seeds of bean (Tanner and Kandler, 1968) and that of leaves of squash (Gaudreault and Webb, 1981), melon (Holthaus and Schmitz, 1991), and common bugle (Bachmann et al., 1994). However, the enzyme from seeds of V. angularis differed markedly from that of melon leaves with respect to molecular mass. The leaf enzyme was reported to consist of two subunits of 45 and 50 kD (Holthaus and Schmitz, 1991). In contrast, SDS-PAGE of the enzyme from V. angularis seeds revealed a single polypeptide of 90.1 kD (Fig. 3A). No native molecular mass data and amino acid sequence information are available for the purpose of a comparison.
The enzyme from V. angularis catalyzed an exchange reaction between galactinol and (labeled) myo-inositol at a rate comparable to that reported for preparations from bean and melon (Tanner and Kandler, 1968; Gaudreault and Webb, 1981). For this galactosyl exchange, a reaction mechanism has been proposed in which galactinol reacts with the enzyme to form a Gal-enzyme complex and myo-inositol that dissociates, making way for (labeled) myo-inositol (Tanner and Kandler, 1968; Dey, 1985). Several other cyclitols could substitute for myo-inositol in the exchange reaction catalyzed by the purified V. angularis enzyme, resulting in a net synthesis of galactosylcyclitols (Table III). The reversible synthesis of galactosylononitol from galactinol and d-ononitol (Fig. 1) deserves special attention, since ononitol is the only naturally occurring O-methyl-inositol in V. angularis and its galactosyl derivative is accumulated in seeds of many legumes (Yasui et al., 1985, 1987; Obendorf, 1997). To our knowledge, this is the only known route for galactosylononitol biosynthesis. The catalytic efficiency for galactosylation of ononitol (calculated as Vmax/Km) was almost 1.9-fold higher compared with that of raffinose (both assayed with galactinol), providing evidence that this pathway is active in vivo (Table IV). It is interesting that galactopinitols, although widespread in legume seeds, were not synthesized by STS from V. angularis to a significant extent.
The involvement of galactinol as a cofactor in the biosynthesis of RFO is firmly established by in vitro and in vivo studies (Senser and Kandler, 1967; Kandler and Hopf, 1980). In addition, purified STS from V. angularis also accepted galactosylononitol as a galactosyl donor in the formation of stachyose, but with a lower catalytic efficiency (Table IV). Although already proposed by others (Beveridge et al., 1977; Dey, 1985), our results provide the first evidence to our knowledge that a galactoside of an inositol O-methyl-ether is involved in the RFO metabolism of a legume seed. We were able to demonstrate that ononitol is a product (arising from galactosylononitol-dependent synthesis of stachyose) and a substrate for the enzyme (GOS activity). Most likely, both reactions are catalyzed at one active site, in agreement with the observed competitive inhibition by myo-inositol and ononitol versus raffinose (Fig. 6), as well as with the proposed mechanism of the above-described exchange reaction.
The enzyme from V. angularis seeds displayed low affinity toward raffinose when assayed with galactinol (Km of 38.6 mm), compared with melon STS (Km between 3.7 and 15 mm) (Huber et al., 1990; Holthaus and Schmitz, 1991). However, the Km value for raffinose was markedly lower (13.2 mm) for STS of V. angularis when assayed with galactosylononitol instead of galactinol (Table IV). It is interesting that the concentration of galactosylononitol in developing V. angularis seeds was consistently higher than that of galactinol and raffinose throughout stachyose accumulation (T. Peterbauer, M. Puschenreiter, and A. Richter, unpublished results), suggesting that galactosyl transfer from galactosylononitol to raffinose may significantly contribute to the formation of stachyose in vivo. Nevertheless, since galactinol is the only known galactosyl donor in the formation of galactosylononitol, the biosynthesis of stachyose via galactosylononitol ultimately seems to depend also on galactinol.
Galactosylcyclitols accumulate alongside RFO during the acquisition of dessication tolerance in legume seeds (Obendorf, 1997). However, the physiological significance of galactosylononitol in seeds is not fully understood at present. We have demonstrated here that the biosynthesis and metabolism of galactosylononitol and stachyose are linked via the enzyme STS in V. angularis seeds. It may therefore be possible that more enzymes are shared by the metabolic pathways of galactosylcyclitols and RFO. However, inhibitory effects of cyclitols on enzymes of RFO synthesis cannot be excluded. A reinvestigation of the RFO metabolic enzymes with respect to cyclitol and galactosylcyclitol specificity is clearly needed.
ACKNOWLEDGMENTS
We wish to thank Prof. M. Popp and Dr. W. Wanek for valuable comments on the manuscript and Dr. R. Prohaska for protein sequencing.
Abbreviations:
- AEC
anion-exchange chromatography
- GOS
galactosylononitol synthase
- HIC
hydrophobic-interaction chromatography
- RFO
raffinose family oligosaccharide(s)
- STS
stachyose synthase
Footnotes
This work was supported by the Austrian Science Foundation (project no. P10917-BIO).
LITERATURE CITED
- Bachmann M, Matile P, Keller F. Metabolism of the raffinose family oligosaccharides in leaves of Ajuga reptans L. Cold acclimation, translocation, and sink to source transition: discovery of a chain elongation enzyme. Plant Physiol. 1994;105:1335–1345. doi: 10.1104/pp.105.4.1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beveridge RJ, Ford CW, Richards GN. Polysaccharides of tropical pasture herbage. VII. Identification of a new pinitol galactoside from seeds of Trifolium subterraneum (subterranean clover) and analysis of several pasture legume seeds for cyclohexitols and their galactosides. Aust J Chem. 1977;30:1583–1590. [Google Scholar]
- Dey PM (1985) d-Galactose-containing oligosaccharides. In PM Dey, RA Dixon, eds, Biochemistry of Storage Carbohydrates in Green Plants. Academic Press, New York, pp 53–129
- Gaudreault P-R, Webb JA. Phytochemistry. 1981;20:2629–2633. [Google Scholar]
- Handley LW, Pharr DM. Ion stimulation, UDP inhibition and effects of sulfhydryl reagents on the activity of galactinol synthase from leaves of cucumber, Cucumis sativus L. Z Pflanzenphysiol. 1982;108:447–455. [Google Scholar]
- Holthaus U, Schmitz K. Stachyose synthesis in mature leaves of Cucumis melo: purification and characterization of stachyose synthase (EC 2.4.1.67) Planta. 1991;184:525–531. doi: 10.1007/BF00197902. [DOI] [PubMed] [Google Scholar]
- Horbowicz M, Obendorf RL. Seed desiccation tolerance and storability: dependence on flatulence-producing oligosaccharides and cyclitols—review and survey. Seed Sci Res. 1994;4:385–405. [Google Scholar]
- Huber JLA, Pharr DM, Huber SC. Partial purification of stachyose synthase in leaves of Cucumis sativus and Cucumis melo: utilization of a rapid assay for myo-inositol. Plant Sci. 1990;69:179–188. [Google Scholar]
- Kandler O, Hopf H (1980) Occurrence, metabolism, and function of oligosaccharides. In J Preiss, ed, The Biochemistry of Plants, Vol. 3. Academic Press, New York, pp 221–270
- Kandler O, Hopf H. Biosynthesis of oligosaccharides in vascular plants. In: Lewis DH, editor. Storage Carbohydrates in Vascular Plants. Cambridge, UK: Cambridge University Press; 1984. pp. 115–131. [Google Scholar]
- Kuo TM. Isolation and identification of galactinol from castor oilseed meal. JAOCS. 1992;69:569–574. [Google Scholar]
- Lehle L, Tanner W. The function of myo-inositol in the biosynthesis of raffinose: purification and characterization of galactinol:sucrose-6-galactosyltransferase from Vicia faba seeds. Eur J Biochem. 1973;38:103–110. doi: 10.1111/j.1432-1033.1973.tb03039.x. [DOI] [PubMed] [Google Scholar]
- Liu JJ, Odegard W, de Lumen BO. Galactinol synthase from kidney bean cotyledon and zucchini leaf. Plant Physiol. 1995;109:505–511. doi: 10.1104/pp.109.2.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obendorf RL. Oligosaccharides and galactosyl cyclitols in seed desiccation tolerance. Seed Sci Res. 1997;7:63–74. [Google Scholar]
- Peterbauer T, Richter A. Stachyose synthase, a multifunctional enzyme in seeds of Vigna angularis (abstract no. P3.56) J Exp Bot. 1997;48:S-31. [Google Scholar]
- Quemener B, Brillouet J-M. Ciceritol, a pinitol digalactoside from seeds of chickpea, lentil and white lupin. Phytochemistry. 1983;22:1745–1751. [Google Scholar]
- Richter A. Phytochemistry. 1992;31:3925–3927. [Google Scholar]
- Richter A, Peterbauer T, Brereton I. Structure of galactosylononitol. J Nat Prod. 1997;60:749–751. [Google Scholar]
- Rudolph FB, Fromm HJ (1979) Plotting methods for analyzing enzyme rate data. In DL Purich, ed, Methods in Enzymology, Vol 63. Academic Press, New York, pp 138–158 [DOI] [PubMed]
- Senser M, Kandler O. Galactinol, ein Galactosyldonor für die Biosynthese der Zucker der Raffinosefamilie in Blättern. Z Pflanzenphysiol. 1967;57:376–388. [Google Scholar]
- Smith PT, Kuo TM, Crawford CG. Purification and characterization of galactinol synthase from mature zucchini squash leaves. Plant Physiol. 1991;96:693–698. doi: 10.1104/pp.96.3.693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanner W, Kandler O. myo-Inositol, a cofactor in the biosynthesis of stachyose. Eur J Biochem. 1968;4:233–239. doi: 10.1111/j.1432-1033.1968.tb00199.x. [DOI] [PubMed] [Google Scholar]
- Wanek W, Richter A. Purification and characterization of myo-inositol 6-O-methyltransferase from Vigna umbellata Ohwi et Ohashi. Planta. 1995;197:427–434. [Google Scholar]
- Webb JA. Partial purification of galactinol synthase from leaves of Cucurbita pepo. Can J Bot. 1982;60:1054–1059. [Google Scholar]
- Yasui T, Endo Y, Ohashi H. Infragenic variation of the low molecular weight carbohydrate composition of the seeds of the genus Vicia (Leguminosae) Bot Mag Tokyo. 1987;100:255–272. [Google Scholar]
- Yasui T, Tateishi Y, Ohashi H. Distribution of low molecular weight carbohydrates in the subgenus Ceratotropis of the genus Vigna (Leguminosae) Bot Mag Tokyo. 1985;98:75–87. [Google Scholar]