

Abstract
Background/Aim:
To evaluate the clinical manifestations, diagnostic features, disease course and response to treatment among Saudi adults with predominantly hepatic Wilson's disease. A retrospective cohort study of 40 adult patients diagnosed with predominantly hepatic Wilson's disease between 1994 and 2008 at King Abdulaziz Medical City, Riyadh was carried out.
Patients and Methods:
The diagnosis was based on varying combinations of clinical and laboratory evidence of liver disease, presence of Kayser Fleisher rings, low serum ceruloplasmin levels, elevated 24 hour urinary copper excretion and histopathological findings on liver biopsy.
Results:
The most frequent clinical presentation was decompensated chronic liver disease in 19 (47.5%), followed by chronic hepatitis in 15 (37.5%) and fulminant hepatic failure (FHF) in 5 (12.5%) patients. Eight (20%) patients with end-stage liver disease had liver transplantation, while 24 (60%) patients followed up on medical treatment for a variable period of 1-12 years showed clinical and laboratory improvement. One patient was lost early in follow up. Eight (20%) patients died during the study period, 5 with FHF, and 2 with advanced hepatic and neurological disease and one seven years after liver transplantation. Mortality rate was 100% in FHF without liver transplantation.
Conclusion:
A predominantly hepatic Wilson's disease has varied clinical presentations with decompensated chronic liver disease being the most common among adult patients. Majority of the patients show stabilization of the disease on medical treatment. FHF in Wilson's disease has a grave prognosis without liver transplantation, the later remains a definitive treatment option for decompensated cirrhotics and patients with FHF.
Keywords: Adult, Hepatic Wilson's disease, diagnosis, outcome, presentation
Wilson's disease (WD) is an Autosomal recessive hereditary disease that was initially described as Progressive Lenticular Degeneration associated with cirrhosis.[1] The gene responsible for WD was located to the long arm of chromosome 13 that encodes a copper-transporting p-type adenosine triphosphatase, ATP7b.[2,3] The disease is observed with a prevalence of approximately 1:30,000, with a corresponding gene frequency of 0.56% and a carrier frequency of about 1/90.[4] WD is characterized pathophysiologically by failure of ATP7b gene function that impairs incorporation of copper with apoceruloplasmin resulting in copper accumulation within hepatocytes. This results into copper-mediated hepatocyte damage and leakage of copper into plasma and eventual copper overload in most tissues accounting for variable clinical presentation of WD.[5] Early diagnosis is essential in order to prevent the progression of liver injury and the occurrence of disabling neurological complications. Medical therapy of WD includes dietary copper restriction, zinc[6] and chelators like D-penicillamine,[7] Trientine.[8] Advanced liver failure with decompensation and fulminant hepatitis remain major indications for liver transplantation.[9,10]
Review of literature revealed only few studies of small sample size among adults with hepatic Wilson's disease in Saudi Arabia.[11,12] We conducted a retrospective chart review of 40 patients among adults with predominantly hepatic Wilson's disease, to study various clinical presentations, biochemical and histopathological abnormalities, diagnostic features, disease course, complications and response to medical treatment and liver transplantation.
PATIENTS AND METHODS
We reviewed medical charts of 65 patients diagnosed as Wilson's disease between January 1994 and July 2008 at King Fahad National Guard Hospital. The study was approved by the Hospital research and ethical committee. All charts carrying ICD-9-CM Code No 275.1 allocated for Wilson's disease were retrieved from medical records section. 19 children (age less than 13 years) and 6 patients with pure neurological disease were excluded from study. Forty patients (22 females and 18 males) with age range 13-49 years and diagnosis of WD with predominant liver disease were included in the study. Patients were followed up for a period of 6.3 ± 2.7 years ranging from 1-12 years. Each chart was reviewed for various clinical features and laboratory abnormalities at initial presentation and during follow up, disease course, complications, and response to either medical or surgical treatment. History and physical findings of jaundice, ascites, hepatic encephalopathy, abnormal speech, tremors and abnormal movements documented in each chart were recorded. Slit lamp examination report on Kayser Fleischer (KF) rings was available in 35 patients. All patients had baseline complete blood counts, liver profile and later serial blood counts and liver profile during follow up. Blood samples were tested for hepatitis B and C serology, immune markers Anti-nuclear antibody (ANA), Anti-smooth muscle antibody (ASMA), Anti-mitochondrial antibody (AMA), Anti-liver kidney microsomal 1 antibody (Anti-LKM1), serum immmunoglobulins, alpha-1 antitrypsin levels and iron profile to exclude viral, autoimmune, and other metabolic liver diseases respectively. Serum ceruloplasmin levels were measured by quantitative immunoturbidimetric method with reference range (0.20-0.60g/L) and 24 hour urinary copper by atomic absorption spectrophotometry with reference range (0.047-0.55 μmol/ day). A transcutaneous or trans-jugular liver biopsy was performed in 35 patients, four patients refused liver biopsy and one patient died before obtaining liver biopsy. Liver biopsy slides were reviewed and reported by a single histopathologist who was not blinded to the diagnosis. Quantitative estimation of copper in liver tissue was not available at the time of study. MRI brain was performed in all patients who presented with mixed hepatic and neurological disease. Siblings of all index cases were screened by physical examination including slit lamp examination for KF rings, liver profile, serum ceruloplasmin levels and 24-hour urinary copper estimation. Liver biopsy was performed on subjects with suspected Wilson's disease. Genetic mutation analysis was done where ever necessary by isolating genomic deoxyribonucleic acid (DNA) from peripheral blood using standard method of QIAamp DNA protocol. The ATP7B coding sequence was screened for mutation by the mutation detection enhancement heteroduplex analysis and single strand conformation polymorphism analysis of the polymerase chain reaction using fluorescent labeled primers. The polymerase chain reaction (PCR) amplicons that showed abnormal migration or band shift relative to normal samples were directly sequenced to identify the mutations and polymorphism. Samples were analyzed on an ALF Express II (pharmacia Biotech) automated DNA sequencer.
All patients were treated with D-penicillamine (500-1500 mg/d) plus dietary restriction of copper rich foods. 10 patients with mixed hepatic and neurological disease were treated with zinc sulphate (100-150 mg/d) as an add-on therapy. Two patients were switched to trientine after developing hematological side effects with D-penicillamine. Adherence to treatment was evaluated by verbal enquiry, physical findings and liver profile on each clinical follow up visit.
Applying the diagnostic algorithm based on Leipzig score,[13] diagnosis of Wilson's disease was based on at least two of the following: Clinical and biochemical evidence of liver disease with or without neuropsychiatric illness, presence of KF rings and or a low serum ceruloplasmin level <0.20g/L. In patients without KF rings or with normal serum ceruloplasmin, diagnosis was based on elevated (>1.53 μmol/day) pre-treatment 24 hour urinary copper excretion, histopathological features of WD and genetic testing. 24 hour urinary copper excretion after D-pencillamine challenge was not estimated as our entire population of patients was adults in whom it is not validated or recommended.[14] Data suitable for analysis by parametric statistics were reported as summary statistics and expressed as percentage, range and mean ± SD. Student's t test was used for statistical analysis of continuous variables. A P value of <0.05 was considered significant.
RESULTS
Clinical features, presentation and biochemical results
The most common clinical presentation of hepatic WD was decompensated liver disease 19 (47.5%) followed by chronic hepatitis in 15 (37.5%) and FHF in 5 (12.5%) patients, [Table 1]. The most frequent clinical signs of decompensation were coagulopathy 23 (57.5%), low serum albumin 17 (42.5%) followed by ascites 14 (35%), jaundice 11 (27.5%) and hepatic encephalopathy 9 (22.5%). 10 (25%) patients had mixed neurological and hepatic disease. The most common neurological signs noted were tremors, slurred speech and rigidity. All Patients with neurological signs had positive KF rings, low serum ceruloplasmin and abnormal brain MRI mainly involving basal ganglia, midbrain, thalami and pons making diagnosis easy to establish on clinical features alone. Wilson's disease was diagnosed by family screening in six (15%) patients by clinical, biochemical testing plus liver histology and genetic testing. Overall KF rings were present in 29/35 (82.5%) and a low (0.043 ± 0.024 g/L) ceroloplasmin in 37 (92.5%) patients, however those with pure hepatic WD KF rings were seen in 19/25 (76%) and low ceruloplasmin in 27/30 (90%). All patients had high (4.48 ± 1.18 μmol/d) 24 hour urinary copper excretion The liver biochemical tests were abnormal in all patients with decompensated liver disease and chronic hepatitis at presentation with serum bilirubin of 171 ± 133 μmol/L, serum ALT 96 ± 80 U/L, AST 98 ± 88U/L, serum albumin 34 ± 11.4g/L and International normalization ratio (INR) 1.65 ± 0.84. Genetic test results were available in 23 patients. Among these 9 patients had a novel mutation in exon 21 (Gln1399Arg), not seen in normal Saudi subjects. No mutations were found in 14 patients. Applying Leipzig score, majority (37 patients) met the diagnostic criteria (score 4 or more) for Wilson's disease having low serum ceruloplasmin and markedly elevated 24 h urinary copper.
Table 1.
Clinical presentation and patient's characteristics

Among the six asymptomatic patients diagnosed by family screening 3 patients had low serum ceruloplasmin, elevated 24 h urinary copper and copper positive stains on liver biopsy. The other 3 patients with normal ceruloplasmin and no KF rings had elevated 24 h urinary copper levels plus gene mutations on genetic testing.
Although six patients had low titres of positive ANA (1:10 to 1:40) and seven patients had positive Anti-smooth muscle antibody (ASMA) (1:40 to 1:80), none of these patients had abnormal serum immunoglobulin and did not meet the criteria for diagnosis of autoimmune hepatitis. All patients were negative for AMA and for serological evidence of hepatitis B or C infection.
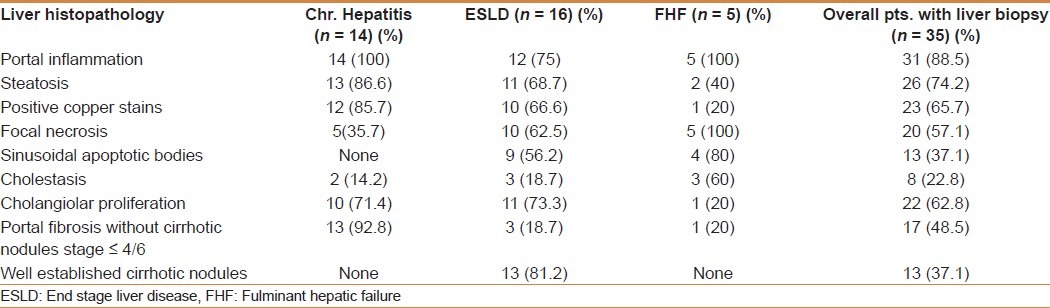
Liver histology among 35 patients showed portal fibrosis in 31 (88.5%), steatosis in 26 (74.2%), positive copper stains in 23 (65.7%), sinusoidal apoptotic bodies in 13 (37.1%), cholestasis in 8 (22.8%), focal necrosis in 20 (57.1%), cholangiolar proliferation in 22 (62.8%), portal fibrosis in 17 (48.5%) and cirrhotic nodules in 13 (37.1%) patients.
Correlating the results of the various histological findings with clinical presentation, the patients with clinical chronic hepatitis had portal inflammation in 100%, steatosis in 86.6%, positive copper stains in 85.7%, focal necrosis in 35%, cholangiolar proliferation in 71.4%, and varying stages of portal fibrosis in 92.8%. Among patients with clinical end stage liver disease portal inflammation was present in 75%, steatosis in 68.7%, positive copper stains in 66.6%, focal necrosis in 62.5%, cholangiolar proliferation in 73.3%, portal fibrosis without cirrhosis in 18.7% and cirrhotic nodules in 81.2%. Patients who presented with FHF had portal inflammation in 100%, focal necrosis in 100%, sinusoidal apoptotic bodies in 80%, cholestasis in 60%, and portal fibrosis in 20%. However, positive copper stains were seen in only 20% and cirrhotic nodules in none. The results are summarized in Table 2.
Table 2.
Correlation of clinical presentation to liver histology findings in (35) patients
Response to medical/surgical treatment and outcome
Among 24 (9 patients with decompensated liver disease and 15 patients with chronic hepatitis) non-fulminant and non-transplanted patients who responded to medical treatment, normalization of ALT was documented in 18 (75%), AST in 22 (92%), serum albumin in 21 (87.5%), bilirubin and INR in 24 (100%) patients. The mean time to normalization of transaminases was 12.4 ± 6.1 months (range 4-32 months) while the mean time to normalization of bilirubin, albumin and INR was 24.2 ± 13.4 months (range 11-60 months).
Eight decompensated cirrhotic patients (4 males and 4 females) who failed to respond to medical treatment had successful deceased donor liver transplantation. During post-transplantation follow up of 68.0 ± 43.1 months, the clinical symptoms and signs of Wilson's disease including KF rings resolved after a period of 9.4 ± 6.1months (3.5-18 months) and patients had consistently normal serum ceruloplasmin and liver profile without additional copper chelating therapy. One patient died seven years post-transplantation from a motor vehicle accident.
The 5 patients (2 males and 3 females) who presented with FHF had various grades of hepatic encephalopathy, jaundice and coagulopathy and died in ICU while receiving medical therapy and awaiting liver transplantation. All 5 patients developed acute renal failure requiring hemodialysis. Three of them had documented bacterial sepsis. One patient died from intrapulmonary hemorrhage.
Among patients with non-fulminant presentation, 2 patients who presented with advanced neurological disease with severe dysphagia and decompensated liver cirrhosis died with liver failure, pneumonia and sepsis. One patient was lost early in follow-up.
DISCUSSION
We studied only adult patients (> 13 years age) of Wilson's disease with predominant hepatic presentation at initial diagnosis. In our study, clinical evidence of end stage liver disease was seen in majority of the patients followed by chronic hepatitis, mixed hepatic and neurological disease and FHF. These observations are consistent with those of Steindl et al.[15] In a series of 55 patients reported by Steindl et al, 17 patients presented with clinical manifestations of chronic liver disease; 5 with fulminant hepatic failure, 20 with neurological disease, and 10 patients were detected by family screening.[15] Our youngest patient was 13 years and oldest one 49 years. Although WD is predominantly a disease of the young, many patients are first diagnosed in middle or even older age[16] and this was observed in our study as well. Patients with mixed decompensated liver disease and neurological disease were significantly older when compared with patients with FHF (30.8 ± 7.0 vs. 18.0 ± 3.0 P < 0.05). Fulminant liver failure was seen more often in young females. Several different manifestations associated with hepatic copper accumulation in WD have been reported.[15,17] We observed various complications related to liver failure as part of ESLD or FHF. Coagulopathy and ascites was the most common followed by jaundice and hepatic encephalopathy. About 37 (92.5%) of our patients had low ceruloplasmin at initial diagnosis which is again consistent with the results of other studies.[18–20] We identified novel genetic mutations in a minority of patients with Wilson's disease while a majority of those tested had no specific genetic mutations.
Overall histologically documented cirrhotic nodules were seen in 37.1% and fibrosis without cirrhosis in 48.5% of patients which is consistent with results of Stremmel study[18] that reported cirrhosis in 37% and fibrosis in 34% of patients. A significant degree of steatosis was seen in majority (74.2%) of patients mimicking that of nonalcoholic fatty liver disease. The same observation has been made recently in an Italian study on WD patients without metabolic co morbidities.[21] In our study patients who presented with ESLD, majority (81.2%) had established cirrhosis on liver biopsy, while patients with clinical chronic hepatitis, majority had variable degree of portal fibrosis on liver biopsy. The higher proportion of patients with positive histochemical copper stains on liver biopsy in our study not reported in other studies could be because of older age of our patients and advanced stage of liver disease. In cirrhotic patients, all changes in the pre-cirrhotic stage were seen with variable degree of copper deposition. The pathological changes in fulminant stage were variable degrees of fibrosis, predominant ballooning of hepatocytes, necrosis, apoptotic bodies, cholestasis, and a sparse lymphocytic and mononuclear infiltrate with negligible copper deposition. In our study, early in the course of the disease, patients had predominantly steatosis and portal inflammation (100%).
The observation of normalization of synthetic function and transaminases on medical treatment is indicative of reversible nature of the disease process, despite large number of patients who were histologically fibrotics and/or cirrhotics, although pathological confirmation of reversibility is lacking in this study. Medical management with chelating agents was effective in most of the patients with clinical chronic hepatitis and in almost 50% of patients with clinical ESLD. Normalization of liver enzymes occurred earlier followed by improvement in liver synthetic functions. This underscores the importance of early diagnosis and early administration of medical treatment in Wilson's disease.[22]
The result of medical therapy for patients who presented in fulminant hepatic failure was rather disappointing as none of our patients with FHF survived while awaiting liver transplantation. This (100%) mortality rate of patients with FHF in Wilson's disease warrants an early consideration of liver transplantation and early referral to a transplant centre. Sepsis was a leading cause of mortality in all patients.
CONCLUSION
In conclusion, hepatic Wilson's disease has varied clinical presentations with decompensated chronic liver disease being the most common among adult patients. Diagnosis still remains a clinical, biochemical and pathological correlation with no single test sufficient enough to make the diagnosis. Long term treatment with D-penicillamine/trientrine along with dietary copper restriction is effective in majority of patients in resolving the clinical and biochemical markers of the disease and improves prognosis and possibly survival. Patients who meet current criteria for liver transplantation should be listed and transplanted if there is deterioration or no improvement while on medical treatment. Fulminant liver failure in Wilson's disease without urgent transplantation has a dismal prognosis.
ACKNOWLEDGEMENT
The authors are grateful to Mrs. Lourdes Maristela for her help in preparing the final manuscript.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Wilson SA. Progressive lenticular degeneration: A familial nervous disease associated with cirrhosis of the liver. Lancet. 1912;1:1115–9. [Google Scholar]
- 2.Bearn A. A genetical analysis of 30 families with Wilson's disease. Ann Hum Genet. 1960;24:33–9. doi: 10.1111/j.1469-1809.1959.tb01713.x. [DOI] [PubMed] [Google Scholar]
- 3.Frydman M, Bonné-Tamir B, Farrer LA, Conneally PM, Magazanik A, Ashbel S, et al. Assignment of the gene for Wilson's disease to chromosome 13: Linkage to the esterase D locus. Proc Natl Acad Sci U S A. 1985;82:1819–21. doi: 10.1073/pnas.82.6.1819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Thomas GR, Forbes JR, Roberts EA, Walshe JM, Cox DW. The Wilson disease gene: Spectrum of mutations and their consequences. Nat Genet. 1995;9:210–7. doi: 10.1038/ng0295-210. [DOI] [PubMed] [Google Scholar]
- 5.Brewer GJ. Recognition, diagnosis and management of Wilson's disease. Proc Soc Exp Bio Med. 2000;223:39–46. doi: 10.1046/j.1525-1373.2000.22305.x. [DOI] [PubMed] [Google Scholar]
- 6.Brewer GJ. Treatment of Wilson's disease with Zinc. J Lab Clin Med. 1999;134:322–4. doi: 10.1016/s0022-2143(99)90213-5. [DOI] [PubMed] [Google Scholar]
- 7.Walshe JM. Penicillamine, a new oral therapy for Wilson's disease. Am J Med. 1956;21:487–92. doi: 10.1016/0002-9343(56)90066-3. [DOI] [PubMed] [Google Scholar]
- 8.Walshe JM. Treatment of Wilson's disease with trientine dihychrochloride. Lancet. 1982;1:643–7. doi: 10.1016/s0140-6736(82)92201-2. [DOI] [PubMed] [Google Scholar]
- 9.Bellamy S, Hassanein T, Van Thiel HD. Liver transplantation for Wilson's disease: Indications and outcomes. Hepatology. 1994;19:583–7. doi: 10.1002/hep.1840190307. [DOI] [PubMed] [Google Scholar]
- 10.Emre S, Atillasoy EO, Ozdemir S, Schilsky M, Rathna Varma CV, Thung SN, et al. Orthotopic liver transplantation for Wilson's disease: A single centre experience. Transplantation. 2001;72:1232–6. doi: 10.1097/00007890-200110150-00008. [DOI] [PubMed] [Google Scholar]
- 11.al Mofleh IA, al Rashed RS, Ayoola EA, Sabah DM, Hafeez MA. Hepatic manifestations of Wilson's disease: Frequency and pattern in Saudi patients. Trop Gastroenterol. 1993;14:91–8. [PubMed] [Google Scholar]
- 12.Bahemuka M, Karrar ZA, Mofleh IA, Bahakim H, Hafeez MA. Protean manifestations of Wilson's disease: A review of seven Saudi patients. Trop Geogr Med. 1988;40:131–8. [PubMed] [Google Scholar]
- 13.Ferenci P, Caca K, Loudianos G, Mieli-Vergani G, Tanner S, Sternlieb I, et al. Diagnosis and phenotypic classification of Wilson's disease. Liver Int. 2003;23:139–42. doi: 10.1034/j.1600-0676.2003.00824.x. [DOI] [PubMed] [Google Scholar]
- 14.Ferenci P, Czlonkowska A, Stremmel W, Houwen R, Rosenberg W, Schilsky M, et al. EASL clinical practice guidelines: Wilson's disease. J Hepatol. 2012;56:671–85. doi: 10.1016/j.jhep.2011.11.007. [DOI] [PubMed] [Google Scholar]
- 15.Steindl P, Ferenci P, Dienes HP, Grimm G, Pabinger I, Madl C, et al. Wilson's disease in patient presenting with liver disease: A diagnostic challenge. Gastroenterology. 1997;113:212–8. doi: 10.1016/s0016-5085(97)70097-0. [DOI] [PubMed] [Google Scholar]
- 16.Ferenci P, Członkowska A, Merle U, Ferenc S, Gromadzka G, Yurdaydin C, et al. Late onset Wilson disease. Gastroenterology. 2007;132:1294–8. doi: 10.1053/j.gastro.2007.02.057. [DOI] [PubMed] [Google Scholar]
- 17.Sternlieb I. Perspectives on Wilson's disease. Hepatology. 1990;12:1234–9. doi: 10.1002/hep.1840120526. [DOI] [PubMed] [Google Scholar]
- 18.Brewer GJ, Yuzbasiyan-Gurkan V. Wilson's disease. Medicine (Baltimore) 1992;71:139–64. doi: 10.1097/00005792-199205000-00004. [DOI] [PubMed] [Google Scholar]
- 19.Stremmel W, Meyerrose KW, Niederau C, Hefter H, Kreuzpaintner G, Strohmeyer G. Wilson's disease: Clinical presentation, treatment and survival. Ann Intern Med. 1991;115:720–6. doi: 10.7326/0003-4819-115-9-720. [DOI] [PubMed] [Google Scholar]
- 20.Gibbs K, Walshe JM. A study of caeruloplasmin concentration found in 75 patients with Wilson's disease, their kinships and various control groups. Q J Med. 1979;48:447–63. [PubMed] [Google Scholar]
- 21.Liggi M, Murgia D, Civolani A, Demelia E, Sorbello O, Demelia L. The relationship between copper and steatosis in Wilson's disease. Clin Res Hepatol Gastroenterol. 2012 doi: 10.1016/j.clinre.2012.03.038. [In press] [DOI] [PubMed] [Google Scholar]
- 22.Roberts EA, Schilsky ML. Diagnosis and treatment of Wilson disease: An update. Hepatology. 2008;47:2089–111. doi: 10.1002/hep.22261. [DOI] [PubMed] [Google Scholar]