

Abstract
Aims
Age-related diastolic dysfunction has been attributed to an increased passive stiffness, which is regulated by extracellular matrix (ECM). We recently showed that matrix metalloproteinase (MMP)-9, an ECM mediator, increases in the left ventricle (LV) with age. The aim of this study, accordingly, was to determine the role of MMP-9 in cardiac ageing.
Methods and results
We compared LV function in young (6–9 months), middle-aged (12–15 months), old (18–24 months) and senescent (26–34 months) wild-type (WT) and MMP-9 null mice (n ≥ 12/group). All groups had similar fractional shortenings and aortic peak velocities, indicating that systolic function was not altered by ageing or MMP-9 deletion. The mitral ratios of early to late diastolic filling velocities were reduced in old and senescent WT compared with young controls, and this reduction was attenuated in MMP-9 null mice. Concomitantly, the increase in LV collagen content was reduced in MMP-9 null mice (n = 5-6/group). To dissect the mechanisms of these changes, we evaluated the mRNA expression levels of 84 ECM and adhesion molecules by real-time qPCR (n = 6/group). The expression of pro-fibrotic periostin and connective tissue growth factor (CTGF) increased with senescence, as did transforming growth factor-β (TGF-β)-induced protein levels and Smad signalling, and these increases were blunted by MMP-9 deletion. In senescence, MMP-9 deletion also resulted in a compensatory increase in MMP-8.
Conclusion
MMP-9 deletion attenuates the age-related decline in diastolic function, in part by reducing TGF-β signalling-induced periostin and CTGF expression and increasing MMP-8 expression to regulate myocardial collagen turnover and deposition.
Keywords: Ageing, Collagen, Diastolic function, Matrix metalloproteinase, Extracellular matrix
1. Introduction
Ageing increases the prevalence of cardiovascular disease and is a major risk factor for cardiac morbidity and mortality.1,2 Even in the absence of concomitant cardiovascular disease, ageing results in declined diastolic function, whereas systolic function is relatively preserved.3
Diastolic function is regulated by the combined active relaxation properties of the myocyte and the passive stiffness properties of the myocardium. Extracellular matrix (ECM) provides structural support to the heart, and ECM quantity and quality are the major determinants of myocardial passive stiffness. Matrix metalloproteinases (MMPs) are a key family of zinc-dependent enzymes that degrade ECM and regulate ECM turnover. MMPs, therefore, play an important role in regulating diastolic function. Martos et al.4 demonstrated increased collagen turnover in 32 patients with diastolic heart failure, compared with 54 control patients, and the increase in collagen turnover was concomitant with increased MMP-9 levels in the plasma of these patients.
MMP-9 processes both ECM substrates, including denatured collagens, fibronectin and laminin, as well as non-ECM substrates, including interleukin (IL)-1β, IL-6, and latent transforming growth factor-beta (TGF-β).5–7 By acting on a wide range of substrates, MMP-9 regulates the pathogenesis of many diseases, including cardiac remodelling.6 Ducharme et al.8 showed that MMP-9 null mice have attenuated dilation of the left ventricle (LV) and reduced collagen accumulation after myocardial infarction, despite compensatory increases in other MMPs. However, whether MMP-9 deletion regulates collagen accumulation in diastolic heart failure or cardiac ageing models is unclear.
Our group has recently showed that MMP-9 levels increase in the LV and plasma of ageing mice.9 Although we know that LV MMP-9 levels increase and diastolic function decreases with age, the link between MMP-9 and diastolic function in the normal ageing process has not been established. We hypothesized that MMP-9 deletion will reduce collagen accumulation and attenuate diastolic dysfunction with ageing. Cardiac ageing phenotypes in four age groups of wild-type (WT) and MMP-9 null mice were compared in this study to address our hypothesis.
2. Methods
2.1. Animals
All animal procedures were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Research Council, National Academy Press, Washington, DC, 1996) and were approved by the Institutional Animal Care and Use Committee at the University of Texas Health Science Center at San Antonio.
2.2. Mice
C57BL/6J wild-type (WT) young (6–9 months), middle-aged (MA; 12–15 months), old (18–24 months), and senescent (26–34 months) male and female mice were used (n ≥ 12/group). MMP-9 null male and female mice of the four corresponding ages were used (n ≥ 12/group) to determine the effect of MMP-9 on cardiac ageing. MMP-9 null mice were a gift from Dr Zena Werb through Dr Lynn Matrisian, whose laboratory backcrossed the mice onto the C57/BL6J strain.10,11
2.3. Blood pressure
Systolic and diastolic blood pressures in mice were measured with the MC4000 Blood Pressure Analysis System (Hatteras Instruments). Each mouse was trained and acclimated to the machine, and training sessions were repeated until the standard deviations of both systolic and diastolic blood pressures were <10 mmHg and at least 6 of the 10 measurement cycles were recorded. Blood pressure was measured by the tail-cuff system, with five preliminary cycles measured, followed by 10 measurement cycles. The average systolic and diastolic pressures of the measurement cycles were determined.
2.4. Doppler echocardiography
To measure the transmitral inflow (the blood flow from the left atrium into the LV via the mitral valve) and aortic outflow, Doppler echocardiography was performed using the Doppler Signal Processing Workstation (Indus Instruments). The mice were anaesthetized with isoflurane (0.5–2% in a 100% oxygen mix), and the four limbs were taped to the electrode plates on a board for monitoring electrocardiograms and heart rates. Here, and elsewhere anaesthesia was used, the depth of anaesthesia was monitored through two methods. A periodic tail pinch was used to assess the pain response of the mouse and confirm a sufficient depth of anaesthesia. Also, while under anaesthesia, the heart rate, respiratory rate, and temperature were monitored to ensure the mouse was not too deeply anaesthetized to alter physiological variables. These methods were recommended by the Guide for the Care and Use of Laboratory Animals. The transmitral inflow and the aortic outflow were recorded. The E/A ratio, which is an indicator of LV diastolic function, was calculated from the early (E) filling velocity and the atrial (A) filling velocity.
2.5. Two-dimensional echocardiography
Echocardiograms were acquired with a Vevo 770™ High-Resolution In Vivo Imaging System (Visual Sonics). Isoflurane (0.5–2% in a 100% oxygen mix) was used to anaesthetize the mouse, which was stationed on an isothermal pad. Electrocardiograms and heart rates were continuously monitored throughout the procedure, and heart rates were maintained at ≥400 b.p.m. for baseline imaging. From a transthoracic approach, two-dimensional-targeted M-mode echocardiographic recordings were obtained.
For a subset of mice, after acquiring baseline values, temporary cardiac stress was induced by administering dobutamine, a β-adrenergic receptor agonist that increases the heart rate by stimulating myocyte contractility. Dobutamine was given at a dose of 3 µg/g body weight (BW) by ip injection.12 Echocardiograms were recorded at 30 min after injection, to determine the magnitude of response.
2.6. Tissue collection and characterization
At least 24 h after dobutamine echocardiography, to allow ample time for the dobutamine to fully wash out, the mice were sacrificed and the LV was harvested. Mice were sacrificed under isoflurane anaesthesia (2–5% in a 100% oxygen mix). At sacrifice, heparin (100 μL of 1000 USP Units/mL) was injected ip, and 5 min after heparin injection, blood was collected from the carotid artery of the mouse. The heart was flushed with cardioplegic solution and removed. The LV was separated from the right ventricle, each ventricle was weighed separately, and the LV was sectioned into three slices: base, mid, and apex. The base was snap frozen and used for RNA extraction; the mid was fixed in zinc formalin and used for histology; and the apex was snap frozen and used for protein extraction. The largest lobe of the lung was weighed before and after drying overnight at 50°C to determine the percentage of the water content in the lung. The BW and the tibia length were also recorded, and the LV/tibia ratio was calculated.
2.7. Picrosirius red staining
Paraffin-embedded mid-LV sections were deparaffinized with Citric-Solv (Fisherbrand) and rehydrated in distilled water. The sections were incubated in 0.2% of phosphomolybdic acid (Electron Microscopy Sciences) for 2 min to eliminate cytoplasmic staining. After rinsing with distilled water, the sections were stained with 0.1% Siris Red in saturated picric acid (Electron Microscopy Sciences) for 90 min. The stained sections were washed in 0.01 N hydrochloric acid (Electron Microscopy Sciences) for 2 min, rinsed in 70% ethanol for 45 s, dehydrated and mounted. For each LV section, five ×40 magnification images were captured. The percentage of the collagen area was measured by Image-Pro Plus version 6.2.
2.8. LV RNA extraction and real-time qPCR analysis
Total RNA was isolated from the LV base using the TRIzol reagent plus Total RNA purification kit (Invitrogen), and cDNA was synthesized using the SABiosciences RT2 first strand kit (C-03). To assess mRNA expression of ECM molecules in the LV, the RT2 qPCR Primer Array for Extracellular Matrix and Adhesion Molecules (SABiosciences PAMM-013A) was used for quantitative real-time PCR analysis. The relative expression of individual target genes was calculated by normalization of the Ct values of the target gene to the average Ct of five housekeeping genes (HKG), which were Gusb, Hprt1, Hsp90ab1, Gapdh, and Act, and the data were reported as the 2−ΔCT value. Before being accepted for normalization, all five HKGs among the eight groups were analyzed by ANOVA. No differences were seen between any of the groups, which were therefore all used for normalization.
2.9. LV protein extraction and immunoblotting
Total protein was extracted from the LV base by homogenizing in 400 μL of protein extraction buffer (Sigma; Protein Extraction Reagent Type 4; 7 M urea, 2 M thiourea, 40 mM Trizma® base and the detergent 1% C7BzO) with 1× Complete Protease Inhibitor Cocktail (Roche). Immunoblotting was performed as previously described with n = 12 for each group.9 Protein samples (10 µg total protein) were resolved on a 4–12% Criterion Bis–Tris gel (Bio-Rad) in XT MES buffer (Bio-Rad) and transferred to a nitrocellulose membrane (Bio-Rad). A 1:5000 dilution of MMP-9 antibody (Abcam ab38898), 1:1000 dilution of MMP-8 antibody (Epitomics), 1:1000 for neutrophil antibody (Cedarlane), 1:1000 of TGF-βi antibody (Abcam), and 1:1000 for Smad2 antibody (Cell Signaling), and 1:500 for p-Smad2 antibody (Abcam) was used for primary antibody detection. A 1:5000 dilution of anti-rabbit IgG (Vector PI-1000) was used for secondary antibody detection. The membrane was incubated in SuperSignal West Pico Chemiluminescent Substrate (Thermo Scientific) for 3 min and exposed to BioMAX MR film (Kodak). The film was scanned on a Kodak Image Station and densitometry of each lane was quantified by Molecular Imaging Software (Carestream). To confirm equal loading, the nitrocellulose membrane was stained with the MemCodeTM Reversible Protein Stain (Thermo Scientific) for total protein, and densitometries across the groups were compared (P = not significant for most groups). For groups that had a P < 0.1, densitometries were normalized using the total protein values.
2.10. Statistical analysis
Data were expressed as mean ± SEM. For necropsy, echocardiographic, collagen area fraction, real-time qPCR, and MMP-9 immunoblotting data, the one-way ANOVA test was used to compare values among all groups, and the Student Newman–Keuls test was used for pairwise comparisons. For MMP-8 immunoblotting, the unpaired Student's t-test was used for the two group comparison. A value of P < 0.05 was considered statistically significant. GraphPad Prism 5 was used for the statistical analyses.
3. Results
3.1. MMP-9 expression increased in old and senescent WT mice
Our group recently demonstrated higher MMP-9 levels in the LV of senescent C57BL/6J mice compared with young counterparts.9 To determine the time-course of MMP-9 expression with ageing, we compared MMP-9 mRNA and protein levels in the four age groups of WT mice. By real-time qPCR, old and senescent mice exhibited increased MMP-9 mRNA levels compared with young and MA controls (Figure 1A). For protein expression, immunoblotting revealed increased MMP-9 protein levels in the LV of senescent mice compared with the other three groups (Figure 1B and C). Of note, we did not see the same increase in MMP-9 protein in old mice that we saw in the senescent mice. This variability in mRNA and protein levels suggests that there might be more MMP-9 protein turnover in the old age group mice compared with the senescent group. MMP-9 levels were similar between male and female mice in all age groups (P = 0.52, 0.79, 0.89, and 0.71 for young, MA, old, and senescent, respectively).
Figure 1.

MMP-9 expression increases with ageing in the LV of WT mice. (A) By real-time qPCR, MMP-9 levels increase in old and senescent LV compared with young and MA controls. (B) A representative immunoblot showing that LV MMP-9 protein levels increase with senescence. (C) A graph showing the densitometry results of the MMP-9 immunoblot. Sample sizes are n = 6 per group for real-time qPCR and n = 12 per group for immunoblotting *P < 0.05 compared with the young control; +P < 0.05 compared with the MA control; #P < 0.05 compared with the old control.
3.2. Ageing WT and MMP-9 null mice did not show heart failure phenotypes
WT and MMP-9 null mice of all four age groups had similar LV mass and LV/tibia ratios (Table 1). Senescent WT and MMP-9 null mice had higher LV/BW ratios compared with corresponding young, MA, and old controls, and senescent MMP-9 null mice had higher LV/BW ratios than senescent WT mice (all P < 0.05). However, these changes in LV/BW were attributed to the reduced BW seen in the older groups, rather than an increase in the LV mass. In addition, all eight groups of mice exhibited similar RV mass (Table 1). These results indicated that there was no difference in LV and RV hypertrophy in these mice. Percentages of the water content in the lung were also similar among all eight groups (Table 1), indicating that there was no overt heart failure phenotype in these mice.
Table 1.
Necropsy data of four age groups of WT and MMP-9 null mice
| WT young (n = 15) | WT MA (n = 14) | WT old (n = 19) | WT senescent (n = 26) | MMP-9 null young (n = 17) | MMP-9 null MA (n = 14) | MMP-9 null old (n = 17) | MMP-9 null senescent (n = 27) | |
|---|---|---|---|---|---|---|---|---|
| LV mass (mg) | 80.6 ± 3.4 | 81.0 ± 3.7 | 87.0 ± 2.9 | 85.9 ± 2.6 | 74.9 ± 3.4 | 84.6 ± 3.7 | 83.9 ± 2.9 | 84.8 ± 3.1 |
| Body weight (mg) | 24.8 ± 0.9 | 25.6 ± 1.0 | 28.0 ± 0.9 | 24.4 ± 0.8 | 25.8 ± 0.9 | 27.3 ± 1.2 | 27.8 ± 1.1 | 22.6 ± 0.8*,** |
| LV/tibia (mg/mm) | 4.64 ± 0.19 | 4.66 ± 021 | 4.85 ± 0.15 | 4.87 ± 0.14 | 4.79 ± 0.23 | 5.06 ± 0.22 | 5.00 ± 0.16 | 4.98 ± 0.17 |
| RV mass (mg) | 16.8 ± 0.9 | 18.3 ± 0.7 | 19.8 ± 0.7 | 17.7 ± 0.9 | 16.9 ± 0.7 | 18.4 ± 0.7 | 19.3 ± 0.6 | 17.8 ± 0.6 |
| Lung % water | 78.0 ± 1.0 | 78.0 ± 0.9 | 78.8 ± 0.8 | 78.8 ± 0.5 | 76.4 ± 0.9 | 76.8 ± 0.9 | 76.2 ± 0.5 | 78.8 ± 0.5 |
Values are mean ± SEM.
LV/tibia, LV mass to tibia length ratio; RV, right ventricle.
*P < 0.05 vs. MA null.
**P < 0.05 vs. Old null.
3.3. Ageing and MMP-9 deletion did not affect blood pressure
WT and MMP-9 null mice of all four age groups had similar systolic and diastolic blood pressures (Table 2). No gender-associated difference was observed in systolic and diastolic pressures in any age group (all P = not significant, n ≥ 5 for each gender). The systolic and diastolic pressures of all eight groups were much lower than the corresponding hypertensive levels seen with angiotensin II infusion, where systolic blood pressure was reported to be over 160 mmHg and diastolic blood pressure was reported to be over 110 mmHg.13,14 This result indicates that both WT and MMP-9 null mice did not develop hypertension with age.
Table 2.
Blood pressure does not change with age in WT or MMP-9 null mice
| WT young (n = 21) | WT MA (n = 14) | WT old (n = 19) | WT senescent (n = 13) | MMP-9 null young (n = 26) | MMP-9 null MA (n = 14) | MMP-9 null old (n = 13) | MMP-9 null senescent (n = 12) | |
|---|---|---|---|---|---|---|---|---|
| Systolic BP | 108 ± 2 | 110 ± 1 | 108 ± 2 | 105 ± 3 | 106 ± 2 | 106 ± 2 | 101 ± 3 | 103 ± 3 |
| Diastolic BP | 94 ± 2 | 95 ± 2 | 95 ± 2 | 90 ± 3 | 89 ± 2 | 88 ± 2 | 88 ± 3 | 87 ± 2 |
Values are mean ± SEM.
BP, blood pressure (mmHg). No blood pressure measurements were significantly different (all P = not significant).
3.4. Systolic function was preserved in ageing mice and was not altered by MMP-9 deletion
Unlike diastolic function, systolic function is relatively preserved with ageing in humans.3 From two-dimensional echocardiography, fractional shortening (FS) and end-diastolic dimension were similar among all age groups of WT and MMP-9 null mice (Table 3). MA WT and MMP-9 null mice showed increased posterior wall thickness compared with their corresponding young controls (both P < 0.05; Table 3). At 30 min after dobutamine injection, senescent WT mice showed reduced FS compared with old WT mice (P < 0.05; Figure 2A), indicating reduced cardiac reserve in senescent WT mice. Systolic function was further evaluated by aortic Doppler echocardiography, and similar aortic peak velocities were detected among all age groups of WT and MMP-9 null mice (Figure 2B).
Table 3.
Baseline echocardiographic parameters of four age groups of WT and MMP-9 null mice
| WT young (n = 15) | WT MA (n = 14) | WT old (n = 19) | WT senescent (n = 24) | MMP-9 null young (n = 17) | MMP-9 null MA (n = 14) | MMP-9 null old(n = 17) | MMP-9 null senescent (n = 26) | |
|---|---|---|---|---|---|---|---|---|
| HR (b.p.m.) | 453 ± 5 | 449 ± 2 | 458 ± 3 | 471 ± 8 | 454 ± 5 | 454 ± 1 | 461 ± 6 | 453 ± 8 |
| EDD (mm) | 3.33 ± 0.09 | 3.34 ± 0.09 | 3.49 ± 0.07 | 3.46 ± 0.09 | 3.51 ± 0.05 | 3.26 ± 0.09 | 3.45 ± 0.07 | 3.56 ± 0.09 |
| ESD (mm) | 1.99 ± 0.09 | 1.98 ± 0.10 | 2.15 ± 0.08 | 2.23 ± 0.09 | 2.20 ± 0.05 | 1.94 ± 0.09 | 2.11 ± 0.06 | 2.26 ± 0.10 |
| FS (%) | 41 ± 1 | 41 ± 2 | 39 ± 2 | 36 ± 1 | 37 ± 1 | 41 ± 2 | 39 ± 1 | 37 ± 1 |
| IVSd (mm) | 0.80 ± 0.03 | 0.93 ± 0.04* | 0.81 ± 0.03** | 0.84 ± 0.02 | 0.78 ± 0.03 | 0.97 ± 0.03* | 0.85 ± 0.04** | 0.84 ± 0.02** |
Values are mean ± SEM.
HR, heart rate; EDD, end-diastolic dimension; ESD, end-systolic dimension; FS, fractional shortening; IVSd, LV interventricular septal wall thickness at diastole.
*P < 0.05 vs. corresponding young controls.
**P < 0.05 vs. corresponding MA.
Figure 2.

MMP-9 deletion attenuates the age-related decline in diastolic function. (A) At 30 min after dobutamine injection, FS of MMP-9 null mice are similar to their age-matched WT, whereas senescent WT mice have reduced FS compared with old WT mice. Sample sizes are n ≥ 11/group. (B) Aortic peak velocities are not statistically different with age and are similar between WT and MMP-9 null mice (all P = not significant). (C) E/A ratios reduce with age in WT but not MMP-9 null mice, and old and senescent MMP-9 null mice have higher E/A ratios compared with age-matched WT mice. *P < 0.05 compared with the young control; +P < 0.05 compared with the MA control; #P < 0.05 compared with the old control; ^P < 0.05 compared with age-matched WT controls. Sample sizes are n ≥ 12/group.
3.5. MMP-9 deletion attenuated decline in diastolic function with ageing
Ageing is associated with diastolic functional decline in both humans and mice.15–18 To evaluate the effect of MMP-9 deletion on diastolic properties of the LV, transmitral Doppler echocardiography was performed. The mitral ratios of early to late diastolic filling velocities (E/A ratios) were reduced in old and senescent WT mice, compared with young WT mice (both P < 0.05). Senescent WT mice also showed reduced E/A ratios, compared with MA and old WT mice (both P < 0.05). Strikingly, MMP-9 null mice of all age groups showed similar E/A ratios, and old and senescent MMP-9 null mice had higher E/A ratios than their age-matched WT controls (both P < 0.05; Figure 2C). No gender-associated difference in the E/A ratio was observed (all P = not significant, n ≥ 6 for each gender).
3.6. Senescent MMP-9 null mice showed reduced collagen deposition in the LV
To evaluate if changes in diastolic function by ageing and MMP-9 were due to changes in collagen deposition, picrosirius red staining was performed to evaluate the collagen content in the LV of young and senescent WT and MMP-9 null mice. The two age groups, young and senescent, were studied because they displayed the most prominent difference in diastolic function. Picrosirius red staining revealed increased collagen deposition in the LV of senescent WT and MMP-9 null mice, compared with their corresponding young controls. Surprisingly, the collagen content in the LV of senescent MMP-9 null mice was significantly lower compared with senescent WT control (n = 5 or 6/group; Figure 3A and B).
Figure 3.

MMP-9 deletion attenuates the increase in the LV collagen content with senescence. (A) Representative pictures of picrosirius red staining of LV mid-cavity sections of young and senescent WT and MMP-9 null mice. (B) A graph of percentages of the collagen area showing an increased collagen content in the senescent WT and MMP-9 null LV compared with corresponding young controls. Additionally, senescent MMP-9 null LV have less collagen content than senescent WT LV. Sample sizes are n = 5 or 6/group. *P < 0.05 compared with the young control; ^P < 0.05 compared with age-matched WT control. Sample sizes are n = 6/group.
3.7. MMP-9 deletion attenuated the age-related increase in periostin and CTGF expression
To dissect the underlying mechanism of how MMP-9 deletion regulates the LV collagen content and diastolic function, the age-related changes in expression levels of 84 ECM and adhesion molecules in WT and MMP-9 null LV were evaluated by the RT2 qPCR array (see the Supplementary material online, Table S1 for the complete data set). Despite the increased total collagen deposition seen in the LV with ageing, mRNA levels of type I and type III collagen did not increase in senescent WT and MMP-9 null mice, compared with their corresponding young controls (Figure 4A and B). By real-time qPCR, periostin mRNA levels increased 85% in senescent WT (n = 5) compared with young WT (n = 6; P < 0.05), and senescent MMP-9 null LV had a 48% reduction in periostin mRNA expression compared with senescent WT LV (P < 0.05; Figure 4C). Connective tissue growth factor (CTGF) mRNA levels increased 57% in senescent WT (n = 5) compared with young WT (n = 6; P < 0.05). Senescent MMP-9 null LV expressed 26% lower levels of CTGF than senescent WT LV (P < 0.05; Figure 4D). This indicates that the increased collagen with ageing observed by picrosirius red is more likely due to translational or post-translational regulation, rather than increased transcription.
Figure 4.

MMP-9 deletion attenuates the age-related increases in periostin and CTGF mRNA expression. (A) Type I collagen mRNA expression levels decrease, rather than increase, with senescence in WT mice. (B) Senescent MMP-9 null mice express reduced levels of type III collagen mRNA compared with the corresponding young, MA and old controls. (C and D) Senescent WT mice express increased levels of periostin (C) and CTGF (D) compared with young WT mice, and senescent MMP-9 null mice express reduced levels of periostin and CTGF compared with senescent WT mice. *P < 0.05 compared with the young control; +P < 0.05 compared with the MA control; #P < 0.05 compared with the old control; ^P < 0.05 compared with age-matched WT control. Sample sizes are n = 6/group.
3.8. MMP-9 deletion attenuated the age-related increase in TGF-β-induced protein transcription and protein levels
MMP-9 deletion attenuated the age-associated increases in periostin and CTGF expression. Periostin and CTGF expression are both TGF-β-inducible, and MMP-9 can cleave and activate latent TGF-β.19–22 By real-time qPCR, mRNA levels of transforming growth factor β (TGF-β)-induced protein (tgfbi) increased with senescence in WT mice, and the increase was attenuated in MMP-9 null mice (Figure 5A). Similar results were seen for protein levels of tgfbi. Levels were increased in senescent WT mice compared with all younger WT groups, while this increase was attenuated in the senescent null mice (Figure 5B).
Figure 5.

(A) By real-time qPCR, the gene expression of tgfbi increased with senescence in WT mice, but this increase was attenuated in MMP-9 null mice. (B) The increase of tgfbi protein levels in WT senescent mice was attenuated in the null senescent mice. (C) By immunoblotting, the psmad2/smad2 ratio increased in the WT senescent group, and this increase was attenuated in the null senescent LV. *P < 0.05 compared with the young control; +P < 0.05 compared with the MA control; #P < 0.05 compared with the old control; ^P < 0.05 compared with age-matched WT control. Sample sizes are n = 6/group.
3.9. MMP-9 deletion attenuated Smad2 phosphorylation
The ratio of phosphorylated Smad2 to Smad2 was increased in the senescent WT group, compared with each of the three younger age groups, and this increase was attenuated in the senescent null group (P < 0.05 for all; Figure 5C).
3.10. MMP-9 deletion resulted in a compensatory increase in MMP-8 expression
MMP-9 deletion has previously been shown to lead to a compensatory increase in MMP-2 and MMP-13 post-myocardial infarction.8 By real-time qPCR, there was no change in MMP-2 levels in any of the groups, but there was a compensatory increase in MMP-13 mRNA levels in young MMP-9 null mice (2.8-fold increase compared with young WT mice). Despite having similar levels in the young mice, MMP-8 mRNA levels were significantly higher in the LV of senescent MMP-9 null mice compared with that of age-matched WT mice (Figure 6A). Additionally, MMP-8 immunoblotting revealed a similar increase in MMP-8 protein levels in senescent MMP-9 LV compared with WT controls (Figure 6B and C). To explore the possibility that increased neutrophil infiltration explained the MMP-8 increase, we evaluated levels of the neutrophil marker Ly-6G by immunoblotting. Although there were no statistical differences among the age groups or genotypes, there was a small but positive linear correlation between age and neutrophil infiltration in the null mice (R = 0.13, P = 0.01). Neutrophils, therefore, in addition to macrophages, are likely a source of the increase in MMP-8 in the MMP-9 null mice. The increase in MMP-8 would increase collagen degradation rates and contribute to reduced LV collagen deposition.
Figure 6.

MMP-9 deletion led to a compensatory increase in MMP-8 expression in senescence. (A) MMP-8 mRNA levels increased with senescence in MMP-9 null mice, and senescent MMP-9 null mice had higher MMP-8 mRNA levels than age-matched WT control. (B) Immunoblotting comparing MMP-8 protein levels in senescent WT and MMP-9 null mice. (C) Densitometry results showed higher MMP-8 levels in senescent MMP-9 null LV compared with WT senescent LV. Sample sizes are n = 12/group. (D) Ly-6G immunoblotting showed no significant changes in neutrophil levels with age (WT P = 0.78; Null P = 0.09). Sample sizes are n = 6/group. *P < 0.05 compared with the young control; +P < 0.05 compared with the MA control; #P < 0.05 compared with the old control; ^P < 0.05 compared with age-matched WT controls.
4. Discussion
Cardiac ageing is associated with a decline in diastolic function.15–18 LV ECM composition regulates LV passive stiffness, one of the major determinants of diastolic function. Further, MMP-9 levels increase in the plasma and LV of ageing mice.9 The goal of this study, accordingly, was to delineate the role of MMP-9 in mediating cardiac ageing responses. The major findings of this study were (i) MMP-9 deletion prevented the age-related decline in LV diastolic function; (ii) the age-related increases in collagen accumulation and expression of pro-fibrotic genes, periostin, and CTGF were attenuated by MMP-9 deletion; and (iii) there was a compensatory increase in MMP-8 levels in the LV of senescent MMP-9 null mice. Together, our results indicate that MMP-9 regulates the cardiac phenotypic changes seen with normal ageing by altering collagen homoeostasis. A proposed mechanistic model of these changes is summarized in Figure 7.
Figure 7.
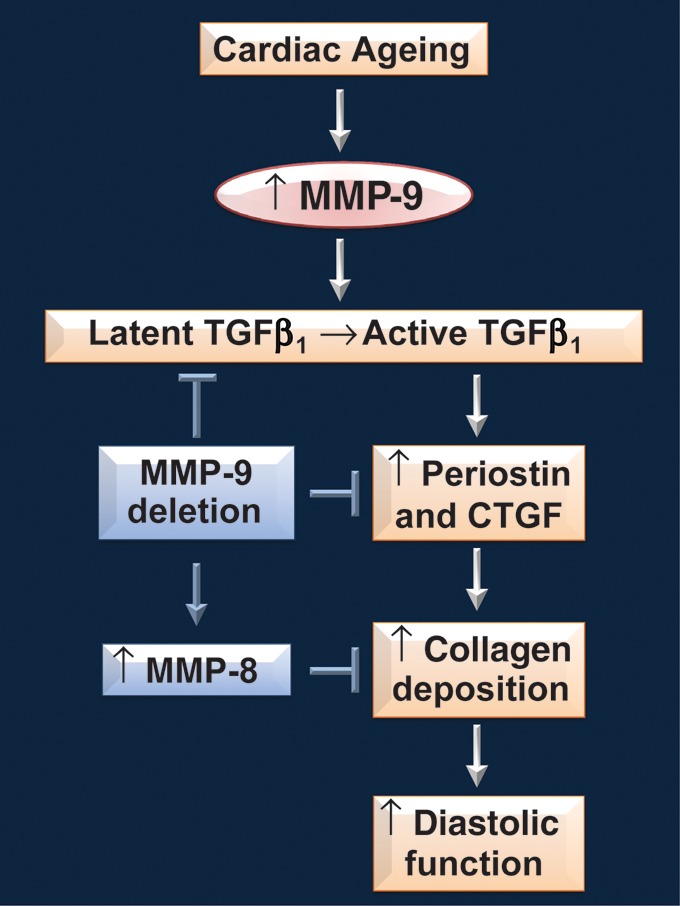
A proposed simplified overview of MMP-9 involvement in cardiac ageing. Cardiac ageing is associated with increased LV collagen deposition and a decline in diastolic function. With ageing, MMP-9 expression increases in the LV. The increased MMP-9 levels increases TGF-β activation, and therefore, increases transcription of TGF-β inducible genes. Periostin and CTGF are two TGF-β inducible genes that are shown to be pro-fibrotic. The increased periostin and CTGF expressions promote collagen deposition in the ageing LV, in turn causing a decline in diastolic function. MMP-9 deletion reduces TGF-β activation and TGF-β inducible transcription, resulting in decreased levels of periostin and CTGF in senescent null mice. Additionally, there is a compensatory increase in MMP-8 levels in senescent MMP-9 null mice, which promotes collagen degradation. The reduced pro-fibrotic signals and increased collagen degradation explain the lower LV collagen content and improved diastolic function in senescent MMP-9 null mice compared with age-matched WT mice.
Diastolic filling consists of two phases, the early diastolic filling (E) phase, which is attributed to relaxation of the LV, and the late diastolic filling (A) phase, which is attributed to the contraction of the atrium. Previous studies in humans and mice have shown that E/A ratios decrease with age, indicating impaired relaxation of the ageing LV.15–17 We also observed a decrease in E/A ratios in old and senescent WT mice, but this decrease was attenuated in MMP-9 null mice. MMP-9 null mice did not exhibit reduced E/A ratios even at senescence. Additionally, old and senescent MMP-9 null mice exhibited higher E/A ratios than age-matched WT mice. These results indicate that MMP-9 deletion can slow the age-related progressive decline in diastolic function.
Systolic function is usually relatively preserved in human ageing, despite an age-related decline in diastolic function.3 In this study, we also saw preserved FS in the ageing mice. During dobutamine stress, a slight but significant reduction in FS was observed in senescent WT mice, compared with old WT mice. This small reduction in FS suggests a reduced ability of the heart to handle stress, which can lead to reduced exercise tolerance or impaired response to a cardiovascular disease such as myocardial infarction. Future studies comparing post-MI remodelling response in ageing WT and MMP-9 null mice will provide more insights in this area.
Increased myocardial fibrosis increases myocardial stiffness and is associated with diastolic dysfunction in hypertensive disease.23,24 Increased plasma MMP-9 levels and increased turnover of collagen have been shown to be associated with diastolic heart failure in hypertensive patients.4 MMP-9 regulates collagen degradation and MMP-9 deletion results in reduced, not increased, collagen accumulation in LV post-myocardial infarction.8 To investigate whether MMP-9 deletion attenuates the age-related decline in diastolic function by reducing LV collagen deposition, we compared the collagen content in the LV of WT and MMP-9 null mice. We studied young and senescent mice, the two age groups with the most prominent difference in diastolic function. As expected, the senescent WT LV showed increased interstitial collagen deposition compared with young WT LV, consistent with an impaired diastolic function. Interestingly, the LV of senescent MMP-9 null mice had reduced interstitial collagen compared with senescent WT mice, which agreed with the improved diastolic function.
MMP-9 deletion reduced LV collagen deposition following myocardial infarction and during cardiac ageing.8 However, the mechanism underlying this observation is unclear. We used a qPCR array to profile the mRNA expression levels of 84 ECM and adhesion molecules genes. The mRNA levels of type I and type III collagen did not increase with age and were not different between senescent WT and MMP-9 null mice, suggesting that the changes in LV collagen deposition observed were not due to the changes in transcription of collagen genes. This agrees with a previous study from Besse et al.25 showing differential changes in myocardial collagen mRNA and protein levels with ageing.
Periostin mRNA expression increased with age in WT mice and was reduced in senescent MMP-9 null mice, compared with senescent WT mice. Periostin is a matricellular protein that increases in pathophysiological conditions, such as myocardial infarction and heart failure.26,27 Periostin regulates collagen fibrillogenesis and is associated with increased collagen deposition.27,28 Periostin null mice show reduced collagen diameter,26 whereas periostin overexpression results in an increased LV collagen content and LV dilation.27 The reduced levels of periostin, therefore, may partially contribute to the attenuation of diastolic functional decline in senescent MMP-9 null mice by a direct effect on collagen deposition.
CTGF expression increases with ageing and in age-associated heart failure.29 In a mouse model, cardiomyocyte-specific CTGF overexpression results in age-dependent cardiac dysfunction as early as 7 months of age.30,31 We also observed an increase in CTGF expression in WT senescent mice, but this increase was attenuated in MMP-9 null mice. These changes in CTGF expression may, in part, contribute to the changes in collagen deposition and diastolic function observed by stimulating cardiac fibroblast function.
In vitro studies have demonstrated that transforming growth factor β1 (TGF-β1) can induce periostin mRNA expression in periodontal ligament fibroblasts, and a TGF-β1-blocking antibody can blunt angiotensin-II-induced periostin expression.21,22 At the same time, CTGF expression is induced two to three-fold in human cardiac fibroblasts and 1.8-fold in rat cardiac myocytes by 10 ng/mL of TGF-β1.19 Because MMP-9 can process and activate latent TGF-β1,20 the reduced periostin and CTGF expression observed in the senescent MMP-9 null mice is potentially due to a decrease in TGF-β1 signalling. We have shown in this study that both mRNA and protein levels of transforming growth factor-beta-induced protein increased in senescent WT, but not MMP-9 null, mice, which supports this hypothesis. We also observed that the increased Smad2 phosphorylation in senescent WT was attenuated in senescent MMP-9 null mice. The phosphorylation of Smad2 to phospho-Smad2 is mediated by TGF-β132. The decrease of phosphorylated Smad2 in the absence of MMP-9 further supports a role for MMP-9 in activating latent TGF-β1. These results indicate that reduced TGF-β signalling blunts the increase in periostin and CTGF levels in senescent MMP-9 null mice.
In MMP-9 null mice, collagen deposition is reduced following myocardial infarction compared with WT mice, and this decrease is accompanied by increased levels of MMP-2 and MMP-13.8 Increased MMP activity would stimulate collagen degradation and explain the reduced collagen deposition. We likewise demonstrated a compensatory increase in MMP-8 levels in the LV of senescent MMP-9 null mice, concomitant with reduced collagen deposition. MMP-8 preferentially degrades type I collagen over type III collagen, which may explain why MMP-8 is overexpressed with ageing, whereas MMPs 2 and 13 are overexpressed following myocardial infarction.33,34 Although the scheme shown in Figure 7 is likely to be an oversimplification, it highlights a role of MMP-9 in cardiac ageing.
No sex-associated differences were observed in MMP-9 expression levels, LV collagen content, and E/A ratios. This suggests that MMP-9 regulates LV collagen deposition and diastolic function by sex-independent mechanisms. The MMP-9 null mice utilized in this study are whole body gene-deleted mice. As cardiomyocytes are not a primary source of MMP-9, cardiomyocyte-specific null mice would not be a relevant cardiac-specific model for MMP-9 deletion. Macrophage-specific MMP-9 overexpressing mice have only recently become available, and their cardiac ageing responses have not yet been studied.35 Whether macrophage-specific MMP-9 overexpression will accelerate or exacerbate cardiac ageing remains to be determined. When available, it would also be helpful to study cardiac ageing in fibroblast-specific MMP-9 null or transgenic mice, as cardiac fibroblasts also express MMP-9. One limitation of this study is that we used mice over 6 months of age for this study as our focus is the ageing response in mature adult mice; however, it is possible that ECM changes that present before this youngest age group could affect the ageing response in later stages of life.
In conclusion, we showed that MMP-9 deletion attenuates the age-related increase in collagen deposition and a decline in diastolic function of the LV. The suppression of the age-related increases in TGF-β signalling-induced periostin and CTGF and the compensatory increase in MMP-8 levels in the LV of MMP-9 null mice likely contribute to the improved cardiac function.
Supplementary material
Supplementary material is available at Cardiovascular Research online.
Conflict of interest: none declared.
Funding
This work was supported by the Translational Science Training (TST) Across Disciplines program at the University of Texas Health Science Center at San Antonio, with funding provided by the University of Texas System's Graduate Programs Initiative, to Y.A.C.; NSF 0649172, NIHEB009496, and NIH1SC2 HL101430 to Y-FJ; and NIH HHSN 268201000036C (N01-HV-00244) for the San Antonio Cardiovascular Proteomics Center and R01 HL-075360, the Max and Minnie Tomerlin Voelcker Fund, and the Veteran's Administration (Merit) to M.L.L.
Supplementary Material
References
- 1.Cheitlin MD. Cardiovascular physiology-changes with aging. Am J Geriatr Cardiol. 2003;12:9–13. doi: 10.1111/j.1076-7460.2003.01751.x. doi:10.1111/j.1076-7460.2003.01751.x. [DOI] [PubMed] [Google Scholar]
- 2.Ferrari AU. Modifications of the cardiovascular system with aging. Am J Geriatr Cardiol. 2002;11:30–33. doi: 10.1111/1467-8446.00044-i1. doi:10.1111/1467-8446.00044-i1. [DOI] [PubMed] [Google Scholar]
- 3.Lakatta EG, Levy D. Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation. 2003;107:346–354. doi: 10.1161/01.cir.0000048893.62841.f7. doi:10.1161/01.CIR.0000048893.62841.F7. [DOI] [PubMed] [Google Scholar]
- 4.Martos R, Baugh J, Ledwidge M, O'Loughlin C, Conlon C, Patle A, et al. Diastolic heart failure: evidence of increased myocardial collagen turnover linked to diastolic dysfunction. Circulation. 2007;115:888–895. doi: 10.1161/CIRCULATIONAHA.106.638569. doi:10.1161/CIRCULATIONAHA.106.638569. [DOI] [PubMed] [Google Scholar]
- 5.Egeblad M, Werb Z. New functions for the matrix metalloproteinases in cancer progression. Nat Rev Cancer. 2002;2:163–176. doi: 10.1038/nrc745. [DOI] [PubMed] [Google Scholar]
- 6.Sternlicht M, Werb Z. How matrix metalloproteinases regulate cell behavior. Annu Rev Cell Dev Biol. 2001;17:463–516. doi: 10.1146/annurev.cellbio.17.1.463. doi:10.1146/annurev.cellbio.17.1.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cauwe B, Van den Steen PE, Opdenakker G. The biochemical, biological, and pathological kaleidoscope of cell surface substrates processed by matrix metalloproteinases. Crit Rev Biochem Mol Biol. 2007;42:113–185. doi: 10.1080/10409230701340019. doi:10.1080/10409230701340019. [DOI] [PubMed] [Google Scholar]
- 8.Ducharme A, Frantz S, Aikawa M, Rabkin E, Lindsey M, Rohde LE, et al. Targeted deletion of matrix metalloproteinase-9 attenuates left ventricular enlargement and collagen accumulation after experimental myocardial infarction. J Clin Invest. 2000;106:55–62. doi: 10.1172/JCI8768. doi:10.1172/JCI8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chiao YA, Dai Q, Zhang J, Lin J, Lopez EF, Ahuja SS, et al. Multi-analyte profiling reveals matrix metalloproteinase-9 and monocyte chemotactic protein-1 as plasma biomarkers of cardiac aging. Circ Cardiovasc Genet. 2011;4:455–462. doi: 10.1161/CIRCGENETICS.111.959981. doi:10.1161/CIRCGENETICS.111.959981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vu TH, Shipley JM, Bergers G, Berger JE, Helms JA, Hanahan D, et al. MMP-9/gelatinase B is a key regulator of growth plate angiogenesis and apoptosis of hypertrophic chondrocytes. Cell. 1998;93:411–422. doi: 10.1016/s0092-8674(00)81169-1. doi:10.1016/S0092-8674(00)81169-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martin MD, Carter KJ, Jean-Philippe SR, Chang M, Mobashery S, Thiolloy S, et al. Effect of ablation or inhibition of stromal matrix metalloproteinase-9 on lung metastasis in a breast cancer model is dependent on genetic background. Cancer Res. 2008;68:6251–6259. doi: 10.1158/0008-5472.CAN-08-0537. doi:10.1158/0008-5472.CAN-08-0537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wiesmann F, Ruff J, Engelhardt S, Hein L, Dienesch C, Leupold A, et al. Dobutamine-stress magnetic resonance microimaging in mice: acute changes of cardiac geometry and function in normal and failing murine hearts. Circ Res. 2001;88:563–569. doi: 10.1161/01.res.88.6.563. doi:10.1161/01.RES.88.6.563. [DOI] [PubMed] [Google Scholar]
- 13.Vecchione C, Patrucco E, Marino G, Barberis L, Poulet R, Aretini A, et al. Protection from angiotensin II-mediated vasculotoxic and hypertensive response in mice lacking PI3Kgamma. J Exp Med. 2005;201:1217–1228. doi: 10.1084/jem.20040995. doi:10.1084/jem.20040995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muir A, Greenspan DS. Metalloproteinases in Drosophila to humans that are central players in developmental processes. J Biol Chem. 2011;286:41905–41911. doi: 10.1074/jbc.R111.299768. doi:10.1074/jbc.R111.299768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Choi SY, Chang HJ, Choi SI, Kim KI, Cho YS, Youn TJ, et al. Long-term exercise training attenuates age-related diastolic dysfunction: association of myocardial collagen cross-linking. J Korean Med Sci. 2009;24:32–39. doi: 10.3346/jkms.2009.24.1.32. doi:10.3346/jkms.2009.24.1.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Downes TR, Nomeir AM, Smith KM, Stewart KP, Little WC. Mechanism of altered pattern of left ventricular filling with aging in subjects without cardiac disease. Am J Cardiol. 1989;64:523–527. doi: 10.1016/0002-9149(89)90433-5. doi:10.1016/0002-9149(89)90433-5. [DOI] [PubMed] [Google Scholar]
- 17.Kitzman DW. Diastolic heart failure in the elderly. Heart Fail Rev. 2002;7:17–27. doi: 10.1023/a:1013745705318. doi:10.1023/A:1013745705318. [DOI] [PubMed] [Google Scholar]
- 18.Reddy AK, Amador-Noguez D, Darlington GJ, Scholz BA, Michael LH, Hartley CJ, et al. Cardiac function in young and old little mice. Gerontology. 2007;62:1319–1325. doi: 10.1093/gerona/62.12.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen MM, Lam A, Abraham JA, Schreiner GF, Joly AH. CTGF expression is induced by TGF- beta in cardiac fibroblasts and cardiac myocytes: a potential role in heart fibrosis. J Mol Cell Cardiol. 2000;32:1805–1819. doi: 10.1006/jmcc.2000.1215. doi:10.1006/jmcc.2000.1215. [DOI] [PubMed] [Google Scholar]
- 20.Yu Q, Stamenkovic I. Cell surface-localized matrix metalloproteinase-9 proteolytically activates TGF-beta and promotes tumor invasion and angiogenesis. Genes Dev. 2000;14:163–176. [PMC free article] [PubMed] [Google Scholar]
- 21.Li L, Fan D, Wang C, Wang JY, Cui XB, Wu D, et al. Angiotensin II increases periostin expression via Ras/p38 MAPK/CREB and ERK1/2/TGF-beta1 pathways in cardiac fibroblasts. Cardiovasc Res. 2011;91:80–89. doi: 10.1093/cvr/cvr067. doi:10.1093/cvr/cvr067. [DOI] [PubMed] [Google Scholar]
- 22.Wen W, Chau E, Jackson-Boeters L, Elliott C, Daley TD, Hamilton DW. TGF-β1 and FAK regulate periostin expression in PDL fibroblasts. J Dent Res. 2010;89:1439–1443. doi: 10.1177/0022034510378684. doi:10.1177/0022034510378684. [DOI] [PubMed] [Google Scholar]
- 23.Diez J, Lopez B, Gonzalez A, Querejeta R. Clinical aspects of hypertensive myocardial fibrosis. Curr Opin Cardiol. 2001;16:328–335. doi: 10.1097/00001573-200111000-00003. doi:10.1097/00001573-200111000-00003. [DOI] [PubMed] [Google Scholar]
- 24.Querejeta R, Lopez B, Gonzalez A, Sanchez E, Larman M, et al. Increased collagen type I synthesis in patients with heart failure of hypertensive origin: relation to myocardial fibrosis. Circulation. 2004;110:1263–1268. doi: 10.1161/01.CIR.0000140973.60992.9A. [DOI] [PubMed] [Google Scholar]
- 25.Besse S, Robert V, Assayag P, Delcayre C, Swynghedauw B. Nonsynchronous changes in myocardial collagen mRNA and protein during aging: effect of DOCA-salt hypertension. Am J Physiol. 1994;267:H2237–2244. doi: 10.1152/ajpheart.1994.267.6.H2237. [DOI] [PubMed] [Google Scholar]
- 26.Oka T, Xu J, Kaiser RA, Melendez J, Hambleton M, Sargent MA, et al. Genetic manipulation of periostin expression reveals a role in cardiac hypertrophy and ventricular remodeling. Circ Res. 2007;101:313–321. doi: 10.1161/CIRCRESAHA.107.149047. doi:10.1161/CIRCRESAHA.107.149047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Katsuragi N, Morishita R, Nakamura N, Ochiai T, Taniyama Y, Hasegawa Y, et al. Periostin as a novel factor responsible for ventricular dilation. Circulation. 2004;110:1806–1813. doi: 10.1161/01.CIR.0000142607.33398.54. doi:10.1161/01.CIR.0000142607.33398.54. [DOI] [PubMed] [Google Scholar]
- 28.Norris RA, Damon B, Mironov V, Kasyanov V, Ramamurthi A, Moreno-Rodriguez R, et al. Periostin regulates collagen fibrillogenesis and the biomechanical properties of connective tissues. J Cell Biochem. 2007;101:695–711. doi: 10.1002/jcb.21224. doi:10.1002/jcb.21224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang M, Zhang J, Walker SJ, Dworakowski R, Lakatta EG, Shah AM. Involvement of NADPH oxidase in age-associated cardiac remodeling. J Mol Cell Cardiol. 2010;48:765–772. doi: 10.1016/j.yjmcc.2010.01.006. doi:10.1016/j.yjmcc.2010.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Panek AN, Posch MG, Alenina N, Ghadge SK, Erdmann B, Popova E, et al. Connective tissue growth factor overexpression in cardiomyocytes promotes cardiac hypertrophy and protection against pressure overload. PLoS One. 2009;4:e6743. doi: 10.1371/journal.pone.0006743. doi:10.1371/journal.pone.0006743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van Almen GC, Verhesen W, van Leeuwen RE, van de Brie M, Eurlings C, et al. MicroRNA-18 and microRNA-19 regulate CTGF and TSP-1 expression in age-related heart failure. Aging Cell. 2011;10:769–779. doi: 10.1111/j.1474-9726.2011.00714.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heldin CH, Miyazono K, ten Dijke P. TGF-β signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390:465–471. doi: 10.1038/37284. doi:10.1038/37284. [DOI] [PubMed] [Google Scholar]
- 33.Hasty KA, Jeffrey JJ, Hibbs MS, Welgus HG. The collagen substrate specificity of human neutrophil collagenase. J Biol Chem. 1987;262:10048–10052. [PubMed] [Google Scholar]
- 34.Horwitz AL, Hance AJ, Crystal RG. Granulocyte collagenase: selective digestion of type I relative to type III collagen. Proc Natl Acad Sci U S A. 1977;74:897–901. doi: 10.1073/pnas.74.3.897. doi:10.1073/pnas.74.3.897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Foronjy R, Nkyimbeng T, Wallace A, Thankachen J, Okada Y, Lemaitre V, et al. Transgenic expression of matrix metalloproteinase-9 causes adult-onset emphysema in mice associated with the loss of alveolar elastin. Am J Physiol Lung Cell Mol Physiol. 2008;294:L1149–1157. doi: 10.1152/ajplung.00481.2007. doi:10.1152/ajplung.00481.2007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.