

Abstract
Left ventricular (LV) hypertrophy is a strong independent predictor of increased cardiovascular morbidity and mortality in clinical and population-based samples. Clinical and hemodynamic stimuli to LV hypertrophy induce not only an increase in cardiac mass and wall thickness but also a fundamental reconfiguration of the protein, cellular and molecular components of the myocardium. Several studies have indicated that LV mass is influenced by genetic factors. The substantial heritability (h2) for LV mass in population-based samples of varying ethnicity indicates robust genetic influences on LV hypertrophy. Genome-wide linkage and association studies in diverse populations have been performed to identify genes influencing LV mass, and although several chromosomal regions have been found to be significantly associated with LV mass, the specific genes and functional variants contained in these chromosomal regions have yet to be identified. In addition, multiple studies have tried to link single-nucleotide polymorphisms (SNPs) in regulatory and pathway genes with common forms of LV hypertrophy, but there is little evidence that these genetic variations are functional. Up to this point in time, the results obtained in genetic studies are of limited clinical value. Much of the heritability remains unexplained, the identity of the underlying gene pathways, genes, and functional variants remains unknown, and the promise of genetically-based risk prediction and personalized medicine remain unfulfilled. However, molecular biological technologies continue to improve rapidly, and the long-term potential of sophisticated genetic investigations using these modern genomic technologies, coupled with smart study designs, remains intact. Ultimately, genetic investigations offer much promise for future prevention, early intervention and treatment of this major public health issue.
Keywords: Left ventricular (LV) hypertrophy, genetic epidemiology, Genome-wide linkage and association studies
Introduction
Heart disease is a major cause of morbidity and mortality worldwide and is of major public health importance. Heart disease affects an estimated 82.6 million Americans and accounts for 1 in every 3 deaths in the United States [1]. Of these, 76.4 million Americans have hypertension and 5.7 million Americans have heart failure. Hypertension had an overall death rate of 17.8 in 2007 and cost an estimated 43.5 billion in direct and indirect costs. Heart failure had an overall death rate of 84.6 in 2008 and cost an estimated $29.6 billion in direct and indirect costs in 2006.
Heart failure is a condition arising when myocardial performance is insufficient to meet the metabolic demand of vital tissues and organs. This complex pathophysiological state represents the final common pathway of a variety of cardiovascular conditions that include hypertension, coronary heart disease, valvular heart disease, cardiomyopathy and congenital heart disease [2]. In response to these cardiovascular insults, the left ventricle (LV) compensates by either hypertrophy or dilatation to preserve and maintain cardiac pump performance. Conditions that cause pressure overload, such as hypertension and aortic stenosis, induce concentric LV hypertrophy (Figure 1) while conditions that promote volume overload, such as valvular regurgitation, lead to LV dilatation and eccentric LV hypertrophy (Figure 2).
Figure 1.
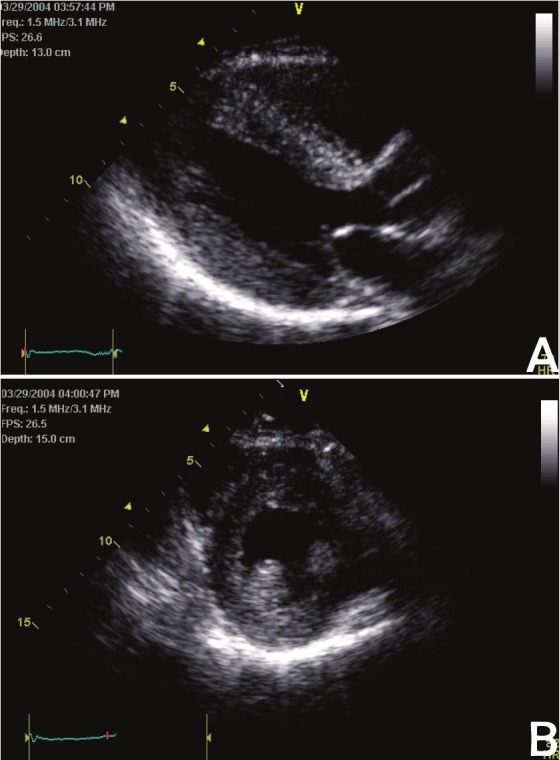
Concentric left ventricular hypertrophy. Parasternal long- (A) and short-axis (B) echocardiographic images.
Figure 2.
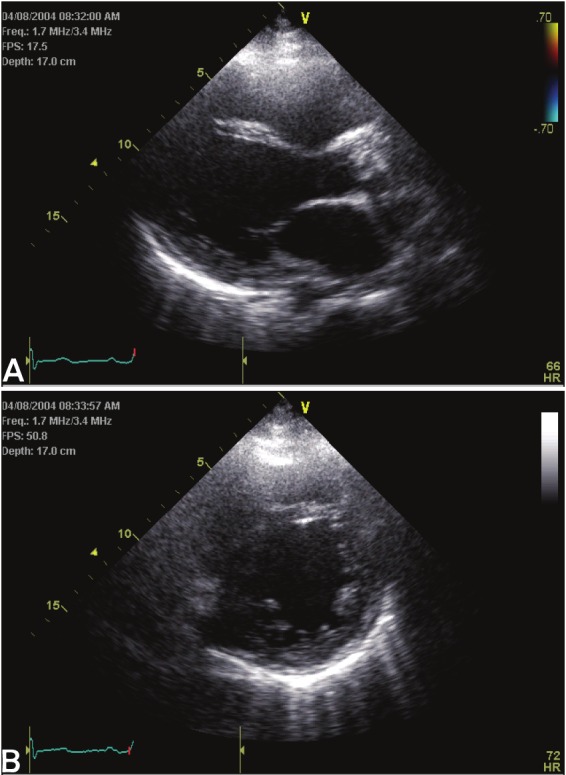
Eccentric left ventricular hypertrophy. Parasternal long- (A) and short-axis (B) echocardiographic images.
LV hypertrophy and prognosis
While compensatory LV hypertrophy or dilatation may initially preserve cardiac pump function, they ultimately become maladaptive. Indeed, LV hypertrophy has been shown to be an independent predictor of increased cardiovascular morbidity and mortality in clinical and population-based samples [3-15]. As may be seen in Table 1, individuals with LV hypertrophy consistently have ³ 2-fold rates of adverse events, as indicated by the odds-ratios.
Table 1.
Prognostic significance of left ventricular hypertrophy by various methods.
| Reference | Method of Diagnoisis | Number of Patients Analysed | End-Point | Odds Ratio |
|---|---|---|---|---|
| Sokolow et al. (3) | ECG | 439 | Death | 8.0 |
| Breslin et al. (4) | ECG | 631 | Death | 4.8 |
| Kannel et al. (5) | ECG | 5,055 | CV Events | 2.5 |
| Casale et al. (6) | Echo | 140 | Death or CV Events | 4.0 |
| Silverberg et al. (7) | Echo | 119 | Death | 3.7 |
| Aronow et al. (8) | Echo | 554 | Ventricular Fibrillation or Sudden Death | 4.7 |
| Levy et al. (9) | Echo | 3,220 | All-cause mortality and All CV Events | 2.4 and 2.5 |
| Koren et al. (10) | Echo | 280 | CV Death and All CV Events | 14.2 and 3.0 |
| Ghali et al. (11) | Echo | 785 | All-cause and Cardiac Mortality | 2.1 |
| Bikkina et al. (12) | Echo | 447 | Stroke, TIA | 2.7 |
| Gardin et al. (13) | Echo | 5,888 | Incident CHD, CHF and Stroke | 3.4 |
| Quinones et al. (15) | Echo | 1,172 | Death | 1.4 |
In the 1960’s, the association of electrocardiographic LV hypertrophy with cardiovascular events was first described in clinical and population-based studies [3-5]. In the first study to relate direct measurements of echocardiographic LV mass to prognosis, 140 men with uncomplicated essential hypertension were followed for five years to determine the incidence of “hard” cardiovascular events, i.e. cardiac death, myocardial infarction, stroke or angina pectoris requiring coronary bypass surgery [6]. The 20% (n=29) of patients with LV mass exceeding a predefined partition value had approximately 4-fold higher rate of morbid events (24%) than the patients without LV hypertrophy (6%).
Other studies have subsequently extended these findings by demonstrating that increased LV mass strongly predicts cardiac and cerebrovascular morbidity and mortality, independent of traditional risk factors [7-15]. A report from the Framingham Heart Study showed that increased LV mass strongly predicted all-cause and cardiac mortality and coronary heart disease events in adults over 40 years, independently of conventional risk factors [9]. Furthermore, they found that age-adjusted incidence of stroke or transient ischemic attack was substantially higher in the highest quartile of LV mass (18.4% in men and 12.2% in women) than in the lowest quartile (5.2% and 2.2%, respectively) [12]. Similarly, among older adults (age>65 years) in the Cardiovascular Heath Study, the highest quartile of LV mass conferred a hazards ratio of 3.36 for incident congestive failure compared with the lowest quartile [13]. In adults with coronary artery disease, Liao et al found that the attributable risk of death from LV hypertrophy was greater (2.4%) than that of multivessel coronary artery disease (1.6%) or low LV ejection fraction at catheterization (2.0%) [14]. Quinones et al. [15] extended this result to the patients enrolled in the Studies of Left Ventricular Dysfunction (SOLVD) Trials, finding higher LV mass to be independently associated with higher age-adjusted risks of death (risk-ratio= 2.75) and cardiovascular hospitalization (risk-ratio=1.81).
Stimuli to LV hypertrophy
The LV normally grows continuously from infancy to adulthood, with cardiomyocyte enlargement or hypertrophy accounting for most of the increase in size [15]. In apparently normal children and adults, LV mass is closely correlated with body size [16,17]. Traditionally, body size has been taken to account by adjusting LV mass for body surface area. However, this method of indexing LV mass may misclassify obesity-induced LV hypertrophy as normal. Height-based indexations of LV mass, which identify both blood pressure and obesity-associated increases in myocardial mass, has been shown to maintain and perhaps enhance prediction of cardiac risk [18].
After puberty, men have higher LV mass than women in relation to body size. LV mass/body surface area is 10-20% greater in men than in women, which parallels the sex-difference in fat-free mass and may reflect genetic, hormonal or exercise effects that influence both skeletal and cardiac muscle [15,19]. Thus, sex and age need to be taken into account when establishing upper normal limits for LV mass.
In addition to demographic factors, hemodynamic variables play an important role in determining LV mass. Our understanding of the full impact of blood pressure on the heart has been enhanced by the use of 24-hour ambulatory blood pressure monitoring. LV mass or wall thicknesses are more closely related to 24-hour than casual blood pressures, most likely because the latter is a poor assessment of the average blood pressure over an extended period of time, as a blood pressure assessment at a single point in time is greatly influenced by many temporary factors that may not have any long-term duration and effects [20]. In a study of normotensive and hypertensive adults, patients with concentric LV hypertrophy had the highest ambulatory systolic and diastolic blood pressures, while those with eccentric LV hypertrophy had lower ambulatory than clinic blood pressures [20]. Exaggerated blood pressure increase during exercise may also contribute to the development of LV hypertrophy [21,22].
Numerous studies have shown that obesity is associated with increased LV mass [17,23]. High salt diets have also been linked to hypertensive LV hypertrophy [24]. Both these factors may increase stroke volume, thereby increasing chamber volume predisposing to eccentric LV hypertrophy [25]. The important role of volume load in the pathogenesis of LV hypertrophy is underscored by the fact that chamber size and stroke volume are more closely related to left ventricular mass than systolic blood pressure in normotensive and hypertensive adults and in population-based samples [25-29]. In addition to hemodynamic pressure and volume load, LV mass is also affected by a negative relation between LV contractility and myocardial mass [27,29]. In a recent population-based report, almost half of the variability in LV mass was associated with inter-individual differences in stroke volume, contractility, systolic blood pressure, body mass indices and gender [29].
Another stimulus to LV hypertrophy is abnormal glucose metabolism. Several epidemiologic studies have shown that adults with diabetes mellitus have higher LV mass, independently of other stimuli to LV hypertrophy [30-32]. This relationship may be important in view of the increasing prevalence of diabetes mellitus in the United States [33].
These stimuli to LV hypertrophy induce not only an increase in cardiac mass and wall thickness but also a fundamental reconfiguration of the protein, cellular and molecular components of the myocardium.
Genetic epidemiology of LV hypertrophy
Heritability analyses
Only one-half to two-thirds of the inter-individual variability of LV mass can be explained by its clinical and hemodynamic correlates [16-32]. Several studies have indicated that LV mass is influenced by genetic factors. Monozygotic twins have substantially more similar LV mass than dizygotic twins [34-36]. Adams et al. [34] evaluated within-pair differences in LV wall thicknesses and LV dimensions in 31 monozygotic twin pairs, 10 dizygotic twin pairs, 6 siblings and 30 unrelated individuals. They found that there were lower within twin-pair differences for LV chamber size and posterior wall thickness, but not for septal wall thickness, suggesting that familial influences, whether genetic and/or shared environmental factors, are important determinants of cardiac size. In contrast, Bielen et al. [36] found significant heritability for LV wall thickness (0.29 and 0.28 for septal wall thickness and posterior wall thickness, respectively), but not for LV chamber size in 32 monozygotic and 21 dizygotic twin pairs, after adjusting for age, weight, blood pressure (BP) and skinfold thicknesses. In 22 African-American normotensive twin pairs, LV mass/body surface area, adjusted for sex and systolic BP, had an estimated heritability of 0.58 [35].
Family-based studies have confirmed that electrocardiographic and echocardiographic measures of LV hypertrophy, after adjustment for covariates, are heritable (Table 2) [37-43]. Mayosi et al. found the heritability of Sokolow-Lyon voltage criteria for LV hypertrophy was 0.39-0.41 while those for electrocardiographic LV mass, Cornell voltage and Cornell product were were 0.12-0.18, 0.19-0.25 and 0.28-0.32, respectively [38]. The Framingham Heart Study, using intraclass correlation methods, assessed the heritability of echocardiographic LV mass in their adult Caucasian population [38]. The estimated heritability of adjusted LV mass was between 0.24 (from aunt/uncle-niece/nephew correlation) and 0.32 (sibling-sibling correlation), with an intermediate estimate of 0.30 from parent-child correlation. A report from the Hypertension Genetic Epidemiology Network (HyperGEN) Study, in a population-based sample of Caucasian and African-American hypertensives, indicated that sibling correlations for echocardiographic LV mass among African-Americans ranged from 0.22 (brother-sister) to 0.44 (brother-brother) compared to 0.05 (brother-sister) to 0.22 (sister-sister) among Caucasians while sibling correlations for relative wall thickness, a measure of LV concentricity, were lower in African-American siblings (0.04-0.12) than their Caucasian counterparts (0.19-0.28) [39], suggesting ethnic heterogeneity among genes and/or environmental conditions influencing LV geometry. Among adult American Indians participating in the Strong Heart Study, the heritability of echocardiographic LV mass and relative wall thickness were both 0.17 after adjusting for a comprehensive set of covariates that included age, sex, body size, blood pressure, heart rate, diabetes and medications [40]. The Northern Manhattan Study reported somewhat higher estimates for echocardiographic LV mass and relative wall thickness (0.49 and 0.26, respectively) after adjusting for similar covariates in Caribbean Hispanic families [41]. Among Chinese families in Taiwan, echocardiographic LV mass heritability was estimated as 0.15 after adjusting for blood pressure [42]. In contrast, the heritability of echocardiographic LV mass was 0.61 in Japanese-American families participating in the Stanford Asian and Pacific Program for Hypertension and Insulin Resistance (SAPPHIRe) Study after adjusting for age, sex, height, body mass index, treated diabetes, valvular heart disease, clinic blood pressure and hypertension treatment [43]. A more recent report from the Genetic Epidemiology Network of Arteriopathy (GENOA) Study showed heritability of echocardiographic LV mass index as 0.34 after adjusting for age, body mass index, stroke volume, systolic blood pressure and diabetes mellitus [44].
Table 2.
Heritability of electrocardiographic and echocardiographic left ventricular hypertrophy in selected populations
| Reference | Method | Ethnicity | Heritability |
|---|---|---|---|
| Mayosi et al (37) | ECG | European Caucasian | 0.12-0.41 |
| Post et al. (38) | Echo | Caucasian | 0.24-0.32 |
| Arnett et al. (39) | Echo | African-American | 0.22-0.44 |
| Caucasian | 0.05-0.22 | ||
| Bella et al. (40) | Echo | American Indians | 0.17-0.27 |
| Juo et al. (41) | Echo | Caribbean Hispanics | 0.49 |
| Chien et al. (42) | Echo | Chinese | 0.15 |
| Assimes et al. (43) | Echo | Japanese | 0.43-0.61 |
| Fox et al. (44) | Echo | African-American | 0.34 |
It is not clear whether the variation in the heritabilities between ethnicities are indicative of differences in the genetic architecture underlying LV mass variation between populations, or whether differences in study design, adjustment for covariates, characteristics of study participants besides ethnicity, and/or environmental and cultural factors, and/or statistical fluctuation, are the main reason for the variability in heritability estimates. In any case, despite the difference in heritability estimates between studies, there is robust and consistent support for the genetic influence on LV hypertrophy across study designs, datasets and ethnicities.
Genome-wide linkage analyses
The search for genomic regions harboring genetic factors that influence LV mass has intensified over the previous decade or so. Both candidate gene/variant studies as well as more comprehensive genome-wide investigations have been conducted using both linkage analysis and/or association analysis to tie genomic regions to LV mass. Genome-wide linkage analyses in extended families have been successful in identifying regions harboring major loci for LV mass [46-49]. These linkage scans were conducted on families that were not ascertained on the basis of LV mass or related traits (and, hence, these family samples can be assumed to be representative of the studied populations overall rather constitute a subset with specific heart-related clinical symptoms), and the studies have shown genome-wide significant evidence for linkage of echocardiographic LV mass to chromosome 7 [46], chromosome 22 [47] and chromosome 12 [48,49]. However, except for the chromosome 12 locus, the identified chromosomal regions appear to not overlap between different studies, suggesting either that the major loci are population-specific to a substantial degree or that (some of) the identified linkage signals are, in fact, false positive findings. Like is the case for most significant linkage signals observed for complex diseases and quantitative traits, the specific genes responsible for the observed linkage results have yet to be identified.
Candidate gene association studies
Many investigators have focused on specific candidate genes to evaluate whether variation in these genes are indeed associated with LV mass. Potential candidate genes include ones encoding proteins regulating cardiac structure, hemodynamic load, calcium homeostasis, hormones, substrate metabolism, growth factors, energy metabolism and cell signaling [50]. Considerable attention has been devoted to polymorphisms in enzymes and hormones involved in the renin-angotensin-aldosterone system. The angiotensin-converting enzyme insertion deletion polymorphism [51,52], angiotensin II type I receptor gene A1166C polymorphism [53] and angiotensinogen gene M235T polymorphism [54,55] have been implicated in exerciseinduced LV hypertrophy. A report from the HyperGEN Study suggested that angiotensin-converting enzyme insertion deletion polymorphism and angiotensinogen gene variants may interact to modulate effects of LV hypertrophy and may vary by ethnicity [55]. The angiotensin II type 2 receptor gene (+1675 G/A) polymorphism has been associated with abnormal LV geometry in young men with mild hypertension [56]. A SNP in the aldosterone synthase gene ([CYP11B2] -344 C/T) has also been found to be associated with eccentric LV hypertrophy in essential hypertension [57,58]. A recent study suggests that a polymorphism of the β-1 adrenergic receptor gene (glycine for arginine at position 389) affects LV mass in patients with renal failure [59]. Moreover, the G-protein β3 subunit (C825T) polymorphism has been associated with LV mass in hypertension [60,61].
Other candidate genes have been selected based on their role in myocardial fatty acid oxidation. Recent studies have shown that LV mass and dilated cardiomyopathy are associated with abnormal fatty acid metabolism [62,63]. Jamshidi et al. [64] reported that a SNP within an intron of the peroxisome proliferator-activated receptor alpha (PPARα) influenced LV growth in response to exercise and hypertension. In their report, the Framingham investigators found statistically significant associations of the angiotensin receptor type 2 (AGTR2) gene with LV mass (p=0.05) and LV chamber size (p=0.007), β-2 adrenergic receptor (ADRB2) gene with LV mass (p=0.02) and LV wall thickness (p=0.005) and cardiac troponin T (TNNT2) gene with LV chamber diameter (p=0.0005) [65].
Despite the apparent success of some of these studies, it should be kept be mind that most of these candidate gene studies were based on small sample sizes, and differ from one another in many aspects, including in ethnicity of study subjects, ascertainment scheme, and covariate adjustment. Many of the reported candidate gene association findings have not been replicated (and several replication attempts failed), and the reported association results, while appearing to meet rigorous statistical significance criteria in the context of candidate gene studies (with a very limited multiple testing burden, i.e. number of SNPs examined), generally do not reach significance levels that would be required in genome-wide association studies with its much larger multiple testing threshold.
Genome-wide association studies
In addition to candidate gene-based association studies, association studies have also been conducted in a systematic, genome-wide manner. Genome-wide association studies (GWAS) utilize high throughput genotyping techniques to assay hundreds of thousands of the most common form of genetic variant, the single-nucleotide polymorphism (SNP), and relate genotypes at these variants to the phenotypes of study participants of diseases or other traits [66]. This approach permits the interrogation of much of the common variation in the entire human genome in thousand (or even hundreds of thousands) of unrelated individuals, achieving a much higher positional resolution than is generally possible in linkage analysis [67].
In a initial genome-wide joint linkage and association study, conducted on up to 1238 individuals participating in the Framingham Heart Study based on only ~71,000 SNPs, significant linkage was obtained for echocardiographic LV mass on chromosome 5 [68]. The study found interesting SNP associations on chromosome 2 and chromosome 11 (with a SNP near heatshock 70-KD protein 8, HSPA8), but these associations did not reach strict genome-wide significance. A recent report from Shah et al. found loci for electrocardiographic LV hypertrophy indices in chromosome 3p22.2 (sodium-channel, voltage gated, type 5, α-sub-unit, SCN5A, rs6797133), chromosome 12q13.3 (p23 or telomerase reverse transcriptase (TERT) binding protein, PTGES3, rs2290893), chromosome 15q25.2 (neuromedin B, NMB. rs2292462) and chromosome 15q26.3 (insulin-growth factor, IGFIR, rs4966014) [69].
Genome-wide association studies conducted on complex traits generally suffer from a lack of power. The main reason is that major loci with common functional variants, which are generally well tagged by the common SNPs included in modern genome-wide SNP panels, do not appear to exist for most complex diseases [70]. In order to detect more minor loci attributable to common functional variants – those loci that explain only a very small proportion of the variance in a trait – very large sample sizes are required. For this reason, investigators have often combined forces in various disease-focused consortia and conducted combined analyses of multiple datasets, including mega- and meta-analyses. In some cases, these studies now involve in excess of 100,000 individuals [71].
Two meta-analyses of LV mass and other cardiac structure and function phenotypes were published recently. The EchoGen Consortium [72] included 7 cohorts of European ancestry (5 used in the discovery stage, comprising ~13,000 individuals, and 2 in the replication stage, consisting of ~4000 individuals). In the first stage, 16 loci were found to be associated with the 5 different echocardiographic traits examined, including 3 for LV mass. However, the LV mass SNPs were not successfully replicated in stage 2. It is clear that any observed loci explain only a very small proportion of variance in the examined cardiac traits. A more recent, smaller-scale study was conducted on both African American (~1300 and ~1000 individuals in two stages) and European American hypertensive families (~1300 individuals) [73]. No genome-wide significant results were obtained for LV mass, but SNPs in NCAM1 were found to be associated with relative wall thickness.
Perspective
Since the completion of the human genome project, many clinicians, scientists, politicians and lay people alike have been predicting that genetic investigations would shortly lead to dramatic advances in prevention and cure for many human diseases, including personalized risk prediction and personalized medicine. In 2000, then-President Bill Clinton stated that “We are here to celebrate the completion of the first survey of the entire human genome. With this profound new knowledge, humankind is on the verge of gaining immense, new power to heal. Genome science will have a real impact on all our lives - and even more, on the lives of our children. It will revolutionize the diagnosis, prevention, and treatment of most, if not all, human diseases.” As a whole, these predictions appear to have been premature and perhaps overly optimistic. For LV mass, like for the vast majority of complex traits, progress to identify functional genetic variants, genes, and gene networks has been proven exceedingly difficult. While robust statistical associations have been obtained for many diseases, for the most part the functional genetic variants and genes are not yet known. Personalized risk prediction and medicine is not yet possible in a meaningful manner for most conditions, despite an increasing number of commercial enterprises hawking a variety of genetic tests.
However, while the future has not come as quickly as predicted by many, this state of affairs does not mean that it will never come. Francis Collins, the current director of the National Institutes of Health, continues to champion the future of personalized medicine due to breakthroughs in genetic epidemiological research. The community of clinicians and genetic researchers are divided among each those in their views as to how quickly functional genes and variants will be identified and what the ramifications of this will be personalized medicine and other areas of disease prevention and treatment. Likely the reality will fall somewhere in the middle between the most optimistic and pessimistic forecasts. It may well be that many have underestimated the time that it takes to see real-life benefits, but at the same time also underestimate the long-term impact due to the genetics revolution. It is clearly the case that the improvement in molecular biological techniques, including the rapid and affordable sequencing of the genome in parts (such as exome) or increasingly also in its entirety, continues unabatedly, enabling scientists to scan the genome in ever more detail. While it seems that for most complex traits common SNPs explain only a relatively small proportion of variance, investigators now increasingly begin to focus on rare variation, structural variants (such as copy number variants) and epigenetic phenomena (such as gene methylation levels). As our knowledge of the genetic underpinnings of our manifold conditions related to health and disease increases, it behooves us not to underestimate the long-term consequences for our health and the healthcare system.
In summary, LV mass is a very complicated phenotype that is influenced by many factors of our own genetic constitution and our lifestyle and environmental exposures alike. Different study designs, such as focused on specific forms of LV hypertrophy or attributable to specific conditions, coupled with new genomic technologies, may be required for us to finally begin to understand the underlying genetic architecture in much more detail. However, it seems that we are only at the early stages of the genetics revolution. Ultimately, it seems likely that clinical outcome studies will characterize the implications of these genetic variations for the natural history of LV hypertrophy and identify its impact on progression or regression of LV hypertrophy. As novel genes responsible for LV hypertrophy are identified, our understanding of the inciting events triggering cardiac remodeling and heart failure will expand. Although the identification and characterization of genes contributing to LV hypertrophy are challenging, genetic investigations still offer much promise for future prevention, early intervention and treatment of this major public health issue, even though this future may come later than hoped and predicted.
References
- 1.Roger VL, Go AS, Lloyd-Jones DM, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Makuc DM, Marcus GM, Marelli A, Matchar DB, Moy CS, Mozaffarian D, Mussolino ME, Nichol G, Paynter NP, Soliman EZ, Sorlie PD, Sotoodehnia N, Turan TN, Virani SS, Wong ND, Woo D, Turner MB. Heart disease and stoke statistics 2012 update: A report from the American Heart Association. Circulation. 2012;125:e2–e220. doi: 10.1161/CIR.0b013e31823ac046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cohn JN, Bristow MR, Chien KR, Colucci WS, Frazier OH, Leinwand LA, Lorell BH, Moss AJ, Sonnenblick EH, Walsh RA, Mockrin SC, Reinlib L. Report of the National Heart, Lung and Blood Institute Special Emphasis Panel on Heart Failure Research. Circulation. 1997;95:766–770. doi: 10.1161/01.cir.95.4.766. [DOI] [PubMed] [Google Scholar]
- 3.Sokolow M, Perloff D. The prognosis of essential hypertension treated conservatively. Circulation. 1961;23:697–706. [Google Scholar]
- 4.Breslin DJ, Gifford RW, Fairburn JF. Essential hypertension: a twenty-year follow-up study. Circulation. 1966;33:87–97. doi: 10.1161/01.cir.33.1.87. [DOI] [PubMed] [Google Scholar]
- 5.Kannel WB, Gordon T, Offutt D. Left ventricular hypertrophy by electrocardiogram: prevalence, incidence and mortality in the Framingham Heart study. Ann Intern Med. 1969;71:89–105. doi: 10.7326/0003-4819-71-1-89. [DOI] [PubMed] [Google Scholar]
- 6.Casale PN, Devereux RB, Milner M, Zullo G, Harshfield GA, Pickering TG, Laragh JH. Value of echocardiographic assessment of LV mass in predicting cardiovascular morbid events in hypertensive men. Ann Intern Med. 1986;105:173–178. doi: 10.7326/0003-4819-105-2-173. [DOI] [PubMed] [Google Scholar]
- 7.Silberberg JS, Barre PE, Prichard SS, Sniderman AD. Impact of left ventricular hypertrophy on survival in end-stage renal disease. Kidney Int. 1989;36:286–290. doi: 10.1038/ki.1989.192. [DOI] [PubMed] [Google Scholar]
- 8.Aronow WS, Epstein S, Koenigsberg M, Schwartz KS. Usefulness of echocardiographic left ventricular mass, ventricular tachycardia and complex ventricular arrhythmias in predicting ventricular fibrillation and sudden death in elderly patients. Am J Cardiol. 1988;62:1124–1125. doi: 10.1016/0002-9149(88)90562-0. [DOI] [PubMed] [Google Scholar]
- 9.Levy D, Garrison RJ, Savage DD, Kannel WB, Castelli MP. Prognostic implication of echocardiographically determined left ventricular mass in the Framingham Heart Study. N Engl J Med. 1990;322:1561–1566. doi: 10.1056/NEJM199005313222203. [DOI] [PubMed] [Google Scholar]
- 10.Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH. Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med. 1991;114:345–352. doi: 10.7326/0003-4819-114-5-345. [DOI] [PubMed] [Google Scholar]
- 11.Ghali JK, Liao Y, Simmons B, Castaner A, Cao G, Cooper RS. The prognostic role of left ventricular hypertrophy in patients with or without coronary artery disease. Ann Intern Med. 1992;117:831–836. doi: 10.7326/0003-4819-117-10-831. [DOI] [PubMed] [Google Scholar]
- 12.Bikkina M, Levy D, Evans JC, Larson MG, Benjamin EJ, Wolf PA, Castelli WP. Left ventricular mass and risk of stroke in an elderly cohort. JAMA. 1994;272:33–36. [PubMed] [Google Scholar]
- 13.Gardin JM, McClelland R, Kitzman D, Lima JAC, Bommer W, Klopfenstein S, Wong ND, Smith VE, Gottdiener J. M-mode echocardiographic predictors of six- to seven-year incidence of coronary heart disease, stroke, congestive heart failure and mortality in an elderly cohort (The Cardiovascular Health Study) Am J Cardiol. 2001;87:1051–1057. doi: 10.1016/s0002-9149(01)01460-6. [DOI] [PubMed] [Google Scholar]
- 14.Liao Y, Cooper RS, McGee DL, Mensah GA, Ghali JK. The relative effects of left ventricular hypertrophy, coronary artery disease and ventricular dysfunction on survival among black adults. JAMA. 1995;273:1592–1597. [PubMed] [Google Scholar]
- 15.de Simone G, Devereux RB, Daniels SR, Meyer R. Gender differences in left ventricular growth. Hypertension. 1995;26:979–983. doi: 10.1161/01.hyp.26.6.979. [DOI] [PubMed] [Google Scholar]
- 16.Hammond IW, Devereux RB, Alderman MH, Laragh JH. Relation of blood pressure and body build to left ventricular mass in normotensive and hypertensive adults. J Am Coll Cardiol. 1988;12:996–1004. doi: 10.1016/0735-1097(88)90467-6. [DOI] [PubMed] [Google Scholar]
- 17.de Simone G, Devereux RB, Roman MJ, Alderman MH, Laragh JH. Relation of obesity and gender to left ventricular hypertrophy in normotensive and hypertensive adults. Hypertension. 1994;23:600–606. doi: 10.1161/01.hyp.23.5.600. [DOI] [PubMed] [Google Scholar]
- 18.de Simone G, Devereux RB, Daniels SR, Koren MJ, Meyer RA, Laragh J. Effect of growth on variability of left ventricular mass: assessment of allometric signals in adults and children and their capacity to predict cardiovascular risk. J Am Coll Cardiol. 1995;25:1056–1062. doi: 10.1016/0735-1097(94)00540-7. [DOI] [PubMed] [Google Scholar]
- 19.Bella JN, Devereux RB, Roman MJ, O’Grady M, Welty TK, Lee ET, Fabsitz RR, Howard BV. Relations of left ventricular mass to fat-free and adipose body mass: The Strong Heart Study. Circulation. 1998;98:2538–2544. doi: 10.1161/01.cir.98.23.2538. [DOI] [PubMed] [Google Scholar]
- 20.Devereux RB, James GD, Pickering TG. What is normal blood pressure? comparison of ambulatory pressure level and variability in patients with normal or abnormal left ventricular geometry. Am J Hypertens. 1993;6:211s–215s. [PubMed] [Google Scholar]
- 21.Gonzales IJ, Bella JN, Blackshear J, Chesebro J, Weigand S, Castello R. Abnormal left ventricular mass and geometry in normotensive patients with hypertensive response to exercise. J Am Soc Echocardiogr. 2002;15:509. [Google Scholar]
- 22.Castello R, Gonzales IJ, Bella JN, Cheseboro J, Blackshear J, Weigand S. Importance of diastolic and systolic blood pressure response to exercise on left ventricular mass and geometry. J Am Coll Cardiol. 2003;41:217A. [Google Scholar]
- 23.Gottdiener J, Reda D, Materson B, Massie B, Notargiacomo A, Hamburger R, Williams DW, Henderson WG. Importance of obesity, race and age to cardiac structural and functional effects of hypertension. J Am Coll Cardiol. 1994;24:1492–1498. doi: 10.1016/0735-1097(94)90145-7. [DOI] [PubMed] [Google Scholar]
- 24.Schmieder RE, Messerli FH, Garavaglia GE, Nunez BE. Dietary salt intake: a determinant of cardiac involvement in essential hypertension. Circulation. 1988;108:7–13. doi: 10.1161/01.cir.78.4.951. [DOI] [PubMed] [Google Scholar]
- 25.Ganau A, Devereux RB, Pickering TG, Roman MJ, Schnall PL, Santucci S, Spitzer MC, Laragh JH. Relation of left ventricular hemodynamic load and contractile performance to left ventricular mass in hypertension. Circulation. 1990;81:25–36. doi: 10.1161/01.cir.81.1.25. [DOI] [PubMed] [Google Scholar]
- 26.Jones EJ, Devereux RB, O’Grady MJ, Schwartz JE, Liu J, Pickering TG, Roman MJ. Hemodynamic volume load stimulates arterial and cardiac enlargement. J Am Coll Cardiol. 1997;29:1303–1310. doi: 10.1016/s0735-1097(97)82755-6. [DOI] [PubMed] [Google Scholar]
- 27.Bella JN, Wachtell K, Palmieri V, Liebson PR, Gerdts E, Ylitalo A, Koren MJ, Pedersen OL, Dahlof B, Roman MJ, Devereux RB. Relation of left ventricular geometry and function to systemic hemodynamics in hypertension: The LIFE Study. J Hypertens. 2001;19:127–134. doi: 10.1097/00004872-200101000-00017. [DOI] [PubMed] [Google Scholar]
- 28.Chen CH, Ting CT, Lin SJ, Hsu TL, Ho SJ, Chou P, Chang MS, O’Connor F, Spurgeon H, Lakatta E, Yin FC. Which arterial and cardiac parameters best predict left ventricular mass? Circulation. 1998;98:422–428. doi: 10.1161/01.cir.98.5.422. [DOI] [PubMed] [Google Scholar]
- 29.Devereux RB, Roman MJ, de Simone G, O’Grady MJ, Paranicas M, Yeh J-L, Welty TK, Lee ET, Fabsitz RR. Relations of left ventricular mass to demographic and hemodynamic variables in American Indians: The Strong Heart Study. Circulation. 1997;96:1416–1423. doi: 10.1161/01.cir.96.5.1416. [DOI] [PubMed] [Google Scholar]
- 30.Devereux RB, Roman MJ, Paranicas M, O’Grady MJ, Lee ET, Welty TK, Fabsitz RR, Robbins D, Rhoades ER, Howard BV. Impact of diabetes on cardiac structure and function: The Strong Heart Study. Circulation. 2000;101:2271–2276. doi: 10.1161/01.cir.101.19.2271. [DOI] [PubMed] [Google Scholar]
- 31.Palmieri V, Bella JN, Arnett DK, Liu JE, Oberman A, Schuck M-Y, Kitzman DW, Hopkins P, Rao DC, Morgan D, Devereux RB. Effect of type 2 diabetes mellitus on left ventricular geometry and systolic function in hypertensive subjects: Hypertension Genetic Epidemiology Network (HyperGEN) Study. Circulation. 2001;103:102–107. doi: 10.1161/01.cir.103.1.102. [DOI] [PubMed] [Google Scholar]
- 32.Bella JN, Devereux RB, Roman MJ, Palmieri V, Liu JE, Paranicas M, Lee ET, Fabsitz R, Welty TK, Howard BV, Devereux RB. Separate and joint effects of systemic hypertension and diabetes mellitus on left ventricular structure and function in American Indians (The Strong Heart Study) Am J Cardiol. 2001;87:1260–1265. doi: 10.1016/s0002-9149(01)01516-8. [DOI] [PubMed] [Google Scholar]
- 33.Mokdad AH, Bowman BA, Ford ES, Vinicor F, Marks JS, Koplan JP. The continuing epidemics of obesity and diabetes in the United States. JAMA. 2001;286:1195–1200. doi: 10.1001/jama.286.10.1195. [DOI] [PubMed] [Google Scholar]
- 34.Adams T, Yanowitz F, Fisher AG, Ridges JD, Nelson AG, Hagan AD, Williams RR, Hunt SC. Heritability of cardiac size: an echocardiographic and electrocardiographic study of monozygotic and dizygotic twins. Circulation. 1985;71:39–44. doi: 10.1161/01.cir.71.1.39. [DOI] [PubMed] [Google Scholar]
- 35.Harshfield GA, Grim C, Hwang C, Savage D, Anderson S. Genetic and environmental influences on echocardiographically determined left ventricular mass in black twins. Am J Hypertens. 1990;3:538–543. doi: 10.1093/ajh/3.7.538. [DOI] [PubMed] [Google Scholar]
- 36.Bielen E, Fagard R, Amery A. The inheritance of left ventricular structure and function assessed by imaging and Doppler echocardiography. Am Heart J. 1991;21:1743–1749. doi: 10.1016/0002-8703(91)90021-9. [DOI] [PubMed] [Google Scholar]
- 37.Mayosi BM, Keavney B, Kardos A, Davies CH, Ratcliffe PJ, Farral M, Watkins H. Electrocardiographic measures of left ventricular hypertrophy show greater heritability than echocardiographic left ventricular mass. Eur Heart J. 2002;23:1963–1971. doi: 10.1053/euhj.2002.3288. [DOI] [PubMed] [Google Scholar]
- 38.Post WS, Larson MG, Myers RH, Galderisi M, Levy D. Heritability of left ventricular mass. The Framingham Heart Study. Hypertension. 1997;30:1025–1028. doi: 10.1161/01.hyp.30.5.1025. [DOI] [PubMed] [Google Scholar]
- 39.Arnett DK, Hong Y, Bella JN, Oberman A, Kitzman DW, Hopkins PN, Devereux RB. Sibling correlations of left ventricular mass and geometry in hypertensive African Americans and whites: The HyperGEN Study. Am J Hypertens. 2001;14:1226–1230. doi: 10.1016/s0895-7061(01)02200-2. [DOI] [PubMed] [Google Scholar]
- 40.Bella JN, MacCluer JW, Roman MJ, Almasy L, North KE, Best LG, Lee ET, Fabsitz RR, Howard BV, Devereux RB. Heritability of left ventricular dimensions and mass in American Indians: The Strong Heart Study. J Hypertens. 2004;22:281–286. doi: 10.1097/00004872-200402000-00011. [DOI] [PubMed] [Google Scholar]
- 41.Juo SHH, DiTullio M, Lin HF, Rundek T, Boden-Albala B, Homma S, Sacco RL. Heritability of left ventricular mass and other morphologic variables in Caribbean Hispanic subjects: The Northern Manhattan Study. J Am Coll Cardiol. 2005;46:730–742. doi: 10.1016/j.jacc.2005.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chein KL, Hsu HC, Su TC, Chen MF, Lee YT. Heritability and major gene effects on left ventricular mass in the Chinese population: a family study. BMC Cardiovasc Disor. 2006;6:37. doi: 10.1186/1471-2261-6-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Assimes TL, Narasimhan B, Beto TB, Yoon S, Curb JD, Olshen RA, Quertermous T. Heritability of left ventricular mass in Japanese Families living in Hawaii: the SAPPHIRe Study. J Hypertens. 2007;25:985–992. doi: 10.1097/HJH.0b013e32809bd740. [DOI] [PubMed] [Google Scholar]
- 44.Fox ER, Klos KL, Penman AD, Blair GJ, Blossom BD, Arnett D, Devereux RB, Samdarshi T, Boerwinkle E, Mosley TH. Heritability and genetic linkage of left ventricular mass, systolic and diastolic function in hypertensive African Americans (from the GENOA Study) Am J Hypertens. 2010;23:870–875. doi: 10.1038/ajh.2010.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Arnett DK, Lynch A, Kitzman D, Rao DC, Miller MB, Province M, Devereux RB. Linkage of left ventricular (LV) structural phenotypes to chromosome 7. Circulation. 2002;105:e102. [Google Scholar]
- 46.Benjamin EJ, DeStefano A, Larson MG, O’Donnel CJ, Vasan RS, Levy D. Genetic linkage analyses for left ventricular mass phenotypes in the Framingham Heart Study. Circulation. 2000;102:4131. [Google Scholar]
- 47.Wang L, Beecham A, Di Tullio M, Slifer S, Blanton SH, Rundek T, Sacco RL. Novel quantitative trait locus is mapped to chromosome 12p11 for left ventricular mass in Dominican families: the Family Study of Stroke Risk and Carotid Atherosclerosis. BMC Med Genet. 2009;10:74. doi: 10.1186/1471-2350-10-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cole SA, Haack K, Laston S, Almasy L, Comuzzie AG, Roman MJ, Best LG, Howard BV, Lee ET, MacCluer JW, Bella JN, Devereux RB, Göring HHH. Left ventricular mass is influenced by genetic variation in chromosome 12p. Circulation. 2011 in press. [Google Scholar]
- 49.Arnett DK, de las Fuentes L, Broeckel U. Genes for left ventricular hypertrophy. Curr Hypertens Rep. 2004;6:36–41. doi: 10.1007/s11906-004-0009-5. [DOI] [PubMed] [Google Scholar]
- 50.Montgomery HE, Clarkson P, Dollery CM, Prasad K, Losi MA, Hemingway H, Statters D, Jubb M, Girvain M, Varnava A, World M, Deanfield J, Talmud P, McEwan JR, McKenna WJ, Humphries S. Association of angiotensinconverting enzyme I/D polymorphism with change in left ventricular mass in response to physical training. Circulation. 1997;96:741–747. doi: 10.1161/01.cir.96.3.741. [DOI] [PubMed] [Google Scholar]
- 51.Karjalainen J, Kujala UM, Stolt A, Mantysaari M, Viitasalo M, Kainulainen K, Kontula K. Angiotensinogen gene M235T polymorphism predicts left ventricular hypertrophy in endurance athletes. J Am Coll Cardiol. 1999;34:494–499. doi: 10.1016/s0735-1097(99)00199-0. [DOI] [PubMed] [Google Scholar]
- 52.Diet F, Graf C, Mahnke N, Wassmer G, Predel HG, Palma-Hohmann I, Rost R, Bohn M. ACE and angiotensinogen gene phenotypes and left ventricular mass in athletes. Eur J Clin Invest. 2001;10:836–842. doi: 10.1046/j.1365-2362.2001.00886.x. [DOI] [PubMed] [Google Scholar]
- 53.Schmieder RE, Erdmann J, Delles C, Jacobi J, Fleck E, Hilgers K, Regitz-Zagrosek V. Effect of angiotensin II type 2 receptor (+1675 G/A) on left ventricular structure in humans. J Am Coll Cardiol. 2001;37:175–182. doi: 10.1016/s0735-1097(00)01063-9. [DOI] [PubMed] [Google Scholar]
- 54.Lynch AI, Tang W, Shi G, Devereux RB, Eckfeldt JH, Arnett DK. Epistatic effects of ACE I/D and AGT gene variants on left ventricular mass in hypertensive patients. J Hum Hypertens. 2011;26:133–140. doi: 10.1038/jhh.2010.131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Kupari M, Hautanen A, Lankinen L, Koskinen P, Virolainen J, Nikkila H, White PC. Associations between human aldosterone (CYP11B2) gene polymorphisms and left ventricular size, mass and function. Circulation. 1998;97:569–575. doi: 10.1161/01.cir.97.6.569. [DOI] [PubMed] [Google Scholar]
- 56.Delles C, Erdmann J, Jacobi J, Hilgers KF, Fleck E, Regitz-Zagrosek V, Schmieder RE. Aldosterone synthase (CYP11B2) -344 C/T polymorphism is associated with left ventricular structure in human arterial hypertension. J Am Coll Cardiol. 2001;37:878–884. doi: 10.1016/s0735-1097(00)01174-8. [DOI] [PubMed] [Google Scholar]
- 57.Stella P, Bigatti G, Tizzoni L, Barlassina C, Lanzani C, Bianchi G, Cusi D. Association between aldosterone synthase (CYP11B2) polymorphism and left ventricular mass in essential hypertension. J Am Coll Cardiol. 2004;43:265–270. doi: 10.1016/j.jacc.2003.08.034. [DOI] [PubMed] [Google Scholar]
- 58.Stanton T, Inglis GC, Padmanabhan S, Dominiczak AF, Jardine AG, Connell JM. Variation at the beta-1 adrenoceptor gene locus affects left ventricular mass in renal failure. J Nephrol. 2002;15:512–518. [PubMed] [Google Scholar]
- 59.Poch E, Gonzalez D, Gomez-Angelats E, Enjuto M, Pare JC, Rivera F, De La Sierra A. G-protein beta (3) subunit gene variant and left ventricular hypertrophy in essential hypertension. Hypertension. 2000;35:214–218. doi: 10.1161/01.hyp.35.1.214. [DOI] [PubMed] [Google Scholar]
- 60.Semplicini A, Siffert W, Sartori M, Monari A, Naber C, Frigo G, Santonastaso M, Cozzutti E, Winnicki M, Palatini P. G-protein beta3 subunit gene 825T allele is associated with increased left ventricular mass in young subjects with mild hypertension. Am J Hypertens. 2001;14:1191–1195. doi: 10.1016/s0895-7061(01)02227-0. [DOI] [PubMed] [Google Scholar]
- 61.de las Fuentes L, Herrero P, Peterson LR, Kelly DP, Gropler RJ, Davila-Roman VG. Myocardial fatty acid metabolism: independent predictor of left ventricular hypertrophy in hypertensive heart disease. Hypertension. 2003;41:83–87. doi: 10.1161/01.hyp.0000047668.48494.39. [DOI] [PubMed] [Google Scholar]
- 62.Davila-Roman VG, Vedala G, Herrero P, de las Fuentes L, Rogers JG, Kelly DP, Gropler RJ. Altered myocardial fatty acid and glucose metabolism in idiopathic dilated cardiomyopathy. J Am Coll Cardiol. 2002;40:271–277. doi: 10.1016/s0735-1097(02)01967-8. [DOI] [PubMed] [Google Scholar]
- 63.Jamshidi Y, Montgomery HE, Hense HW, Myerson SG, Torra IP, Staels B, World MJ, Doering A, Erdmann J, Hengstenberg C, Humphries SE, Schunkert H, Flavell DM. Peroxisome proliferator-activated receptor alpha gene regulates left ventricular growth in response to exercise and hypertension. Circulation. 2002;105:950–955. doi: 10.1161/hc0802.104535. [DOI] [PubMed] [Google Scholar]
- 64.O’Donnell CJ for the Writing Group of the Cardiogenomics Program. A systematic search for genetic variants associated with left ventricular structure and function phenotypes in the NHLBI Cardiogenomics Program for Genomics Applications. Circulation. 2004;109:e77–e78. [Google Scholar]
- 65.Christensen K, Murray JC. What genome-wide association studies can do for medicine. N Engl J Med. 2007;356:1094–1097. doi: 10.1056/NEJMp068126. [DOI] [PubMed] [Google Scholar]
- 66.Pearson TA, Manolio TA. How to interpret a genome-wide association study. JAMA. 2008;299:1335–13344. doi: 10.1001/jama.299.11.1335. [DOI] [PubMed] [Google Scholar]
- 67.Vasan RS, Larson MG, Aragam J, Wang TJ, Mitchell GF, Kathiseran S, Newton-Chen C, Vita JA, Keyes MJ, O’Donnell CJ, Levy D, Benjamin EJ. Genome-wide association of echocardiographic dimensions, brachial artery endothelial function and treadmill exercise responses in the Framingham Heart Study. BMC Med Genet. 2007;8:S2. doi: 10.1186/1471-2350-8-S1-S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shah S, Nelson CP, Gaunt TR, van der harst P, Barnes T, Braund PS, Lawlor DA, Casas J-P, Padmababhan S, Drenos F, Kivimaki M, Talmud PJ, Humphries SE, Whitaker J, Morris RW, Whincup PH, Dominiczak A, munroe PB, Johnson T, Goodall AH, Cambien F, Diemert P, Hengstenberg C, Tobin WHD, van Venldhuesen DJ, de Boer RA, Navis G, van Gilst WH, Mayosi BM, Thompson JR, Kumari M, MacFaralne PW, Day INM, Hingorani AD, Samani NJ. Four genetic loci influencing electrocardiographic indices of left ventricular hypertrophy. Circ Cardiovasc Genet. 2011;4:626–635. doi: 10.1161/CIRCGENETICS.111.960203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Terwilliger JD, Göring HHH. Gene mapping when rare variants are common and common variants are rare. Hum Biol. 2009;81:729–733. doi: 10.3378/027.081.0617. [DOI] [PubMed] [Google Scholar]
- 70.Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU, Allen HL, Lindgren CM, Luan J, Mägi R, Randall JC, Vedantam S, Winkler TW, Qi L, Workalemahu T, Heid IM, Steinthorsdottir V, Stringham HM, Weedon MN, Wheeler E, Wood AR, Ferreira T, Weyant RJ, Segrè AV, Estrada K, Liang L, Nemesh J, Park JH, Gustafsson S, Kilpeläinen TO, Yang J, Bouatia-Naji N, Esko T, Feitosa MF, Kutalik Z, Mangino M, Raychaudhuri S, Scherag A, Smith AV, Welch R, Zhao JH, Aben KK, Absher DM, Amin N, Dixon AL, Fisher E, Glazer NL, Goddard ME, Heard-Costa NL, Hoesel V, Hottenga JJ, Johansson A, Johnson T, Ketkar S, Lamina C, Li S, Moffatt MF, Myers RH, Narisu N, Perry JR, Peters MJ, Preuss M, Ripatti S, Rivadeneira F, Sandholt C, Scott LJ, Timpson NJ, Tyrer JP, van Wingerden S, Watanabe RM, White CC, Wiklund F, Barlassina C, Chasman DI, Cooper MN, Jansson JO, Lawrence RW, Pellikka N, Prokopenko I, Shi J, Thiering E, Alavere H, Alibrandi MT, Almgren P, Arnold AM, Aspelund T, Atwood LD, Balkau B, Balmforth AJ, Bennett AJ, Ben-Shlomo Y, Bergman RN, Bergmann S, Biebermann H, Blakemore AI, Boes T, Bonnycastle LL, Bornstein SR, Brown MJ, Buchanan TA, Busonero F, Campbell H, Cappuccio FP, Cavalcanti-Proença C, Chen YD, Chen CM, Chines PS, Clarke R, Coin L, Connell J, Day IN, den Heijer M, Duan J, Ebrahim S, Elliott P, Elosua R, Eiriksdottir G, Erdos MR, Eriksson JG, Facheris MF, Felix SB, Fischer-Posovszky P, Folsom AR, Friedrich N, Freimer NB, Fu M, Gaget S, Gejman PV, Geus EJ, Gieger C, Gjesing AP, Goel A, Goyette P, Grallert H, Grässler J, Greenawalt DM, Groves CJ, Gudnason V, Guiducci C, Hartikainen AL, Hassanali N, Hall AS, Havulinna AS, Hayward C, Heath AC, Hengstenberg C, Hicks AA, Hinney A, Hofman A, Homuth G, Hui J, Igl W, Iribarren C, Isomaa B, Jacobs KB, Jarick I, Jewell E, John U, Jørgensen T, Jousilahti P, Jula A, Kaakinen M, Kajantie E, Kaplan LM, Kathiresan S, Kettunen J, Kinnunen L, Knowles JW, Kolcic I, König IR, Koskinen S, Kovacs P, Kuusisto J, Kraft P, Kvaløy K, Laitinen J, Lantieri O, Lanzani C, Launer LJ, Lecoeur C, Lehtimäki T, Lettre G, Liu J, Lokki ML, Lorentzon M, Luben RN, Ludwig B, Manunta P, Marek D, Marre M, Martin NG, McArdle WL, McCarthy A, McKnight B, Meitinger T, Melander O, Meyre D, Midthjell K, Montgomery GW, Morken MA, Morris AP, Mulic R, Ngwa JS, Nelis M, Neville MJ, Nyholt DR, O'Donnell CJ, O'Rahilly S, Ong KK, Oostra B, Paré G, Parker AN, Perola M, Pichler I, Pietiläinen KH, Platou CG, Polasek O, Pouta A, Rafelt S, Raitakari O, Rayner NW, Ridderstråle M, Rief W, Ruokonen A, Robertson NR, Rzehak P, Salomaa V, Sanders AR, Sandhu MS, Sanna S, Saramies J, Savolainen MJ, Scherag S, Schipf S, Schreiber S, Schunkert H, Silander K, Sinisalo J, Siscovick DS, Smit JH, Soranzo N, Sovio U, Stephens J, Surakka I, Swift AJ, Tammesoo ML, Tardif JC, Teder-Laving M, Teslovich TM, Thompson JR, Thomson B, Tönjes A, Tuomi T, van Meurs JB, van Ommen GJ, Vatin V, Viikari J, Visvikis-Siest S, Vitart V, Vogel CI, Voight BF, Waite LL, Wallaschofski H, Walters GB, Widen E, Wiegand S, Wild SH, Willemsen G, Witte DR, Witteman JC, Xu J, Zhang Q, Zgaga L, Ziegler A, Zitting P, Beilby JP, Farooqi IS, Hebebrand J, Huikuri HV, James AL, Kähönen M, Levinson DF, Macciardi F, Nieminen MS, Ohlsson C, Palmer LJ, Ridker PM, Stumvoll M, Beckmann JS, Boeing H, Boerwinkle E, Boomsma DI, Caulfield MJ, Chanock SJ, Collins FS, Cupples LA, Smith GD, Erdmann J, Froguel P, Grönberg H, Gyllensten U, Hall P, Hansen T, Harris TB, Hattersley AT, Hayes RB, Heinrich J, Hu FB, Hveem K, Illig T, Jarvelin MR, Kaprio J, Karpe F, Khaw KT, Kiemeney LA, Krude H, Laakso M, Lawlor DA, Metspalu A, Munroe PB, Ouwehand WH, Pedersen O, Penninx BW, Peters A, Pramstaller PP, Quertermous T, Reinehr T, Rissanen A, Rudan I, Samani NJ, Schwarz PE, Shuldiner AR, Spector TD, Tuomilehto J, Uda M, Uitterlinden A, Valle TT, Wabitsch M, Waeber G, Ware ham NJ, Watkins H, Wilson JF, Wright AF, Zillikens MC, Chatterjee N, McCarroll SA, Purcell S, Schadt EE, Visscher PM, Assimes TL, Borecki IB, Deloukas P, Fox CS, Groop LC, Haritunians T, Hunter DJ, Kaplan RC, Mohlke KL, O'Connell JR, Peltonen L, Schlessinger D, Strachan DP, van Duijn CM, Wichmann HE, Frayling TM, Thorsteinsdottir U, Abecasis GR, Barroso I, Boehnke M, Stefansson K, North KE, McCarthy MI, Hirschhorn JN, Ingelsson E, Loos RJ MAGIC; Procardis Consortium. Association analysis of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010;42:937–948. doi: 10.1038/ng.686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vasan RS, Glazer NL, Felix JF, Lieb W, Wild PS, Felix SB, Watzinger N, Larson MG, Smith NL, Dehghan A, Grosshennig A, Schillert A, Teumer A, Schmidt R, Kathiresan S, Lumley T, Aulchenko YS, König IR, Zeller T, Homuth G, Struchalin M, Aragam J, Bis JC, Rivadeneira F, Erdmann J, Schnabel RB, Dörr M, Zweiker R, Lind L, Rodeheffer RJ, Greiser KH, Levy D, Haritunians T, Deckers JW, Stritzke J, Lackner KJ, Völker U, Ingelsson E, Kullo I, Haerting J, O’Donnell CJ, Heckbert SR, Stricker BH, Ziegler A, Reffelmann T, Redfield MM, Werdan K, Mitchell GF, Rice K, Arnett DK, Hofman A, Gottdiener JS, Uitterlinden AG, Meitinger T, Blettner M, Friedrich N, Wang TJ, Psaty BM, van Duijn CM, Wichmann HE, Munzel TF, Kroemer HK, Benjamin EJ, Rotter JI, Witteman JC, Schunkert H, Schmidt H, Völzke H, Blankenberg S. Genetic variants associated with cardiac structure and function: a meta-analysis and replication of genome-wide association data. JAMA. 2009;302:168–178. doi: 10.1001/jama.2009.978-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Arnett DK, Meyers KJ, Devereux RB, Tiwari HK, Gu CC, Vaughan LK, Perry RT, Patki A, Claas SA, Sun YV, Broeckel U, Kardia SL. Genetic variation in NCAM1 contributes to left ventricular wall thickness in hypertensive families. Circ Res. 2011;108:279–283. doi: 10.1161/CIRCRESAHA.110.239210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Collins F. Has the revolution arrived? Nature. 2010;464:674–675. doi: 10.1038/464674a. [DOI] [PMC free article] [PubMed] [Google Scholar]