

Abstract
Aim:
To investigate the growth potential of keratinocytes derived from the germinative epithelium (GE) of ovine hair follicles. Stem cells from the outer root sheath (ORS) of hair follicles migrate to the GE in the lower follicle where they proliferate and differentiate to form the hair fiber. It has been suggested that the GE comprises transit-amplifying cells and that the duration of anagen is determined by their limited proliferative potential. However, we show here that keratinocytes derived from the GE of ovine follicles grow extensively in vitro, arguing against this hypothesis.
Materials and Methods:
Primary cultures of keratinocytes were initiated from microdissected GE tissue from ovine vibrissae and wool follicles. Clonal lines of keratinocytes were derived by limiting dilution. Their growth potential was determined by exhaustive serial passaging. Expression of differentiation markers was evaluated by real-time polymerase chain reaction.
Results:
Initiation of these cultures required that interaction between the GE and dermal papilla was maintained. However, the keratinocytes could subsequently be cloned and were grown as pure cell populations for 26-52 cell doublings. This proliferative potential is several orders of magnitude greater than required to maintain a single anagen phase. The keratinocytes were indistinguishable from ORS keratinocytes from the same follicles, expressing K14 while undifferentiated, and upregulating the epidermal and inner root sheath markers, loricrin and KRT27 on differentiation. Thus, these cells initially depend on papilla-derived signals to grow, but can revert to an ORS-like phenotype in vitro. Their extensive proliferative capacity shows that the GE is not an exclusively transit-amplifying cell population.
Keywords: Dermal papilla, germinative epithelium, keratinocytes, proliferative potential
INTRODUCTION
The stem cells that ultimately maintain hair follicles have been shown to reside in the bulge region of the outer root sheath (ORS). Progeny of these cells migrate down the ORS to maintain the germinative epithelium (GE) at the base of the anagen follicle.[1–6] However, the extent to which they retain stem cell properties as they do so remains unclear.[7]
It has been suggested that the lower follicle contains only transit-amplifying cells, and that the duration of anagen in the hair cycle is determined by their intrinsic proliferative potential.[1,4,8] Genetic ablation of Sox9 in bulge stem cells inhibits their migration into the GE compartment and results in premature termination of anagen.[4] On the other hand, Lgr5-expressing cells in the anagen ORS have been shown to persist through successive hair cycles, indicating that they are not transit-amplifying cells.[3] When anagen rat vibrissae are plucked, the matrix is repopulated by residual GE cells rather than bulge cells.[9] When vibrissae are repeatedly plucked and allowed to re-grow to mid-anagen, for several iterations, the cumulative fiber production suggests that the proliferative capacity of the GE cells is greater than would be required for a single, unperturbed anagen phase.
The presence of stem cells can be inferred in vitro by determining the number of clonogenic cells that form extensively growing colonies. Clonogenic keratinocytes are concentrated in the ORS of rat vibrissae[10] and human follicles.[11] Keratinocytes from the lower anagen follicle typically show much more limited proliferation in culture.[8] However, the proliferation of GE cells in organ-cultured follicles is inhibited when their interaction with the dermal papilla (DP) is disrupted.[12] Thus, the proliferation of these cells appears to depend on signals from their environment that are not readily reproduced in culture, suggesting that their growth in vitro may not be a true reflection of their intrinsic potential.
Wool follicles of sheep provide a useful model for human hair growth, quite apart from their direct relevance to the wool industry.[13] Like human scalp hair, but unlike mice or rats, sheep follicles cycle asynchronously, remain in anagen for several years, and lack the complex coat structure comprising guard, awl, auchene, and zigzag hair types. When initiating primary cultures of DP cells from ovine vibrissae, we have often seen the outgrowth of keratinocytes that appear to derive from remnants of GE tissue adhering to microdissected papillae. In this paper, we show that these cells provide insights into the cell biology of the anagen follicle bulb. Although their growth is initially dependent on interaction with the DP, they subsequently loose this dependency and revert to a phenotype indistinguishable from ORS keratinocytes. Their proliferative capacity is substantially greater than would be required to maintain wool growth for a single follicle cycle, showing that anagen GE tissue is not an exclusively transit-amplifying cell population.
MATERIALS AND METHODS
Dissection and primary culture
For cell cultures from sheep, upper lip vibrissa- and wool-bearing skin samples from the neck region were collected from lambs at a local abattoir shortly after death, and placed in Minimum Essential Medium, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 12.5 ng/ml amphotericin B, 20% lamb serum (components from Invitrogen, Carlsbad, CA, and Sigma, St. Louis, MO), on ice. Microdissection of the skin was performed in the same medium at room temperature. Follicles were cut from the surrounding dermis, then explants of either GE tissue attached to the DP (DP/GE) or ORS tissue attached to the dermal sheath (DS/ORS) were excised and transferred to 4-well mini-plates (Nunc, Roskilde, Denmark) containing 1 ml per well of culture medium (KSFM, 100 units/ml penicillin, 0.1 mg/ml streptomycin, 12.5 ng/ml amphotericin B, 5% fetal calf serum, 0.18 mM CaCl2; components from Invitrogen and Sigma). Cultures were maintained at 37°C, 5% CO2. For cell cultures from rats, vibrissae were removed from the whisker pad of 2- to 4-month-old male Sprague-Dawley rats. Explants were then isolated and cultured as for sheep explants.
To determine the proportions of each explant type that produced keratinocytes, dissections were repeated in at least three experiments, each using tissue from a different animal. Individual explants were scored as producing keratinocytes or not. Data were analyzed using a generalized linear model with binomial variation, and logit link to allow for extra-binomial variation.
Immunocytochemistry and microscopy
Freshly isolated explants were stained as whole-mounts for expression of proliferating cell nuclear antigen (PCNA). Explants were washed briefly in phosphate buffered saline (PBS), fixed in 90% ethanol, 5% acetic acid, 5% water for 20 minutes, washed in PBS again, then blocked overnight in 5% normal sheep serum at 4°C. They were then incubated with a mouse monoclonal anti-PCNA primary antibody (DAKO, Glostrup, Denmark) at 1:400 dilution for 2 hours, followed by a polyclonal goat-anti-mouse immunoglobulin/horseradish peroxidase conjugate (DAKO) secondary antibody, at 1:100 dilution for 1 hour, both at room temperature, with three 5-minute washes in PBS after each incubation. Negative controls were maintained in blocking solution instead of primary antibody. Staining was visualized by incubation with Sigma Fast DAB with metal cation enhancer (Sigma) for up to 10 minutes followed by thorough washing in water.
Cultures for K14 staining were grown on glass slides (BD Falcon, Bedford, MA) coated with rat type I collagen. They were fixed in 90% ethanol, 5% acetic acid, 5% water for 20 minutes, washed three times in PBS, then blocked in 5% normal sheep serum at 4°C for up to 8 hours. K14 fluorescent primary antibody (Chemicon, Temecula, CA) was applied to each culture at 1:25 dilution, overnight at 4°C. Cultures were then washed in PBS, and mounted with Prolong gold anti-fade fluorescent mountant, containing DAPI nuclear counter-stain (Invitrogen). Fluorescent and DIC images were photographed on a BX50 microscope (Olympus, Tokyo, Japan) with a SPOT digital camera (Diagnostic Instruments, Sterling Heights, MI). Phase contrast images were photographed on an Axiovert 40CFL microscope (Zeiss, Göttingen, Germany) with a DC330E digital camera (MTI, Michigan City, IN).
Keratinocyte cloning
To clone keratinocytes, cells in a 3- to 4-day-old colony were counted from a photograph and then harvested by incubation with 0.25% trypsin in PBS, 0.6 mM ethylenediaminetetraacetic acid at 37°C for 5 to 10 minutes. Detached cells were resuspended in culture medium and seeded at 1 cell/well into 96-well plates. After approximately 2 weeks’ culture, wells containing a single colony were selected, trypsinized as above, and then seeded into a fresh culture vessel. Clones were further amplified by serial passaging. Cells were typically passaged once they reached 70% to 90% confluence, and seeded at a density of 2 500 to 10 000 cells/cm2 of culture substrate. To measure the proliferative potential of keratinocyte clones, cells were counted at consecutive passages on a hemocytometer. All culture vessels used for keratinocyte clones were coated with rat type I collagen. Keratinocytes were stored frozen in liquid nitrogen, suspended in culture medium supplemented with 10% dimethyl sulfoxide.
Keratinocyte differentiation
Ovine keratinocyte clones derived from vibrissa DP/GE, vibrissa DS/ORS, or wool follicle DP/GE were compared. Differentiation was induced by maintaining cells at passage 3 in a confluent state for 6 to 9 days. Undifferentiated cells were at 70% to 80% confluence. RNA was extracted using a Micro-to-Midi RNA kit (Invitrogen) according to the manufacturer's instructions and eluted in 30 μl RNase-free water. RNA was treated with DNase I using a Turbo DNA-free kit (Ambion, Austin, TX). Random hexamer primers were used to synthesize cDNA from 500 ng RNA, with a Superscript III First-Strand cDNA Synthesis kit (Invitrogen, Carlsbad, CA).
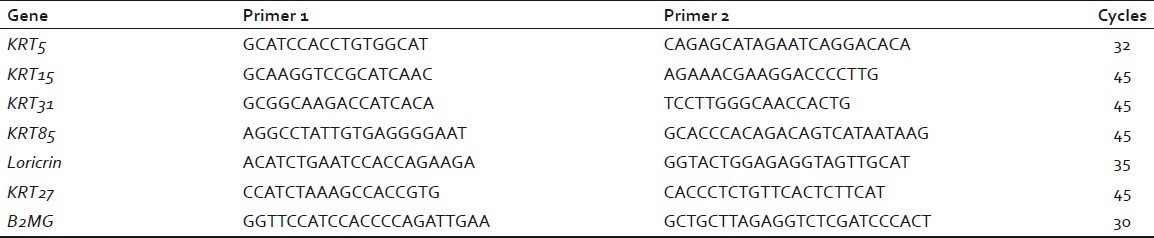
Polymerase chain reaction (PCR) primers were designed using Primer3 software.[14] Quantitative PCR was performed using a Lightcycler 2 real-time PCR instrument and software (Roche, Mannheim, Germany) with a Lightcycler Faststart DNA Master SYBR Green kit (Roche), according to the supplied protocol. cDNA (2 μl) was added to a master mixture containing the appropriate pair of primers (8 μl). PCR conditions used were as follows: activation at 95°C for 10 minutes; followed by 32 to 45 amplifying cycles at 95°C for 5 seconds, 59°C for 5 seconds, 72°C for 10 seconds [Table 1]. For each gene, standard curves were prepared using a two-fold dilution series of cDNA made from 500 ng of ovine skin RNA. Expression values for each gene were normalized to expression of beta 2-microglobulin (B2MG).
Table 1.
Polymerase chain reaction primers and reaction conditions
RESULTS
Primary cultures
To optimize the growth of keratinocytes from ovine vibrissae, we first established microdissection techniques that left as much GE as possible attached to the DP, preserving the physiological interaction between these two tissues [Figure 1a]. A small amount of proximal DS was left attached to minimize disruption to the GE. Some explants were stained immunohistochemically for expression of PCNA, in order to label proliferating cells. Staining was localized to pieces of epithelial tissue around the junction between the DP and the proximal DS [Figure 1b]. This illustrates the presence of GE tissue and confirms that the vibrissae were in the anagen stage of the hair cycle.
Figure 1.
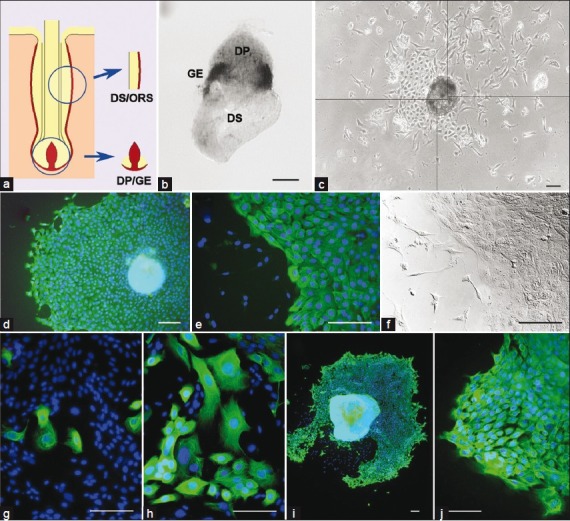
Primary culture of sheep and rat keratinocytes (a) Diagram showing follicle compartments dissected for explant and culture (epithelial tissue, yellow; dermal tissue, red); (b) Intact DP/GE explant immunostained for PCNA to label proliferating cells.Expression (black) is restricted to fragments of GE tissue surrounding the junction between the DP (upper) and DS (lower); (c) Photomontage of primary cells growing from an ovine DP/GE explant (phase contrast view).The original explant is in the center of the image.Two cell types can be seen growing out from it—polygonal keratinocytes clustered on the left side of the colony and scattered, stellate dermal cells concentrated toward the upper right.Note that this colony hasproportionally more dermal cells than is typical; (d) K14 immunostaining of an ovine vibrissa DP/GE colony. K14 expression is seen as green, nuclei are counterstained with DAPI (blue); (e) Higher power view of a second ovine vibrissa DP/GE colony immunostained for K14.Labeled keratinocytes can be seen on the right of the image, with scattered, unlabelled dermal cells on the left; (f) DIC image of the field of view shown in (e); (g, h) Two primary cultures of cells grown from rat DP/GE explants, immunostained for K14 expression (green), nuclei counterstained with DAPI (blue).K14-positive cells are less frequent and more variable in morphology compared with ovine cultures; (i, j) Two primary cultures of cells grown from rat DS/ORS explants, immunostained for K14 expression (green), nuclei counterstained with DAPI (blue). K14-positive cells are similar in abundance and morphology to ovine cultures.In all images, scale bars = 100 μm
Cells growing from these explants were variable in appearance. Often a single explant produced both stellate, fibroblast-like cells (presumably originating from the dermal tissue) and polygonal cells closely juxtaposed in a pavement-like pattern, typical of keratinocytes [Figure 1c]. Only the polygonal cells stained positively with a K14 antibody [Figure 1d–f] confirming their identity as keratinocytes. Extensive outgrowth of keratinocytes was typically seen within a week, and comprised the great majority of cells in most colonies [Figure 1d–f].
Keratinocyte outgrowth was highly reproducible: 85.3%±3.9 (S.E.M.) of explants produced keratinocytes among 18 experiments, with each experiment using vibrissae from a different animal, 4 to 16 explants per experiment [Table 2]. Similar keratinocyte outgrowth was seen from DP/GE explants dissected from wool-type pelage follicles, showing that the phenomenon is not unique to vibrissae. Keratinocyte outgrowth was also seen when explants of ORS with attached DS [Figure 1a] were isolated from vibrissae and cultured under identical conditions. DS/ORS cultures were somewhat slower to initiate, requiring approximately 10 days before cell outgrowth was first seen. Keratinocytes derived from vibrissa GE, vibrissa ORS, and wool follicle GE showed no difference in morphology or K14 staining.
Table 2.
Frequencies of keratinocyte production in primary cultures
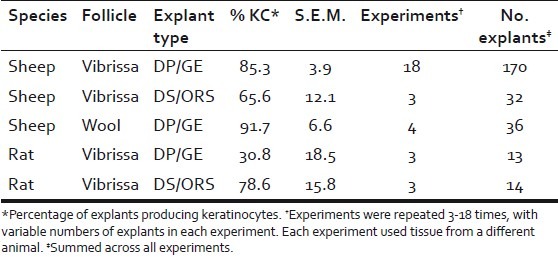
We investigated whether keratinocytes would grow from rat vibrissa explants isolated and maintained under identical conditions. Unlike for sheep, only 30.8%±18.5 (P=0.001) of DP/GE explants produced K14-positive cells [Table 2]. Even when K14-positive cells were produced, they comprised a minority of the total cell outgrowth [Figure 1g–h]. Furthermore, the K14-positive cells that grew from rat DP/GE explants were more variable in morphology than those from sheep, and often indistinguishable from the K14-negative fibroblasts. On the other hand, rat DS/ORS explants produced K14-positive keratinocytes at similar frequencies to the equivalent ovine explants [Table 2], and these cells generally comprised a greater proportion of the outgrowing cells and had a more typical keratinocyte morphology [Figure 1i–j]. Thus, under these culture conditions, rat and sheep follicles appear to differ in their propensity to produce keratinocytes from GE tissue, but not from ORS tissue.
We investigated the role of the DP in promoting ovine keratinocyte outgrowth by comparing DP/GE explants with explants of isolated GE tissue. Although some keratinocyte outgrowth was seen in the absence of the DP, both the size of the keratinocyte colonies and the proportion of explants producing keratinocytes were reduced [Figure 2a–b]. Thus, signals from the DP promote the outgrowth of keratinocytes from GE tissue. We further investigated the effect of separating the GE and DP and then loosely recombining them. Keratinocyte outgrowth was reduced compared with intact DP/GE explants, and similar to isolated GE tissue [Figure 2a]. Thus, the critical signals from the DP seem to require direct physical contact with the GE tissue, suggesting that they are not mediated by diffusible molecules. Furthermore, this signaling is not spontaneously re-established in culture after the interaction between the DP and GE tissue is mechanically disrupted.
Figure 2.
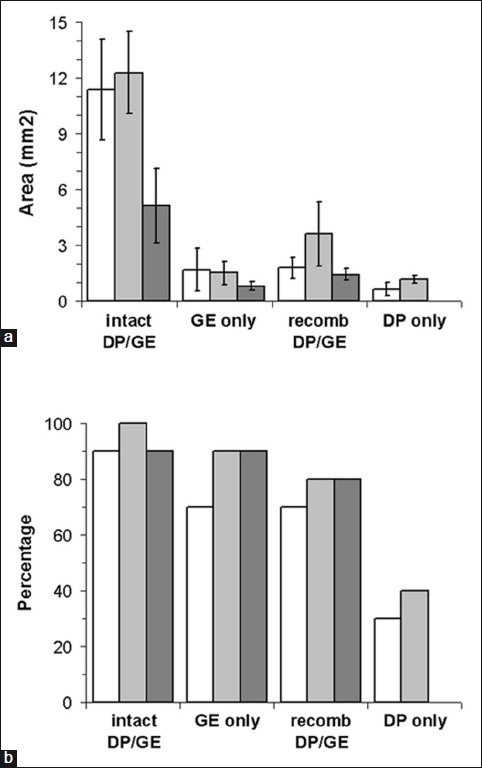
Dependence of keratinocyte outgrowth on the dermal papilla. Keratinocyte outgrowths from four types of explant were quantified. Intact DP/GE explants were compared with isolated GE tissue, DP and GE tissue that was separated and then recombined, and isolated DP tissue.The latter are a control to determine how effectively GE tissue could be removed from the papilla—the limited keratinocyte outgrowth seen from these explants is presumably derived from remnants of GE tissues that were not removed from some papillae.Data from three replicate experiments are represented by white, light grey, and dark grey bars; (a) The areas of polygonal keratinocytes growing from each explant were measured (any stellate dermal cells were excluded).Error bars = S.E.M; (b) The proportions of explants producing keratinocytes were counted
Clones
In order to isolate pure populations of ovine keratinocytes and to better characterize them, the cells were cloned by isolating colonies from multiwell plates seeded at single cell density. The same culture medium was used as for the primary cultures. Cloned cells retained typical keratinocyte morphology [Figure 3a–b] and continued to express K14.
Figure 3.
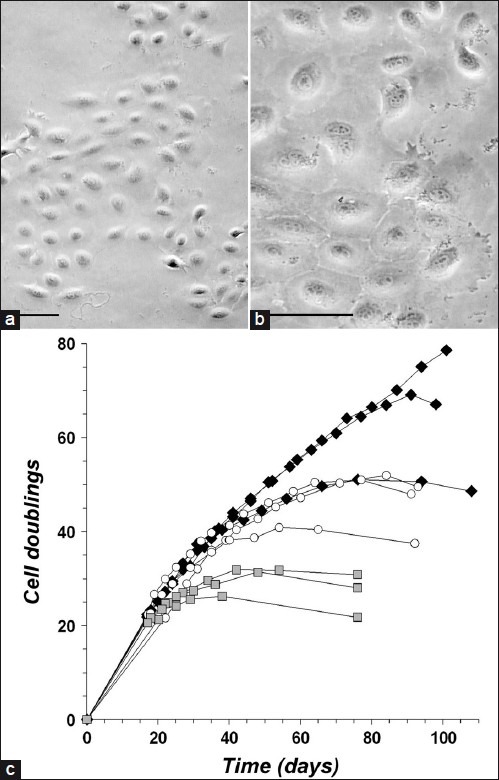
Proliferative potential of keratinocyte clones (a, b) Lower and higher power views of cloned keratinocytes from ovine vibrissa DP/ GE explants (phase contrast view). Scale bars = 50 μm; (c) Growth time courses of ovine keratinocyte clones through multiple passages. Nine clones were compared, 3 from vibrissa DS/ORS explants (black diamonds), 3 from vibrissa DP/GE explants (white circles), and 3 from wool follicle DP/GE explants (grey squares). Each data point represents a passage, when cells were counted and cumulative cell doublings were calculated.Proliferation was considered to have ceased when each growth curve reached a plateau or began to decline. At these times, the rate of production of new cells had become less than the rate of cell death
The proliferative potential of keratinocyte clones was determined by continuing to passage them until proliferation ceased. At this time, the morphology of the cells was indicative of replicative senescence rather than terminal differentiation.[15] The cells appeared enlarged with an extensive but thin-looking cytoplasm and were more circular in shape. Clones derived from vibrissa GE, vibrissa ORS, and wool follicle GE were compared [Figure 3c]. Clones derived from wool follicles ceased proliferating after 26-32 population doublings, sooner than vibrissa-derived clones. Clones from vibrissae, either GE- or ORS-derived, grew at a steady rate of ≥0.5 doublings per day for at least 38 cumulative doublings before the proliferation rate began to slow. One ORS-derived clone was still proliferating at this rate after 75 doublings. The extensive proliferative potential of these keratinocytes implies that they are not derived from transit-amplifying cells.
When maintained in a confluent state, keratinocyte clones underwent morphological changes suggestive of differentiation. The cytoplasm of cells appeared grainy and the nuclei became less distinct [Figure 4a–b]. We used quantitative PCR to investigate the expression of keratinocyte lineage markers, comparing subconfluent with confluent cells. Cytokeratin 5 (KRT5) and loricrin were the most abundantly expressed markers [Figure 4c]. KRT5 was detected in subconfluent cells and was consistently downregulated after confluence [Figure 4d]. Loricrin, an epidermal marker, was expressed only in confluent cells [Figure 4e]. Expression of the inner root sheath acidic intermediate filament KRT27[16] was also consistently detected in confluent cells, albeit at a much lower level than loricrin [Figure 4f]. These results suggest that the keratinocytes might undergo epidermal and/or inner root sheath differentiation when confluent. Expression of wool cortex markers, KRT31 and KRT85, could not be reproducibly detected in any cells. Overall, there was no difference between keratinocytes derived from vibrissa GE, vibrissa ORS, and wool follicle GE, suggesting that these cells are equivalent in their differentiation potential.
Figure 4.
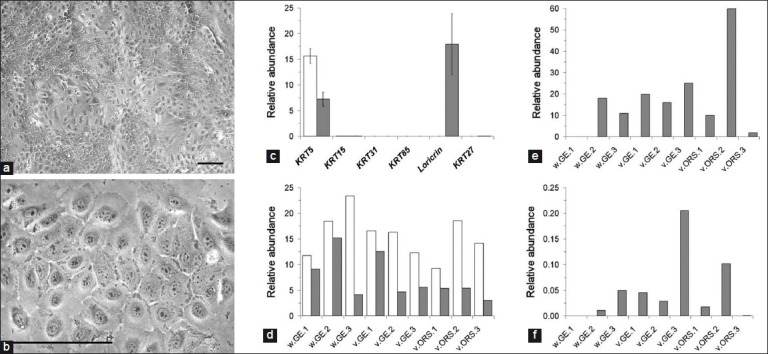
Differentiation of keratinocytes (a, b) Lower and higher power views of differentiating keratinocytes from ovine vibrissa DP/GE explants (phase contrast view). Patches of more and less advanced differentiation can be seen. More differentiated cells appear with a grainy, slightly darker cytoplasm and a less distinct nucleus.Scale bars = 100 μm; (c-f) Quantitative PCR analysis of marker gene expression.The same 9 clones as described in Figure 3 were compared, 3 from wool follicle DP/GE explants (w.GE), 3 from vibrissa DP/GE explants (v.GE), and 3 from vibrissa DS/ORS explants (v.ORS). For each clone, expression was compared in undifferentiated, subconfluent cells (white bars) and differentiated, confluent cells (grey bars); (c) The mean, normalized expression value across all nine clones was plotted for each marker gene and differentiation state.The most abundantly expressed genes were KRT5 in both undifferentiated and differentiated cells and loricrin in differentiated cells only; (d) The normalized expression levels of KRT5 were plotted separately for each clone and differentiation state; (e, f) Normalized expression levels of loricrin and KRT27, respectively, for each clone and differentiation state.Overall, expression levels varied between clones, but there were no consistent differences between tissues of origin
DISCUSSION
We have shown that long-lived keratinocytes can be derived from the GE of both wool follicles and vibrissae from sheep. These cells were very similar to ovine keratinocytes derived from the ORS. They express K14 and KRT5 while proliferating, and express epidermal and inner root sheath markers when they differentiate. We isolated clones derived from single cells to demonstrate that they have the potential to grow for a total of 26-52 cell divisions. Growth for 26 divisions represents the production of 6.7 × 107 cells from a single progenitor. However, a wool fiber contains in the order of 105 cells: a typical fiber is 20 μm diameter and grows to at least 30 cm over an anagen phase of 2 years,[13] giving a volume of 108 μm3; a single cortical cell has a volume of 103 μm3.[17] Thus, the proliferative potential of keratinocytes derived from the wool follicle GE is at least 2-3 orders of magnitude greater than required to maintain fiber growth for a single follicle cycle. In contrast, clones of transit-amplifying keratinocytes isolated from human epidermis by different methods have been reported to grow for no more than 5[18] or 15[19] divisions. Our result suggests that the wool follicle GE does not consist solely of transit-amplifying cells, and contradicts the idea that the duration of anagen is limited by the proliferative potential of the GE.
Panteleyev et al. postulated that the daughters of stem cells from the follicle bulge migrate down the ORS during anagen to form a distinct sub-population within the GE/matrix compartment, termed the lateral disk.[6] These cells remain quiescent during anagen, are preserved during catagen-associated follicle regression to form the telogen hair germ, and are only activated during the subsequent anagen to repopulate the GE/matrix. Our results show that some cells within the GE possess extensive proliferative potential, but do not indicate that all GE cells are equivalent in this respect. We did not investigate heterogeneity within the GE tissue compartment. The culture conditions we employed may have selected for a distinct but uncharacterized subpopulation of cells. Hence, it is possible that the GE-derived keratinocytes we describe originate from a subpopulation of cells with properties similar to the lateral disk.
Primary cultures of keratinocytes from DP/GE explants have been reported for wool follicles[20] and rat vibrissae,[21] but the isolation and characterization of clones was not described. More generally, a variety of methods have been used to culture cells derived from the GE or matrix of the lower follicle.[5,8,11,22,23] There was considerable variation in the duration for which these cells continued to grow, possibly because of differences in species, tissue isolation methods, and culture conditions. We found much poorer outgrowth of keratinocytes from DP/GE explants derived from rat vibrissae, compared with sheep. The low number of keratinocytes in proportion to fibroblasts that grew from rat DP/GE explants suggests that the culture medium was sub-optimal for the keratinocytes. Furthermore, their variable morphology is suggestive of stressed cells, which could also reflect an inappropriate culture medium. It is not clear to what extent differences in the growth of GE-derived keratinocytes from sheep and other species reflect differences in the culture conditions required for optimal growth, rather than intrinsic differences in proliferative potential.
We showed that preserving the interaction between the GE and DP was important for initiating the growth of ovine keratinocytes in culture. Others have also found extensive keratinocyte proliferation from explants in which the GE was still associated with a DP.[20,21] In contrast, when tissue was disaggregated by trypsinization for the culture of clonogenic cells, only limited growth of keratinocytes from the lower anagen follicle was seen.[10,11] It is thought that the proliferation of GE cells in vivo is dependent on direct interaction with the DP.[12] Unlike DP/GE explants, we found similar profuse outgrowth of keratinocytes from DS/ORS explants of both rat and sheep follicles. The difference in the in vitro proliferation of ORS- and GE-derived keratinocytes from rat vibrissae may reflect a requirement for DP-derived signals to support GE cell growth, rather than a difference in the intrinsic potential of the cells. In a study of human scalp follicles, DP/GE explants also produced keratinocytes with less growth potential than keratinocytes from telogen follicles.[8] For some species, the culture conditions required to initiate outgrowth from GE tissue still interacting with the DP may be different from the conditions required to sustain the growth of daughter cells that have ceased to interact with the DP. It would be interesting to compare the growth behavior of rat DP/GE explants in different culture media, supplemented with different sera and/or growth factors, including growth factors expressed in the DP.
Although DP-derived signals are important for the initiation of ovine keratinocyte cultures from GE tissue, the fact that the daughters of these cells can subsequently be cloned shows that they become independent of the DP once established in culture. The long-term proliferative behavior of clones derived from GE tissue was essentially the same as those from the ORS. Both GE- and ORS-derived keratinocytes appeared to differentiate when maintained in a confluent state, and there was no difference between them in the expression of the four differentiation markers that we investigated. Neither of two wool cortex markers was expressed. It would be interesting to compare their differentiated phenotypes more comprehensively, perhaps by microarray. Nevertheless, the available results suggest that their differentiated phenotype is similar to ORS-derived keratinocytes from other species, for example in the expression of loricrin on differentiation.[24] It therefore appears that the daughters of GE cells might acquire a phenotype more similar to ORS cells as they continue to proliferate in culture.
In summary, our results suggest that GE cells in vivo possess extensive proliferative potential, but their growth is strictly regulated by the DP. In the course of normal hair growth, immigrating ORS cells would displace cells from the GE compartment before their proliferative capacity was exhausted. Anagen would cease when trophic signals from the DP are switched off.
Footnotes
Source of Support: Nil
Conflict of Interest: None declared.
REFERENCES
- 1.Cotsarelis G, Sun TT, Lavker RM. Label-retaining cells reside in the bulge area of pilosebaceous unit: implications for follicular stem cells, hair cycle, and skin carcinogenesis. Cell. 1990;61:1329–37. doi: 10.1016/0092-8674(90)90696-c. [DOI] [PubMed] [Google Scholar]
- 2.Hsu YC, Pasolli HA, Fuchs E. Dynamics between stem cells, niche, and progeny in the hair follicle. Cell. 2011;144:92–105. doi: 10.1016/j.cell.2010.11.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jaks V, Barker N, Kasper M, van Es JH, Snippert HJ, Clevers H, et al. Lgr5 marks cycling, yet long-lived, hair follicle stem cells. Nat Genet. 2008;40:1291–9. doi: 10.1038/ng.239. [DOI] [PubMed] [Google Scholar]
- 4.Nowak JA, Polak L, Pasolli HA, Fuchs E. Hair follicle stem cells are specified and function in early skin morphogenesis. Cell Stem Cell. 2008;3:33–43. doi: 10.1016/j.stem.2008.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oshima H, Rochat A, Kedzia C, Kobayashi K, Barrandon Y. Morphogenesis and renewal of hair follicles from adult multipotent stem cells. Cell. 2001;104:233–45. doi: 10.1016/s0092-8674(01)00208-2. [DOI] [PubMed] [Google Scholar]
- 6.Panteleyev AA, Jahoda CA, Christiano AM. Hair follicle predetermination. J Cell Sci. 2001;114:3419–31. doi: 10.1242/jcs.114.19.3419. [DOI] [PubMed] [Google Scholar]
- 7.Waters JM, Richardson GD, Jahoda CA. Hair follicle stem cells. Semin Cell Dev Biol. 2007;18:245–54. doi: 10.1016/j.semcdb.2007.02.003. [DOI] [PubMed] [Google Scholar]
- 8.Roh C, Tao Q, Photopoulos C, Lyle S. In vitro differences between keratinocyte stem cells and transit-amplifying cells of the human hair follicle. J Invest Dermatol. 2005;125:1099–105. doi: 10.1111/j.0022-202X.2005.23958.x. [DOI] [PubMed] [Google Scholar]
- 9.Gharzi A, Robinson M, Reynolds AJ, Jahoda CA. Repeated plucking and proliferative activity of follicle epidermal cells--significance for follicle cycle control. Exp Dermatol. 1999;8:345–7. [PubMed] [Google Scholar]
- 10.Kobayashi K, Rochat A, Barrandon Y. Segregation of keratinocyte colony-forming cells in the bulge of the rat vibrissa. Proc Natl Acad Sci U S A. 1993;90:7391–5. doi: 10.1073/pnas.90.15.7391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rochat A, Kobayashi K, Barrandon Y. Location of stem cells of human hair follicles by clonal analysis. Cell. 1994;76:1063–73. doi: 10.1016/0092-8674(94)90383-2. [DOI] [PubMed] [Google Scholar]
- 12.Link RE, Paus R, Stenn KS, Kuklinska E, Moellmann G. Epithelial growth by rat vibrissae follicles in vitro requires mesenchymal contact via native extracellular matrix. J Invest Dermatol. 1990;95:202–7. doi: 10.1111/1523-1747.ep12478002. [DOI] [PubMed] [Google Scholar]
- 13.Rogers GE. Biology of the wool follicle: an excursion into a unique tissue interaction system waiting to be re-discovered. Exp Dermatol. 2006;15:931–49. doi: 10.1111/j.1600-0625.2006.00512.x. [DOI] [PubMed] [Google Scholar]
- 14.Rozen S, Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol Biol. 2000;132:365–86. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 15.Muller M. Cellular senescence: molecular mechanisms, in vivo significance, and redox considerations. Antioxid Redox Signal. 2009;11:59–98. doi: 10.1089/ars.2008.2104. [DOI] [PubMed] [Google Scholar]
- 16.Langbein L, Rogers MA, Praetzel-Wunder S, Helmke B, Schirmacher P, Schweizer J. K25 (K25irs1), K26 (K25irs2), K27 (K25irs3), and K28 (K25irs4) represent the type I inner root sheath keratins of the human hair follicle. J Invest Dermatol. 2006;126:2377–86. doi: 10.1038/sj.jid.5700494. [DOI] [PubMed] [Google Scholar]
- 17.Hynd P. Effects of nutrition on wool follicle cell kinetics in sheep differing in efficiency of wool production. Aust J Agric Res. 1989;40:409–17. [Google Scholar]
- 18.Jones PH, Watt FM. Separation of human epidermal stem cells from transit amplifying cells on the basis of differences in integrin function and expression. Cell. 1993;73:713–24. doi: 10.1016/0092-8674(93)90251-k. [DOI] [PubMed] [Google Scholar]
- 19.Barrandon Y, Green H. Three clonal types of keratinocyte with different capacities for multiplication. Proc Natl Acad Sci U S A. 1987;84:2302–6. doi: 10.1073/pnas.84.8.2302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bates EJ, Penno NM, Hynd PI. Wool follicle matrix cells: culture conditions and keratin expression in vitro. Br J Dermatol. 1999;140:216–25. doi: 10.1111/j.1365-2133.1999.02652.x. [DOI] [PubMed] [Google Scholar]
- 21.Osada A, Kobayashi K. Characterization of vibrissa germinative cells: transition of cell types. Exp Dermatol. 2001;10:430–7. doi: 10.1034/j.1600-0625.2001.100606.x. [DOI] [PubMed] [Google Scholar]
- 22.Limat A, Breitkreutz D, Thiekoetter G, Noser F, Hunziker T, Braathen LR, et al. Phenotypic modulation of human hair matrix cells (trichocytes) by environmental influence in vitro and in vivo. Epithelial Cell Biol. 1993;2:55–65. [PubMed] [Google Scholar]
- 23.Reynolds AJ, Jahoda CA. Hair follicle stem cells? A distinct germinative epidermal cell population is activated in vitro by the presence of hair dermal papilla cells. J Cell Sci. 1991;99(Pt 2):373–85. doi: 10.1242/jcs.99.2.373. [DOI] [PubMed] [Google Scholar]
- 24.Blanpain C, Lowry WE, Geoghegan A, Polak L, Fuchs E. Self-renewal, multipotency, and the existence of two cell populations within an epithelial stem cell niche. Cell. 2004;118:635–48. doi: 10.1016/j.cell.2004.08.012. [DOI] [PubMed] [Google Scholar]