

Abstract
A major advantage of hippocampal slice preparations is that the cytoarchitecture and synaptic circuits of the hippocampus are largely retained. In neurotoxicology research, organotypic hippocampal slices have mostly been used as acute ex vivo preparations for investigating the effects of neurotoxic chemicals on synaptic function. More recently, hippocampal slice cultures, which can be maintained for several weeks to several months in vitro, have been employed to study how neurotoxic chemicals influence the structural and functional plasticity in hippocampal neurons. This chapter provides protocols for preparing hippocampal slices to be used acutely for electrophysiological measurements using glass microelectrodes or microelectrode arrays or to be cultured for morphometric assessments of individual neurons labeled using biolistics.
Keywords: Acute hippocampal slice, Biolistics, Electrophysiology, Hippocampal slice culture, Microelectrode arrays, Morphometry
1. Introduction
As an experimental model, hippocampal slice cultures provide distinct experimental advantages over other in vitro and in vivo preparations of the hippocampus. The most significant advantages are that, except for the absence of afferent input, hippocampal slice preparations retain the cytoarchitecture and synaptic circuits of the intact hippocampus (1), yet are readily accessible for optical imaging or electrophysiological studies. Hippocampal slices have proven to be a powerful experimental model for investigating the structural and functional features of synaptic connectivity at the molecular, cellular, and circuit levels. The most widely used hippocampal slice preparations are the acute hippocampal slices and the organotypic hippocampal slice cultures. Acute hippocampal slice preparations are most often obtained from the adult rodent brain, are intended to be used for experimentation the same day that they are prepared, and are typically used to study the electrophysiological properties of individual neurons or circuits. In contrast, hippocampal slice cultures are usually derived from neonatal rodent brains and can be maintained in vitro for weeks to months (2, 3). Molecular biology, imaging, electrophysiology, and immunohistochemical techniques are routinely used in slice cultures to investigate molecular and cellular processes of neurodevelopment, synaptic plasticity, and cell death in both physiologic and pathophysiologic contexts.
The advantages of acute slices relative to slice cultures are that they involve less work to prepare and maintain, and the pattern of synaptic connections within the slice is minimally altered relative to the in vivo patterns at the time of harvest. In contrast, hippocampal slice cultures require more effort, particularly if the goal is to maintain the cultures beyond several days, and the neuronal injury and disruption of afferent and efferent connections that occur during the preparation of hippocampal slices induce axonal and dendritic remodeling and reorganization of synapses among the remaining neurons (4). The tissue isolation and slicing procedure also induces glial cell activation, which can lead to the formation of an astrocytic scar that surrounds the healthier tissue in the center of the slice within several weeks after harvest. There are several advantages of the slice cultures relative to the acute slice preparation beyond the obvious advantages afforded by being able to track processes that occur over extended periods of time. First, the dead cells and tissue debris caused by the slicing procedure disappear after 1–2 weeks in vitro (5). Second, the slice has time to recover from the altered metabolic state caused by the enzymes and ions released during the cutting of the tissue (6).
This chapter describes the techniques for preparing acute hippocampal slices for electrophysiological measurements as well as culturing and transfecting hippocampal brain slices for morphometric analyses. The preparation of hippocampal slices involves two major steps: (1) preparation of equipment, substrates, and media and (2) dissection and slicing of the hippocampus. While there are many similarities, the details of these steps vary depending on whether the goal is to generate acute slices or slice cultures. Protocols for each preparation are, therefore, described separately. With regards to hippocampal slice cultures, several methods have been developed over the years, but currently, the two most frequently used are the roller tube technique, which was developed and characterized by Gahwiler (5), and the membrane interface technique introduced by Stoppini et al. (7). With the roller tube technique, slices are attached to glass coverslips and incubated in rotating tissue culture tubes for several weeks. The slow rotation results in periodic alteration of the gas–liquid interface to which the cultures are exposed. This process leads to considerable thinning of the slice, eventually resulting in a monolayer of cells. With the membrane interface technique, slices are placed on porous, transparent membranes and maintained at the interface between air and culture medium. These membrane slice cultures do not thin to the same extent as seen with rollertube cultures, but rather remain five to eight cell layers thick. The preparation and equipment needs are much simpler using the membrane interface technique, making it more accessible to a greater number of laboratories, and this is the technique described for preparing hippocampal slice cultures in this chapter.
Freshly prepared (acute) slices of rat or mouse hippocampus are widely used to study how drugs and/or targeted mutations influence synaptic excitability and changes in neuroplasticity. Traditionally, glass microelectrodes have been used to record electrically evoked extracellular potentials, including the excitatory and inhibitory postsynaptic potentials (EPSPs and IPSPs) and population spikes (PSs). Glass microelectrodes have several advantages, permitting the investigator to precisely place the recording and stimulating electrodes within the subregions of the hippocampus and to a depth beyond the surface of the slice, where cells are likely injured by the slicing protocol. The use of multiple or multibarreled electrodes permit the measurements of evoked potentials simultaneously with the measurements of local pH, oxygen, or neurotransmitter release. Glass microelectrodes also afford the measurements of excitatory and inhibitory currents from a single neuron using an electrode placed intracellularly or using the whole cell voltage clamp configurations. Application of single electrode methods to hippocampal slices has been invaluable in determining how xenobiotics influence the neuronal excitability and plasticity on an acute timescale and have contributed to our knowledge of underlying mechanisms. However, a major limitation of single electrode approaches for toxicological research is that they are not amenable to rapid throughput screening and do not provide information about the patterns of network activity across the entire hippocampus. Recent advances in multielectrode array (MEA) technology and fabrication have resulted in clear advantages over the traditional extracellular pulled-glass electrode approaches. Advances in printed MEA chips have permitted the fabrication of electrodes at high density (arrays of 64, 128, or more electrodes) spaced ≤50 μm, apart with limited interference from cross talk. Moreover, the spatial orientation of individual electrodes within an MEA chip can be customized to measure patterns of neural activity among hippocampal regions in response to stimuli delivered to one or more discrete locations within the slice. The major steps common to both the electrode and MEA methods include: (1) cutting and equilibration of hippocampal slices in ACSF; (2) determining whether the interface or submerged perfusion will be used; (3) placement of recording and stimulating electrodes or positioning of slice on the electrode array; (4) defining the input/output (I/O) relationship for each slice; and (5) recording a stable baseline response to a constant electrical pulse protocol determined from the I/O relationship (stimulus giving ~50% of maximum response) prior to perfusion of the test substance.
Optical imaging of hippocampal slice cultures has proven to be a powerful experimental approach for elucidating cellular and molecular mechanisms contributing to both functional and structural plasticity in the developing hippocampus. Various labeling techniques have been successfully applied to image neuronal and glial cells in hippocampal slice cultures, including: (1) immunohistochemistry; (2) bath application, injection, or ballistic delivery (8) of fluorescent structural or physiological indicators; and (3) biolistic (9) or viral transfection (10) to express cDNAs or siRNAs in a subset of neuronal or glial cells. In this chapter, we describe a method for labeling the soma, dendritic arbors, and axon of a subset of neurons in slice cultures using the biolistic delivery of cDNA encoding a fluorescent protein. The major steps include: (1) preparation of DNA-coated gold microcarriers or DNA bullets; (2) coating of tubing with DNA bullets; and (3) transfection of organotypic slice cultures.
2. Materials
2.1. Preparation of Acute Hippocampal Slices
Tissue slicer to generate 300–400-μm thick slices of the hippocampus. Suitable slicers include the Vibratome (model VT1000S, Leica, Bannockburn, IL), McIlwain tissue chopper (Brinkman, Westbury, NY), and the Dosaka microslicer (Model DTK-1000, Kyoto, Japan).
Artificial cerebral spinal fluid (ACSF): 124 mM NaCl, 2.5 mM KCl, 1.25 mM KH2PO4, 26 mM NaHCO3, 2 mM MgSO4, 2.5 mM CaCl2, 10 mM d-glucose, and 4 mM d-sucrose bubbled with 95% O2/5% CO2 (aka carbogen) (see Notes 1 and 2).
Six- to twelve-week old rats (125–200 g) or mice (20–25 g) (see Note 3).
Vaporizer for the administration of volatile anesthetic.
Guillotine (WPI, Sarasota, FL).
Preincubation chamber (Medical Systems, Corp., New York, NY) (Fig. 1).
Fig. 1.

Illustration of prechamber for holding brain slices before transfer to the recording chamber, such as the Haas chamber (Fig. 2). The prechamber contains a mesh bottom permitted exchange of ACSF once placed inside the outer chamber. The outer chamber possesses an inlet for bubbling carbogen from an external source via tubing.
2.2. Preparation of Hippocampal Slice Cultures
Four- to five-day old rat pups (see Note 4).
Laminar flow tissue culture hood (see Note 5).
Tissue slicer to generate 400-μm thick slices of the hippocampus. Suitable slicers include Vibratome (model VT1000S, Leica, Bannockburn, IL), McIlwain tissue chopper (Brinkman, Westbury, NY), and the Dosaka microslicer (Model DTK-1000, Kyoto, Japan).
Dissecting microscope.
Glass bead sterilizer.
Sterile double-edged razor blades.
Large scissors, small round-ended spatula, scalpel handle and blade, small fine-tipped straight forceps, two pairs of #4 and #5 Dumont forceps, one pair of #7 curved Dumont forceps, and iridectomy scissors (or small spring scissors).
Sterile 6-cm Whatman filters.
Aklar squares are cut to fit the chopping platform of the tissue slicer.
Sterile the plastic transfer pipettes.
Sterile 10-cm diameter glass Petri dishes, 6-cm diameter tissue culture plastic dishes, and 6-well tissue culture plastic plates (NUNC 140685).
Culture the plate inserts: 0.4 μm Millicel membrane, 30-mm diameter (Millipore, PICM0RG50).
Dissection buffer: 1 mM CaCl2, 5 mM MgCl2, 4 mM KCl, 26 mM NaHCO3, 8% sucrose, 0.5% phenol red, and filter-sterilized and bubbled with 95% O2/5% CO2 (aka carbogen) (see Note 1).
Slice culture medium: MEM with Earle’s salts and l-glutamine (Gibco 11095) supplemented with 1 mM CaCl2, 2 mM MgSO4, 1 mg/L insulin, 1 mM NaHCO3, 20% heat-inactivated horse serum, 0.5 mM l-ascorbate, 30 mM HEPES, 2.3 g/L d-glucose, and pH 7.3.
2.3. Measuring Electrical Excitability in Acute Slices Using Glass Microelectrodes
Nichrome wire (50-μm diameter).
Flaming/Brown type – Micropipette/Patch pipette puller – P-97 (Sutter Inst, Novato, CA).
Recording electrodes of approximately 1 MΩ resistance.
Amplifier KS-700 (WPI, New Haven, CT).
Digitizer (DigiData 1200, Axon Instruments, Molecular Devices, Sunnyvale, CA).
Data acquisition and analysis software (pClamp 9, Axon Instruments).
Interface or submerged recording chamber (Medical Systems, Corp., New York, NY) (Fig. 2).
Fig. 2.

Illustration of the perfusion (HAAS) chamber (middle panel) and the interface top (right panel) that holds slices on which stimulating and recording electrodes are placed. Left panel shows the interface chamber assembled and connected to an external temperature controller.
2.4. Measuring Electrical Excitability in Acute Slices Using Microelectrode Arrays
MEA probes and amplifiers: These can be purchased from a number of vendors. The MED64 MEA from Tensor Biosciences (Irvine, CA) is an integrated system that supports 64-channel data acquisition and analysis. For rat hippocampal slices, a MED64 probe 8 × 8 array with 150-μm interelectrode spacing is the best, but other configurations are commercially available.
Microinjection pump (Harvard apparatus, Holliston, MA).
0.1% polyethylenimine (PEI) in 25-mM borate buffer.
ACSF: 124 mM NaCl, 2.5 mM KCl, 1.25 mM KH2PO4, 26 mM NaHCO3, 2 mM MgSO4, 2.5 mM CaCl2, 10 mM d-glucose, and 4 mM d-sucrose bubbled with 95% O2/5% CO2 (carbogen).
Data acquisition and analysis software (Performer® software, Tensor Biosciences, Irvine, CA).
2.5. Labeling Hippocampal Slice Cultures for Morphometric Analyses
Helios Gene Gun system (BioRad, Hercules, CA), including Helios gene gun, tubing preparation station, and tubing cutter.
Clean bench or tissue culture hood.
Nitrogen gas cylinder and nitrogen regulator.
Helium gas cylinder and helium regulator.
Gold-Coat tubing (BioRad).
Transwell® permeable support (3.0-μm polyester membrane 24 mm, Costar).
Gold microcarriers, 1.6-μm diameter (BioRad) (see Note 6).
0.5 M spermidine (Sigma S-0266).
Ethanol (100%).
1 M CaCl2.
50 μg/mL polyvinylpyrrolidone (PVP, BioRad).
Purified cDNA: To visualize dendritic arbors in hippocampal slice cultures, we typically use pCAGGS expressing tomato fluorescent protein (see Note 7).
3. Methods
3.1. Preparation of Acute Hippocampal Slices
Anesthetize rat or mouse by applying isoflurane through a nose cone using a calibrated vaporizer set at 2–3%. Once a deep plane of anesthesia has been reached, euthanize the animal by decapitation, preferably using an appropriately sized guillotine.
Remove the brain quickly and place in chilled (5°C) ACSF (see Notes 8 and 9).
Dissect the hippocampus quickly on an ice-cold platform with plenty of ACSF.
Using a tissue slicer, section hippocampus (transverse plane) at 400-μm intervals beginning at approximately 10 mm from the rostral ends (see Note 10).
Transfer the slices to a preincubation chamber (Fig. 1) and hold them at 35.0 ± 0.5°C in carbogenated ACSF prior to transfer to the recording chamber (see Note 11).
Transverse slices 400 μm thick are cut from each hippocampus and placed, in turn, upon platforms within an interface incubation chamber (four to eight slices per platform) with ACSF that is continuously gassed with carbogen (see Note 12).
3.2. Preparation Of Hippocampal Slice Cultures
Warm a 50-mL aliquot of slice culture medium to 37°C.
Place separate 50-mL aliquot of slice culture medium and 50-mL aliquot of dissection medium on ice. Bubble dissection medium with carbogen.
Sterilize the surgical equipment using the bead sterilizer.
Place the bottom of a 10-cm glass Petri dish on a pan of packed ice and fill with cold dissection medium (see Note 9). Spray a 11-cm Whatman filter with 70% ethanol and place it in overturned lid of the Petri dish on top of the ice. Cover this with cold dissection medium. Place a second 10-cm glass Petri dish on packed ice and place a 6-mm plastic tissue culture dish inside it. Add cold dissection medium to cover the bottom of the 6-mm plastic tissue culture dish.
Prepare the inserts for plating the slices. Place an insert into each well of a six-well plate. Add 800 μL of slice culture medium under each insert. Make sure that the bottom of each insert is completely wet and there are no air bubbles. Place the culture dishes with inserts into a cell culture incubator at 35°C at 5% CO2 until the slices are ready to plate.
Prepare the tissue slicer (this protocol is based on the use of the McIlwain tissue slicer, but can be adapted for other slicers) and wipe the slicer down thoroughly with 70% ethanol. Sequentially sterilize the following with 70% ethanol, dry, and then stack on the stage: the two plastic-cutting platforms provided by the manufacturer, 6-cm Whatman filter, and Aklar square cut to fit the stage. Ensure that the edge of the Aklar extends just beyond the edge of the filter. Insert a sterile razor blade and align it to the chopping platform.
Wipe the head of the pup with 70% ethanol and then decapitate using large scissors. Cut the skin down the midline from the neck up to and between the eyes with a scalpel while gently holding the skull with small forceps. Insert the scissors into the foramen magnum and cut the skull along the midline up to and between the eyes. Make four small incisions perpendicular to the main cut at the ends of the main incision. Peel the skull back with straight forceps, remove the brain using the small spatula, and submerge it immediately in the 10-mm Petri dish with chilled dissection medium, using the transfer pipette to add more dissection medium as necessary.
Place the brain in the Petri dish lid containing the Whatman filter covered with cold dissection medium. Be sure the brain remains covered with dissection medium throughout the remainder of dissection. Using a scalpel or iridectomy scissors, remove the cerebellum by making a coronal cut just behind the inferior colliculi. Make a second cut sagittally down the midline completely through the brain to separate the hemispheres. Place the hemibrain so that the medial surface is facing up. Separate the neocortex with the underlying hippocampus from the midbrain and brain stem, being sure to remove the meninges and blood vessels surrounding the hippocampus. The hippocampus should now be exposed. Using the iridectomy scissors or curved Dumont forceps, disrupt the connection of the hippocampus on the ventral side (fimbria) by gently rotating the hippocampus around its longitudinal axis. Use a wide-bore pipette (precoated with slice culture medium) to transfer the hippocampus to the second 10-mm dish containing chilled dissection medium. Repeat this process until all hippocampi have been removed (see Note 8).
Remove the hippocampi (four to six at a time) with a wide-bore pipette and place them on the chopping block. Position hippocampi, smooth side up, using a pipette filled with dissection medium to push hippocampi into the correct position. Place them so that the long axis of each hippocampus is parallel to the direction of the stage movement, but allow the septal end to curve away from the parallel axis. Remove the excess liquid (see Note 13). Repeat this until there are 10–11 hippocampi lined up on the stage.
Set the chopper to move the stage 400 μm. Position the stage to the edge of hippocampi and turn the chopper on. Transfer the slices from the Aklar square into the 6-mm dish with chilled dissection medium using a wide-bore pipette. Repeat until all the hippocampi are sliced.
Separate the slices under a dissecting microscope either by gentle agitation of the dish or by using Dumont forceps. Be careful to not poke or tear the slices.
Select the slices with an intact structure that display distinct CA1, CA3, and DG cell layers, are of even thickness, and do not have tears or holes.
Use a wide-bore pipette (precoated with slice culture medium) to transfer three to four of the good slices per Millicell insert. Keep the slices distant from each other, but relatively centered in the middle of the membrane. Gently remove the excess medium from the top of the insert, as medium on top of the insert prevents oxygen exchange. Place the cell culture dishes in a cell culture incubator at 35°C with 5% CO2.
To maintain the cultures for several weeks to several months, it is necessary to change the medium the first day after plating and every 3 days thereafter. To change the medium, tilt the plates and aspirate the medium in each well. Slowly add 800 μL of warm slice culture medium to each well, placing the pipette tip along the edge of the well below the insert. Be sure the medium covers the bottom of the filter and there are no air bubbles. An alternative method which accounts for possible conditioning of the medium by the slice culture is to partially replace the medium (see Note 14).
3.3. Measuring Electrical Excitability in Acute Slices Using Glass Microelectrodes
Make recording electrodes of approximately 1 MΩ resistance using a Flaming/Brown type – Micropipette/Patch pipette puller.
Transfer several slices from the medial hippocampus from the preincubation chamber to the nylon netting of either an interface (11) or submerged (12) recording chamber (Fig. 2).
For interface preparations, make sure ACSF contacts the bottom of the slice that is in contact with the nylon mesh support (Fig. 1a). Perfuse carbogenated ACSF at 22°C underneath the slice for 30 min at a flow rate of 4 mL/min. After 30 min, perfuse the slices with carbogenated ACSF warmed to 35 ± 0.5°C for the duration of the experiment using a temperature controller (Medical System Corp.) (see Note 8). Measurements can also be made from a submerged slice preparation in which ACSF flows over the entire slice and carbogen flows over the bath solution. The slices are permitted to recover in perfused ACSF for 2 h prior to placing the electrodes for conventional recording with an extracellular microelectrode.
A bipolar stimulating electrode made from two lengths of nichrome wire, 50 μm in diameter, is directed through holes in the cover of the interface chamber and placed on the surface of the stratum radiatum (S. rad) of area CA1 to orthodromically stimulate Schaffer collateral/commissural fibers (Fig. 3).
Place the glass microelectrode filled with ACSF (1 MΩ resistance) 1–2 mm from the stimulating electrode near the stratum pyramidale to record field excitatory postsynaptic potentials (fEPSPs) or PSs (Fig. 3).
Responses are amplified (KS-700, WPI, New Haven, CT, USA) using a low-pass filter at 1 kHz, digitized with a DigiData 1200 (Axon Instruments, NY, USA) and stored on a personal computer for subsequent analysis with pClamp 9 (Axon Instruments).
During recording, a combination of stimulation intensity and micropipette placement is used to obtain maximal responses.
Stimuli consist of constant current rectangular pulses lasting 200 μs applied at 0.1 Hz, ranging from 10 to 170 μA. Once the stimulus–response curve is obtained at the beginning of each experiment, the stimulation intensity is fixed to obtain an fEPSP amplitude ranging from 50 to 70% of maximum (1.0–1.5 mV).
Begin baseline recording with constant perfusion of ACSF at 3–4 mL/min. Baseline synaptic responses are recorded for a period of 20–30 min to assure the slice is stable. Those slices not exhibiting stable responses should be discarded.
Record the magnitude of the fEPSP and PS (if present) as a function of stimulus intensity ranging from threshold of EPSP to that producing a maximal PS (control input/output curve).
The slices are then perfused at 3–4 mL/min with ACSF containing either vehicle or the test substance dissolved in vehicle for the duration of the experiment (typically 1 h).
After 30 min of perfusion with test substance, Input/Output data are acquired over the same stimulus intensity range.
A high-frequency protocol (tetanic pulse train) is then used to induce long-term potentiation (LTP) in the slice consisting of three trains of 100 Hz each 1 s in duration administered with 20 s intertrain intervals to induce LTP.
Responses are monitored continually for 20 min to access the stability of the LTP.
Record the magnitudes of fEPSP and PS responses at the same stimulus intensity range as the respective control (LTP data).
Digitized data are analyzed with pCLAMP software (Axon Instruments, version 9.2).
The initial slope of the population fEPSP is measured by a linear fit to a 1 ms window immediately before the onset of the PS.
PS amplitudes are measured from extrapolated fEPSP baseline to the peak of the PS. Input/output curves are generated by normalizing the PS amplitudes and EPSP slopes to their respective control maximum (13).
Fig. 3.
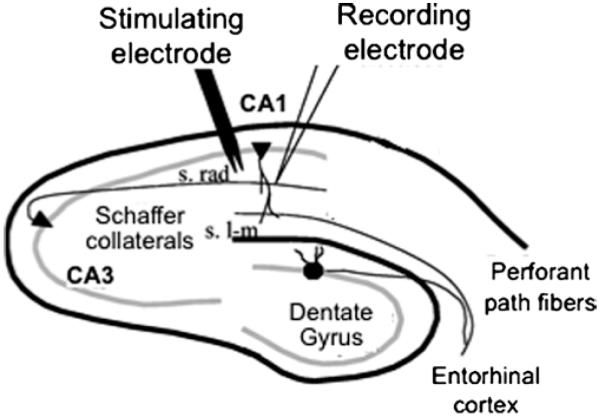
Recording evoked potentials from the hippocampus CA1 using microelectrodes. Diagram showing the placement of stimulating and recording electrodes near the Schaffer collateral/commissural fibers and stratum radiatum (S. rad), respectively. The relative location of stratum lacunosum-moleculare (s. l-m) is also shown.
3.4. Measuring Electrical Excitability in Acute Slices Using Microelectrode Arrays
MED probes are relatively hydrophobic and need to be initially pretreated with PEI, 0.1% in 25-mM borate buffer for 12 h to facilitate tissue adhesion. The coating process is done once to new probes, since subsequent exposures to ACSF and tissue slices enhance slice adhesion.
Transfer the slice to the MED probe chamber, containing ACSF with sera (10% fetal bovine serum + 10% horse serum) using a small paintbrush to position the slice on the electrode array.
Use an inverted microscope equipped with a 4× phase lens to align the slice such that electrodes make contact with Schaffer-commissural fibers and the recording sites contact the pyramidal cell layer as indicated in Fig. 4.
After positioning the slice on the electrode array, completely remove the culture medium with the sera used for incubation and replace with 250 μL of fresh culture medium with sera.
During the adhesion process, it is critical that the slice receives sufficient oxygen. Prepare an airtight container with a secure lid. Flow oxygen into the container, optionally through a Teflon plate with very fine orifices.
Pour a small amount of water into the bottom of the container to ensure sufficient humidity.
Place the MED probe in the container and close the lid tightly.
Place the container securely in a water bath at 38°C with 95% oxygen and 5% carbon dioxide flow for 1 h. Make sure the airflow pressure is positive, typically 20 mmHg.
The incubation period should not exceed 1 h since the culture medium contains 20% sera, which may cause damage to the slices.
Apply a weight at the margins of the slice with several twists of fine wire (100–200 mg). Avoid applying pressure directly on the electrodes.
Take a photomicrograph (using a 4× objective) of the position of the slice on the probe.
Import the photomicrograph to the MED conductor software and confirm the relative placement of the slice on the electrode array.
Place the MED probe on the MED connector, replace the connector cover, and finally place the probe cover on the probe.
Referring to the photomicrograph, select the channels for stimulation. Adjust the stimulation magnitude with the MED amplifier. MED Conductor sends the stimulation signal while simultaneously recording and storing evoked responses from all 64 channels.
During the electrophysiological recording, the slice is perfused with oxygenated 35°C ACSF at a rate of 2–3 mL/min using a microinjection pump.
The stimulation electrode is chosen within the MEA based on its proximity to the Schaffer-Collateral/Commissural pathway (Fig. 4).
Apply field pulses, biphasic stimuli (10–80 mA, 0.1 ms) delivered to Schaffer-Collateral/Commissural pathway at 0.05 Hz (i.e., once every 20 s).
The recording electrode is chosen based on its proximity to the S. rad within the CA1 region of hippocampus (Fig. 4).
The evoked fEPSPs are sampled at 20 kHz using an MED 64 multichannel amplifier, digitized and graphically displayed using Performer® software.
Baseline fEPSPs are experimentally set between 50 and 60% of maximum amplitude for each slice, and the slope of each fEPSP is calculated and displayed by the Performer® software.
Slices that exceed more than 20% fluctuation during the stabilization period (at least 30 min) are discarded.
Once the slice is stabilized, fEPSPs are collected for an additional 10 min period in the absence of test compound to establish a baseline control (i.e., 100% baseline).
Test compounds or solvent controls are added directly into the ACSF after the recording of baseline fEPSP. The volumes of vehicle or text solutions added to the ACSF should not exceed 0.05% (v/v).
Perfusion with vehicle or test compound dissolved in ACSF is initiated at the completion of baseline period, and the effects on fEPSP slope are recorded for an additional 30–40 min minimally depending on the experimental design.
For each experimental condition, we typically record from 5 to 15 slices per experimental paradigm. The mean change in fEPSP slope (normalized to each slice’s baseline period) is calculated. After a 10 min recording to establish a baseline period, the slice is perfused for 30 min (test period) with vehicle (DMSO, ≤0.025% v/v) or test compound dissolved in ACSF. After an additional 30 min of recording during the test period, LTP can be induced using the protocol described above.
Fig. 4.
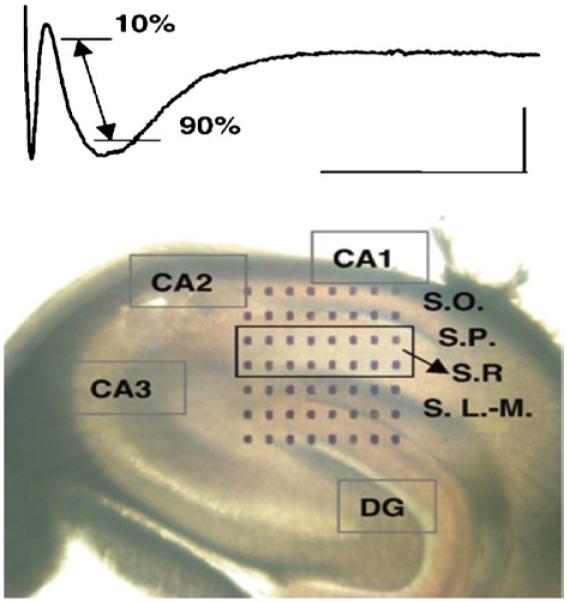
Rat hippocampal slice placed on multielectrode array (MEA) showing the position of 64 microelectrodes relative to the hippocampal slice architecture. Trace depicts a typical waveform indicating the location where the fEPSP slope is measured. Abbreviations: CA Cornu Ammon, DG Dentate Gyrus, S.O. stratum oriens; S.T. stratum pyramidale, S.R. stratum radiatum (the region indicated by the box), and S. L.-M. stratum lucidum–moleculare.
3.5. Morphometric Analyses of Hippocampal Slice Cultures
Make up the PVP solution: Weigh out 20 mg PVP in a 1.5-mL sterile microfuge tube. Add 1 mL of 100% ethanol and mix until dissolved. Dilute the PVP to 50 μg/mL in 100% ethanol.
Weigh out 12.5 mg of gold microcarriers and add to 100 mL of 0.05 M spermidine. Sonicate (in a bath sonicator) for several seconds to resuspend the gold microcarriers.
Add 50 μg of total DNA (typically, we use 16.6 μg fluorescent marker, e.g., tomato fluorescent protein, and 33.3 μg of test plasmid or carrier DNA) and bath sonicate briefly to mix and resuspend the mixture.
While vortexing the DNA mixture at a moderate to slow rate, add 100 μL of 1 M CaCl2 dropwise.
Allow the gold and DNA to precipitate at room temperature for 10 min. Mix gently by flicking the tube periodically during precipitation.
Spin for 15 s at maximal rpm in a microcentrifuge. Remove and discard the supernatant. Wash 3× in 1 ml 100% ethanol; spin for 5 s at maximal rpm between each wash.
Resuspend the DNA-coated gold microcarriers in 500 μL of PVP (50 μg/mL) and transfer to a 15-mL centrifuge tube. Add PVP to a final volume of 3 mL (see Note 15).
Place a 30-in. length of Gold-Coat tubing into the tubing prep station as described by the manufacturer. Turn on the N2 gas and adjust the flow rate to 0.35 L/min and maintain for 30 min to completely dry the tubing. Remove the tubing and attach a syringe to one end using adaptor tubing.
Bath sonicate the DNA-coated gold microcarrier suspension for 5–10 s, invert the suspension several times, and then immediately place the free end of tubing into the suspension and pull the suspension into the tubing using the syringe. Remove the tubing from the 15-mL tube of DNA-coated gold microcarrier suspension, but continue to apply suction with the syringe to leave a 2-in. air gap on the free end.
Holding the syringe and attached tubing horizontally, insert the loaded tubing into the prep station until the end is 1 in. from the O-ring.
Allow the gold microcarriers to settle for 5 min.
Slowly and steadily suck out the fluid from the tubing without disturbing the gold microcarriers. This should take approximately 30 s.
Immediately insert the tubing into the O-ring and turn the tubing 180° using the switch on the tubing prep station, pause for 5 s, and then turn on the switch to rotate the tubing for 30 s. This is to ensure an even spread of gold in the tubing (see Note 16).
Open the valve on the flowmeter of the N2 tank at 0.35 L/min for 5 min to dry the gold to the tubing.
Remove the tubing from the prep station and insert it into the tubing cutter to create cartridges (see Note 17).
Set up the gene gun in a clean bench or tissue culture hood, wiping it down thoroughly with 70% ethanol. Load cartridges into the chamber of the gene gun per the manufacturer’s directions. Attach the gene gun to a tank of helium gas using the high-pressure hose supplied by the manufacturer. Adjust the helium pressure to 200 psi.
Remove the organotypic slice cultures from the incubator and place inside the sterile hood. Remove the lid of the tissue culture plate and set it aside.
Place a sterile Transwell® permeable support (3.0-μm polyester membrane 24 mm, Costar) over the barrel of the gun.
Place the end of the gene gun barrel with the transwell support about 1 cm above the slice cultures and fire the gun.
Replace the lid to the tissue culture plate and return cultures to the incubator (see Note 18).
4. Notes
Unless stated otherwise, all solutions should be prepared in water that has a resistivity of 18.2 MΩ-cm and total organic content of less than five parts per billion. This standard is referred to as “water” in the text.
Glucose concentrations between 1 and 10 mM optimize several electrophysiological properties of the in vitro hippocampal slice preparation, such as the amplitude of population spikes (14, 15). Concentrations in excess of 5-mM exert neuroprotection (16-18). Cerebrospinal fluid (CSF) contains numerous amino acids that are omitted from ACSF. Of these, glutamine, which is a major precursor of glutamate and GABA, is the most abundant in CSF (0.4–0.8 mM). However, systematic analysis of varying concentrations of glutamine added to slices perfused in ACSF containing 10 mM glucose indicated that the inclusion of glutamine in ACSF did not enhance the population spike amplitude and higher concentrations resulted in spreading depression (14).
The majority of electrophysiological studies performed on acute hippocampal slices use slices prepared from adult male rats or mice. Males are typically used because this limits the influence of variable hormonal status inherent in adult females. However, for questions that address how the alteration of hormonal status in vivo impacts hippocampal neuroplasticity ex vivo or to identify gender differences in electrical excitability when hormones are exogenously applied in vitro, hippocampal slices derived from male and female animals can be incorporated in the experimental design. For example, intact and ovariectomized mice have been used to address specific questions of how estrogen influences electrophysiological properties of the hippocampus (19). Similarly, the age of the animal used as the source of hippocampal slices can be an important aspect of the experimental design. For example, electrical recordings from hippocampal slices obtained from juvenile mice (3–4 weeks of age) have been used to probe how neuromodulators differentially affect plasticity at developing versus adult stages (20). Acute hippocampal slices have also been prepared from neonatal (PND 2–4) mice to study the ionic and synaptic basis of periodic population discharges known as interictal bursts, as well as the pacemaker mechanisms underlying interictal rhythmicity (21).
Hippocampal slice cultures fare best when they are derived from early postnatal rats or mice (postnatal days 4–10). The typical experience has been that as the age of the donor increases beyond postnatal day 7, long-term culture success decreases; however, there are reports of healthy organotypic slice cultures being obtained from adult animals (22-24).
All steps in this procedure should be performed in a laminar flow tissue culture hood using sterile equipment and good aseptic technique.
Gold particles come in a variety of sizes, and the size needs to be optimized for each particular instrument and biological system under investigation.
It has been the empirical observation of our colleague, Dr. Gary Wayman (Department of Veterinary and Comparative Anatomy, Pharmacology and Physiology, Washington State University, Pullman), that cDNA constructs which are driven by CMV promoters in hippocampal neurons show dramatic upregulation following stimuli, such as increased synaptic activity (bicuculline or KCl stimulation) or neurotrophic stimulation (BDNF) as well as other stimuli that elevate intracellular Ca2+ in hippocampal neurons. This can greatly complicate the interpretation of experiments in which these stimuli are used. In his experience, the pCAGGS vector gives the most consistent results with very slight to no modulation of the transgene expression under any culture or stimulation condition. The pCAGGS vector consists of the CMV immediate early enhancer and chicken β-actin promoter (25). When expressed in hippocampal or cortical neurons, stimuli, such as bicuculline or BDNF, do not significantly affect the expression of cDNAs contained with the pCAGGS vector. This vector has the added benefit of restricted expression. Within a mixed culture of neurons and glia, this expression vector only detectably expresses cDNA in neurons and not glia. A potential disadvantage of this vector may be that in neurons, it may not express cDNA inserts to the same high level in the first 12–48 h after transfection as seen when using a CMV driving expression system. Tomato fluorescent protein is preferred over other more traditionally used green fluorescent proteins because of its brighter expression which facilitates the imaging of individual neurons by confocal microscopy.
It is imperative that the dissections should be completed quickly to minimize the length of time between euthanasia of the animal and separation and exposure of cut tissue slices to culture medium. Prolonged delays result in deterioration of the tissue and reduced quality of the slices. A reasonable goal is to have cut slices separated in less than 5–10 min after the animal has been euthanized.
Keeping all dissection solutions cold and performing the entire procedure over ice help delay the deterioration of the tissue during dissection.
Depending on the tissue slicer available for your use and your specific application, slices ranging from 250 to 500 μm have been successfully used for electrical recordings. In some applications, the CA1 region can be separated from the CA3 area to minimize the spontaneous action potentials in the Schaffer collaterals in the CA1 area.
Slices can be stored in the prechamber (Fig. 1) for 4–6 h before their integrity is compromised resulting in unstable electrical recordings.
The interface chamber maintains the slice at the interface between the perfused ACSF fluid and the oxygenated atmosphere. With the interface configuration, oxygen is supplied to the slice by the warmed, humidified carbogen flowing immediately over the slice. By contrast, slices completely submerged in ACSF (submerged recording chamber configuration) are oxygenated by the perfused ACSF (26). Field potential recordings have been widely published using both slice configurations to study pharmacological responses and changes in neuroplasticity. However, some notable differences were revealed in systematic comparisons of how anoxia alters ion transport when hippocampal slices are maintained at a gas–liquid interface versus being completely immersed. Some of the factors underlying this difference include heterogeneous rates of glucose diffusion into the tissue slices and washout of accumulating substances, such as extracellular K+ during perfusion (14, 26). It is important to note that the responses typically measured with hippocampal slices maintained in either interface or submerged chambers can be differentially influenced by O2 deprivation and may be of pathophysiologic interest. For example, hippocampal slices maintained in the interface chamber were shown to recover their responses to othodromically evoked stimuli after they were subjected to anoxia, whereas submerged slices did not (26).
If the tissue is too wet, hippocampi tend to move each time the blade is lifted; however, if the tissue is too dry, hippocampi tend to stick to the blade.
Healthy hippocampal slices should have smooth edges and their surface should not appear sandy or grainy. The neuronal cell body layers should remain tight and transparent. If cultures are overfed and become flooded, they will become opaque. After 1 week in vitro, cultures initially cut at 400 μm normally thin down to about 150–200-μm thick. Underfed cultures thin out much sooner and become nearly invisible.
This solution can be stored at −20°C for up to 2 months if capped with Parafilm wrapped around the cap. Let it warm to room temperature before removing the Parafilm and opening the cap.
The gold microcarriers must be evenly distributed within the tubing for efficient transfection. Uneven labeling of the gold suspension inside the tube may be due to the PVP solution, which should be replaced every 6–8 weeks.
Cartridges can be kept for up to 1 year at 4°C if stored in a tightly capped vial wrapped with Parafilm.
The time and level of expression vary depending on the expression vector used, as discussed above in Note 7. With the pCAGGS vector, we typically see the expression of transfected proteins within 12 h with maximal expression at about 48 h. In a typical experiment, we obtain 20–100 transfected neurons per slice.
Acknowledgments
This work was supported by the NIH (grants U01 NS 057993 and R01 ES014901).
References
- 1.Lo DC, McAllister AK, Katz LC. Neuronal transfection in brain slices using particle-mediated gene transfer. Neuron. 1994;13(6):1263–8. doi: 10.1016/0896-6273(94)90412-x. [DOI] [PubMed] [Google Scholar]
- 2.Galimberti I, Gogolla N, Alberi S, Santos AF, Muller D, Caroni P. Long-term rear-rangements of hippocampal mossy fiber terminal connectivity in the adult regulated by experience. Neuron. 2006;50(5):749–63. doi: 10.1016/j.neuron.2006.04.026. [DOI] [PubMed] [Google Scholar]
- 3.Nagerl UV, Eberhorn N, Cambridge SB, Bonhoeffer T. Bidirectional activity-dependent morphological plasticity in hippocampal neurons. Neuron. 2004;44(5):759–67. doi: 10.1016/j.neuron.2004.11.016. [DOI] [PubMed] [Google Scholar]
- 4.Coltman BW, Earley EM, Shahar A, Dudek FE, Ide CF. Factors influencing mossy fiber collateral sprouting in organotypic slice cultures of neonatal mouse hippocampus. Comp Neurol. 1995;362(2):209–22. doi: 10.1002/cne.903620205. [DOI] [PubMed] [Google Scholar]
- 5.Gahwiler BH. Organotypic monolayer cultures of nervous tissue. J Neurosci Methods. 1981;4(4):329–42. doi: 10.1016/0165-0270(81)90003-0. [DOI] [PubMed] [Google Scholar]
- 6.De Simoni A, Yu LM. Preparation of organotypic hippocampal slice cultures: interface method. Nat Protoc. 2006;1(3):1439–45. doi: 10.1038/nprot.2006.228. [DOI] [PubMed] [Google Scholar]
- 7.Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37(2):173–82. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- 8.O’Brien JA, Lummis SC. Diolistic labeling of neuronal cultures and intact tissue using a hand-held gene gun. Nat Protoc. 2006;1(3):1517–21. doi: 10.1038/nprot.2006.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Brien JA, Lummis SC. Biolistic transfection of neuronal cultures using a hand-held gene gun. Nat Protoc. 2006;1(2):977–81. doi: 10.1038/nprot.2006.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kasri NN, Govek EE, Van Aelst L. Characterization of oligophrenin-1, a RhoGAP lost in patients affected with mental retardation: lentiviral injection in organotypic brain slice cultures. Methods Enzymol. 2008;439:255–66. doi: 10.1016/S0076-6879(07)00419-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Haas HL, Schaerer B, Vosmansky M. A simple perfusion chamber for the study of nervous tissue slices in vitro. J Neurosci Methods. 1979;1(4):323–5. doi: 10.1016/0165-0270(79)90021-9. [DOI] [PubMed] [Google Scholar]
- 12.Teyler TJ. The introduction of brain slices to neurophysiology. In: Schurr A, Teyler TJ, Tseng MT, editors. Brain Slices: Fundamentals, Applications and Implications. Karger; Basel: 1987. pp. 1–9. [Google Scholar]
- 13.Wong PW, Joy RM, Albertson TE, Schantz SL, Pessah IN. Ortho-substituted 2,2′,3,5′,6-pentachlorobiphenyl (PCB 95) alters rat hippocampal ryanodine receptors and neuroplasticity in vitro: evidence for altered hippocampal function. Neurotoxicology. 1997;18(2):443–56. [PubMed] [Google Scholar]
- 14.An JH, Su Y, Radman T, Bikson M. Effects of glucose and glutamine concentration in the formulation of the artificial cerebrospinal fluid (ACSF) Brain Res. 2008;1218:77–86. doi: 10.1016/j.brainres.2008.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kirchner A, Veliskova J, Velisek L. Differential effects of low glucose concentrations on seizures and epileptiform activity in vivo and in vitro. Eur J Neurosci. 2006;23(6):1512–22. doi: 10.1111/j.1460-9568.2006.04665.x. [DOI] [PubMed] [Google Scholar]
- 16.Cater HL, Chandratheva A, Benham CD, Morrison B, 3rd, Sundstrom LE. Lactate and glucose as energy substrates during, and after, oxygen deprivation in rat hippocampal acute and cultured slices. J Neurochem. 2003;87(6):1381–90. doi: 10.1046/j.1471-4159.2003.02100.x. [DOI] [PubMed] [Google Scholar]
- 17.Schurr A, Payne RS, Miller JJ, Rigor BM. Study of cerebral energy metabolism using the rat hippocampal slice preparation. Methods. 1999;18(2):117–26. doi: 10.1006/meth.1999.0765. [DOI] [PubMed] [Google Scholar]
- 18.Schurr A, West CA, Rigor BM. Electrophysiology of energy metabolism and neuronal function in the hippocampal slice preparation. J Neurosci Methods. 1989;28(1–2):7–13. doi: 10.1016/0165-0270(89)90004-6. [DOI] [PubMed] [Google Scholar]
- 19.Gureviciene I, Puolivali J, Pussinen R, Wang J, Tanila H, Ylinen A. Estrogen treatment alleviates NMDA-antagonist induced hippocampal LTP blockade and cognitive deficits in ovariectomized mice. Neurobiol Learn Mem. 2003;79(1):72–80. doi: 10.1016/s1074-7427(02)00012-6. [DOI] [PubMed] [Google Scholar]
- 20.Selbach O, Bohla C, Barbara A, Doreulee N, Eriksson KS, Sergeeva OA, Haas HL. Orexins/hypocretins control bistability of hippocampal long-term synaptic plasticity through co-activation of multiple kinases. Acta Physiol (Oxf) 2009 doi: 10.1111/j.1748-1716.2009.02021.x. [DOI] [PubMed] [Google Scholar]
- 21.Agmon A, Wells JE. The role of the hyperpolarization-activated cationic current I(h) in the timing of interictal bursts in the neonatal hippocampus. J Neurosci. 2003;23(9):3658–68. doi: 10.1523/JNEUROSCI.23-09-03658.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Krassioukov AV, Ackery A, Schwartz G, Adamchik Y, Liu Y, Fehlings MG. An in vitro model of neurotrauma in organotypic spinal cord cultures from adult mice. Brain Res Brain Res Protoc. 2002;10(2):60–8. doi: 10.1016/s1385-299x(02)00180-0. [DOI] [PubMed] [Google Scholar]
- 23.Leutgeb JK, Frey JU, Behnisch T. LTP in cultured hippocampal-entorhinal cortex slices from young adult (P25-30) rats. J Neurosci Methods. 2003;130(1):19–32. doi: 10.1016/s0165-0270(03)00228-0. [DOI] [PubMed] [Google Scholar]
- 24.Tom VJ, Doller CM, Malouf AT, Silver J. Astrocyte-associated fibronectin is critical for axonal regeneration in adult white matter. J Neurosci. 2004;24(42):9282–90. doi: 10.1523/JNEUROSCI.2120-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Niwa H, Yamamura K, Miyazaki J. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene. 1991;108(2):193–9. doi: 10.1016/0378-1119(91)90434-d. [DOI] [PubMed] [Google Scholar]
- 26.Croning MD, Haddad GG. Comparison of brain slice chamber designs for investigations of oxygen deprivation in vitro. J Neurosci Methods. 1998;81(1–2):103–11. doi: 10.1016/s0165-0270(98)00023-5. [DOI] [PubMed] [Google Scholar]