

Abstract
The endoribonuclease, Dicer, is indispensable for generating the majority of mature microRNAs (miRNAs), which are posttranscriptional regulators of gene expression involved in a wide range of developmental and pathological processes in the mammalian CNS. Although functions of Dicer-dependent miRNA pathways in neurons and oligodendrocytes have been extensively investigated, little is known about the role of Dicer in astrocytes. Here, we report the effect of Cre-loxP-mediated conditional deletion of Dicer selectively from postnatal astroglia on brain development. Dicer-deficient mice exhibited normal motor development and neurological morphology before postnatal week 5. Thereafter, mutant mice invariably developed a rapidly fulminant neurological decline characterized by ataxia, severe progressive cerebellar degeneration, seizures, uncontrollable movements, and premature death by postnatal week 9–10. Integrated transcription profiling, histological, and functional analyses of cerebella showed that deletion of Dicer in cerebellar astrocytes altered the transcriptome of astrocytes to be more similar to an immature or reactive-like state before the onset of neurological symptoms or morphological changes. As a result, critical and mature astrocytic functions including glutamate uptake and antioxidant pathways were substantially impaired, leading to massive apoptosis of cerebellar granule cells and degeneration of Purkinje cells. Collectively, our study demonstrates the critical involvement of Dicer in normal astrocyte maturation and maintenance. Our findings also reveal non-cell-autonomous roles of astrocytic Dicer-dependent pathways in regulating proper neuronal functions and implicate that loss of or dysregulation of astrocytic Dicer-dependent pathways may be involved in neurodegeneration and other neurological disorders.
Introduction
Astrocytes are crucial for many developmental and physiological functions in the CNS (Barres, 2008; Freeman, 2010). They are essential for promoting neuronal synaptogenesis (Christopherson et al., 2005; Eroglu et al., 2009) and have direct local contacts with neuronal synapses (Bushong et al., 2002). Astrocytes can be activated to release gliotransmitters, including glutamate, GABA, acetylcholine, ATP, adenosine, d-serine, etc. (Halassa and Haydon, 2010). Astrocytes also actively uptake and recycle spilled-over neurotransmitters and maintain ionic balance in the extracellular space (Oliet et al., 2001) and can function as bridging units connecting nearby neurons, endothelial cells, and other glial cells. A growing body of evidence indicates that neuron–glia interactions may contribute to neurological disorders and neurodegeneration, including ischemia, glioma, amyotrophic lateral sclerosis (ALS), AIDS-related neuropathology [for review, see Volterra and Meldolesi (2005) and Sofroniew and Vinters (2010)]. In response to many CNS pathological conditions, astrocytes may become reactive with hallmarks of upregulation of intermediate filament proteins such as glial fibrillary acidic protein (Gfap) and Vimentin. Reactive astrocytes may provide neuroprotective effects in the early stage of the injury, whereas at a later stage the formation of glial scar inhibits CNS regeneration (Sofroniew, 2009).
miRNAs are endogenous noncoding RNAs that regulate gene expression in a sequence-specific manner by either mRNA cleavage or translational repression (Bartel, 2004; He and Hannon, 2004; Rana, 2007). Transcribed nascent pri-miRNAs are processed by Drosha and Dicer in a stepwise manner to produce mature miRNAs (Hutvágner et al., 2001; Lee et al., 2003). The RNase III endoribonuclease Dicer is essential for the majority of mature miRNA biogenesis. Dicer deletion in early embryonic neuroepithelial cells results in dramatic impairment of neural lineage differentiation (Davis et al., 2008; De Pietri Tonelli et al., 2008). Ablation of Dicer in postmitotic neurons results in neuronal dysfunction or apoptosis (Kim et al., 2007; Schaefer et al., 2007). Importantly, dysregulation of miRNAs is implicated in neurological disorders (Saba and Schratt, 2010; Wu et al., 2010). Dicer is also found to be an important regulator of oligodendrocyte differentiation (Shin et al., 2009; Dugas et al., 2010; Zhao et al., 2010). However, the role of Dicer-dependent pathways in astrocytes is yet to be revealed.
By using two transgenic mouse lines in which cre recombinase expression was controlled by a mouse Gfap gene regulatory sequence, which turned on primarily postnatally in the CNS, we devised mouse models in which floxed Dicer alleles were disrupted in astroglia. Here we report that loss of Dicer in astroglia leads to ataxia, cerebellar degeneration, seizures, and premature death. In this study, we focused on the cerebellum, where dramatic neurodegeneration occurred. We found that before the onset of neurological symptoms, Dicer-deficient mature astroglial transcriptome was altered to resemble an immature or reactive state with important mature astroglial functional genes downregulated and immature/developing astrocytic genes upregulated, thereby contributing to excitotoxicity and neurological dysfunction.
Materials and Methods
Generation and characterization of mGFAP-Cre;Dicerflox/flox mice.
Mice were maintained in a 12 h light/dark cycle under standard conditions at the animal facility at the University of California, Los Angeles (UCLA). Experiments were conducted in accordance of protocols approved by the UCLA Office for Protection of Research Subjects. mGfap-Cre transgenic mice line 73.12, line 77.6, and Rosa 26-LacZ mice were provided by M. V. Sofroniew (UCLA). Dicerflox/flox mice (Cobb et al., 2005) were kindly provided by S. T. Smale (UCLA) and M. Merkenschlager (Imperial College London).
Genomic DNA PCR.
Brain tissues or cells were lysed in DNA lysis buffer (100 mm Tris-Cl, pH 8.1, 200 mm NaCl, 5 mm EDTA, 0.2% SDS) with proteinase K. Genomic DNA were extracted and subjected to PCR. Primers used to discriminate floxed and deleted Dicer alleles are listed below, and the numbers refer to position downstream of start codon. 28290: AGTAATGTGAGCAATAGTCCCAG; 31831: AGTGTAGCCTTAGCCATTTGC; 32050 AS: CTGGTGGCTTGAGGACAAGAC; WT allele (259 bp product): 31831 and 32050AS; floxed allele (390 bp product): 31831 and 32050AS; deleted allele (309 bp product): 28290 and 32050AS.
Behavioral analysis.
The motor function and balance of seven control male mice and six mutant male mice were measured by rotarod task at an initial speed of 4 rpm, accelerating up to 40 rpm. The time (in seconds) taken for the mice to fall from the rod was measured. Mice were trained for 2–3 d with three trials per day before testing at postnatal week 5 and week 7. Footprint patterns were obtained by dipping the front paws into nontoxic red paint and the hindpaws into nontoxic green paint and placing mice on top of a piece of white paper at the end of a tunnel. Repeated-measures two-way ANOVA or paired t test was used for statistical analysis.
Western blot analysis.
Whole-cell lysates were prepared using SDS lysis buffer (1% SDS, 10 mm EDTA, 50 mm Tris-HCl, pH 8.1) supplemented with protease inhibitor cocktails (Roche), 1 mm PMSF, and 1 mm DTT. The lysates were immediately pulse sonicated using a microtip (Branson sonifier 450) to break the viscosity caused by chromatin released during the process. Protein concentrations were measured using the BCA Protein Assay Kit (Pierce). The lysates were loaded with SDS sample buffer and size-separated with 12% SDS-PAGE. Primary antibodies used were as follows: GLT-1 (Millipore Bioscience Research Reagents), GLAST (Millipore Bioscience Research Reagents), and β actin (Sigma). Secondary goat anti-mouse or anti-rabbit IgG-horseradish antibodies (Calbiochem) were used, and detection was performed using the ECL Plus chemiluminescence system (PerkinElmer) on X-Omat Blue films (Kodak). Densitometry analysis was done with Quantity One (Bio-rad).
Immunohistochemistry of tissue sections.
Frozen or floating tissue sections were prepared from 4% paraformaldehyde-fixed brains. Fluorescence immunohistochemistry was performed using secondary antibodies. Primary antibodies used were mouse anti-NeuN (Millipore Bioscience Research Reagents); rabbit anti-Gfap (Accurate); mouse anti-Gfap (Sigma); chicken anti-β-galactosidase (Abcam); mouse anti-S100β (Sigma); rabbit anti-calbindin (Cell Signaling Technology). Stained sections were examined and photographed using fluorescence microscopy (Zeiss) and scanning confocal laser microscopy (Leica).
TUNEL assay.
Frozen sections of 10 μm thickness were used for TUNEL assay following the manufacturer's instructions (Roche).
Golgi staining.
Adult mouse brains were quickly dissected out and used for Golgi staining with FD Rapid GolgiStain kit following the manufacturer's instructions (FD Neuro Technologies).
Glutamate uptake assay.
Glutamate uptake assays were performed as previously described (Liévens et al., 2001). Cerebella were rapidly dissected out at 4° from control and mutant animals, immediately homogenized in a 0.32 m isotonic sucrose solution, and diluted 1/80 (w/v) in a Tris-buffered sodium-free physiological medium (16.2 mm Tris, 0.38 m sucrose, 10 mm glucose, 5 mm KCl, 1.2 mm CaCl2, and 1.2 mm MgSO4, pH 7.4). After centrifugation at 10,000 rpm for 10 min, the supernatants were discarded and the pellet was resuspended in twice the initial volume of physiological medium (final dilution 1/160 (w/v)) and kept on ice. One micromolar l-[3H]GLU (GE Healthcare Pharmacia Biotech) was preincubated with 80 μl of the physiological medium in which the sucrose has been replaced by 140 mm NaCl, 1.2 mm NaH2PO4/Na2HPO4 at 25° for 5 min. The uptake assays started by adding 20 μl aliquots of the synaptosomes. The suspension was incubated at 25°C for 2 min and the reaction was stopped on ice. Excessive nonincorporated radio-labeled glutamate was removed by centrifugation at 10,000 rpm for 5 min. The pellet was washed with 400 μl of the sodium-free physiological medium followed by centrifugation at 10,000 rpm for 5 min. The pellet was dissolved by lysis buffer and the radioactivity was assessed by liquid scintillation count (Beckman). The blank values were obtained from samples incubated in sodium-free medium. Protein concentrations were determined by BCA assay (Pierce). The radioactivity readouts were normalized to their corresponding protein concentrations.
Gene expression microarrays.
Total RNA was extracted from four independent pairs of cerebella from postnatal day 30 (P30) control and mutant mice of either sex using Trizol (Invitrogen). Two-color gene expression microarrays (Agilent) were used to directly compare relative changes in mRNA expression levels between wild-type and mutant cerebella. Five hundred nanograms of total RNA were labeled with Cy3- or Cy5-CTP using the Agilent Low RNA Input Fluorescent Linear Amplification Kit. After labeling and cRNA purification (Qiagen), cRNA was quantified using the NanoDrop spectrophotometer (Agilent). Eight hundred twenty-five nanograms of Cy3- or Cy5-labeled cRNA were combined and hybridized to the Agilent 4×44K whole mouse genome microarray (G4122F, ∼44,000 probes per array) for 17 h at 65°C (10 rpm). Four replicate experiments using samples derived from four pairs of littermate mice were performed with a dye-swap experimental design. Data were collected using Agilent microarray scanner and extracted using Feature Extraction 9.5 software (Agilent).
For identification of differentially expressed genes, we used NIA array analysis tool (http://lgsun.grc.nia.nih.gov/ANOVA). Of all the probes present on the microarray, signal intensity of redundant probes was averaged before analysis. The following parameters were used for analyzing statistically significant differential expression: threshold z-value to remove outliers: 10,000; error model: max (average, Bayesian); error variance averaging window: 200; proportion of highest error variances to be removed: 0.05; Bayesian degrees of freedom: 10; false discovery rate (FDR) threshold: 0.05.
For clustering and heat map display, Lowess-normalized signal intensity was log2 transformed and median centered. Heat maps were generated using Cluster3 and Java Treeview.
The transcription profiling data has been deposited in the NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) and is accessible through GEO Series accession number GSE28589.
RNA isolation and quantitative real-time RT-PCR.
Total RNA was isolated using Trizol (Invitrogen). After Turbo DNase (Ambion) treatment, 1 μg of total RNA was used for cDNA synthesis using Superscript III (Invitrogen) following manufacturer's recommendations. Real-time PCR was performed in an iCycler using iQ SYBR Green Supermix (Bio-Rad). PCR efficiency and specificity of each primer pair was examined by standard curve of serially diluted cDNA and melting curve functionality, respectively. Fold change was calculated based on 2−ΔΔCt method after normalization to the transcript level of housekeeping gene Gapdh. Primer sequences used in the real-time RT-PCR are as follows. Tnc forward: tggagtacgagctgcatgac; Tnc reverse: aaacttggtggcgatggtag; Thbs2 forward: acgagtttgggtctgtggac; Thbs2 reverse: tcccagtaggtctgggtcac; Adamts1 forward: gataatggacacggggaatg; Adamts1 reverse: gcagtgcttggattcctctc; Vimentin forward: ccagagaccccagcgctcct; Vimentin reverse: gccggagccaccgaacatcc; Tgfb3 forward: cccaaccccagctccaagcg; Tgfb3 reverse: cctcaacagccactcgcgca; Aqp4 forward: atcatgggcgctgtgctggc; Aqp4 reverse: gctgcgcggctttgctgaag; Dao1 forward: cggcctcaggtccggctaga; Dao1 reverse: aggttggccgcctccattgc; Gstm3 forward: tctgcactgtggctcccggt; Gstm3 reverse: gctgtgaggttgggtctgggc; GLAST forward: gccctccgaccgtataaaat; GLAST reverse: gccattcctgtgacgagact; GLT-1 forward: ccaagctgatggtggagtttc; GLT-1 reverse: gtccttgatggcgatgatct; Gapdh forward: ctgagtatgtcgtggagtctactgg; Gapdh reverse: gtcatattctcgtggttcacacc.
Slice preparation for electrophysiology.
Adult P35–P40 mice (presymptomatic or early symptomatic stage) were anesthetized with halothane and decapitated according to a protocol approved by the UCLA Chancellor's Animal Research Committee. The brains were removed and placed in ice-cold artificial CSF (aCSF, 126 mm NaCl, 2.5 mm KCl, 2 mm CaCl2, 2 mm MgCl2, 1.25 mm NaH2PO4, 26 mm NaHCO3, and 10 mm d-glucose, pH 7.3–7.4 when bubbled with 95% O2 and 5% CO2). Sagittal cerebellar sections (350 μm thick) were cut with a Leica VT1000S Vibratome in aCSF containing 3 mm kynurenic acid (Sigma) and placed into a submerged chamber at 32°C.
Electrophysiological recording.
For whole-cell recordings, slices were transferred to a submerged recording chamber and perfused (4–5 ml/min) at 31–33°C with 95% O2 and 5% CO2 saturated aCSF. Cerebellar granule cells were visually identified by IR-DIC videomicroscopy (custom-made; 40× water-immersion objective) and recorded with an Axopatch 200B amplifier (Molecular Devices). For measurement of NMDA receptor-mediated tonic currents, d-AP5 was injected into bath (final concentration >50 μm) after reaching the stable baseline for 5 min. Series resistance and whole-cell capacitance were estimated and compensated to 70–80% (lag 10 μs). Recordings were discontinued if series resistance increased by >25%.
Data acquisition and analyses.
All recordings were low-pass filtered at 3 kHz and digitized on-line at 10 Hz using a PCI-MIO 16E-4 data acquisition board (National Instruments). Tonic current measurement was determined as described previously (Glykys and Mody, 2007). Detection and analyses were performed using custom-written LabView-based software (EVAN). All data are shown as mean ± SEM. Statistical analysis was performed by Student's t test. Significance was set to p < 0.05. We used bootstrap statistics to compare the potentiation (expressed as fold of baseline) of the tonic NMDA receptor-mediated current by the glutamate uptake blocker TBOA. This was necessary because the full measurement of the tonic current under three different experimental conditions (control, TBOA, and AP5) in the same cell proved to be technically challenging due to the large tonic current induced by TBOA. Therefore, measurements of the tonic NMDA current (AP5-sensitive component of the holding current) were done by subtracting the holding current in the presence of AP5 from that before its perfusion. This current was converted to conductance and was normalized to cell capacitance. The values were (in siemens per ampere; mean + SEM) as follows: WT: 50.8 + 9.5 (n = 7), mutant (Mut): 262.1 + 31.1 (n = 7). In another set of recordings, the increase in holding current produced perfusion of TBOA was measured, and it was assumed that it represented the additional NMDA current following blockade of glutamate uptake. Other glutamate receptors were unlikely to participate in this current as they were either desensitized or inwardly rectifying at the holding potential (+30 mV). The recorded currents were also converted to conductance and normalized to cell capacitance. The obtained values were (in siemens per ampere; mean + SEM) as follows: WT: 776.0 + 68.5 (n = 7), Mut: 633.1 + 70.6 (n = 8). The bootstrap statistics for each group (WT and Mut) were calculated as follows. A 7 × 7 and a 7 × 8 table were generated from the WT and Mut data, respectively. In each row, the 7 [control (Con)] or 8 (Mut) TBOA values were added to one of the 7 tonic NMDA current values to yield the total of all possible combinations of 49 (Con) and 56 (Mut) summed values. These summed were considered the total NMDA receptor-mediated tonic current (baseline + TBOA) in the presence of TBOA. Then 7 (Con) and 8 (Mut) values were selected at random from this table and were each divided by a randomly selected basal NMDA current value to yield 7 (Con) and 8 (Mut) (basal+TBOA)/basal ratios, which represented the fold potentiation by TBOA. The average value of these 7 (Con) or 8 (Mut) ratios was calculated. The procedure was repeated 1103 times, yielding as many average ratios. These ratios were then plotted as frequency histograms to yield the average bootstrap TBOA potentiation ratios (Con: 20.08 and Mut: 3.94) and the 95% confidence levels.
Results
mGfap-Cre transgenic lines target astroglia in cerebellum
Glial fibrillary acidic protein (Gfap) is a widely used marker for astroglia. In the most commonly used 2.2 kb human GFAP promoter-driven cre transgenic mouse line (hGFAP-cre), cre activity turns on in embryonic multipotent neural precursor cells (Zhuo et al., 2001) as early as embryonic day 13.5 (E13.5), thereby leading to widespread lacZ reporter expression in neurons and astroglia throughout the brain of hGFAP-cre;Rosa26-LacZ reporter mouse. In search for a more astroglial lineage-restricted transgenic mouse line, we characterized two independent lines [line B6.Cg-Tg (Gfap-cre)73.12Mvs/J (mGfap-cre 73.12) and line B6.Cg-Tg(Gfap-cre)77.6Mvs/J (mGfap-cre 77.6)], in which cre recombinase expression was driven by the 15 kb mouse Gfap gene regulatory sequence. Previous studies using single-cell evaluations of reporter gene expression have shown that cre activity in line 73.12 mice was present in postnatal astrocytes throughout the CNS, as well as in adult neural stem/progenitor cells localized in subventricular zone (SVZ) and subgranular zone (SGZ) (Garcia et al., 2004; Herrmann et al., 2008). In line 77.6, cre recombinase activity was also targeted to postnatal astrocytes throughout the CNS, and a subpopulation of adult neural stem/progenitor cells in SVZ, but to a much lesser extent in SGZ (Gregorian et al., 2009) and not to nonastrocytes in other CNS regions (unpublished observations, M. V. Sofroniew).
To further examine expression patterns of mGfap-driven Cre recombinase, both transgenic lines were crossed with a reporter line in which expression of β-galactosidase (β-gal) protein was under the regulation of an ubiquitous promoter (Rosa-26) that contained a loxP-flanked stop sequence (Fig. 1A). As expected, while β-gal expression was detected in astrocytes throughout the CNS as well as in small populations of neurons in olfactory bulb and hippocampus that are derived from Gfap-expressing adult neural/stem progenitors, cerebellum was among the most densely labeled brain regions with β-gal expression mostly restricted to astrocytes. In the cerebellum, cre was first activated in line 73.12 at ∼P4 and was first detected in line 77.6 at ∼P7. As β-gal staining of P35 mouse cerebella showed, in line 77.6, the vast majority of labeled cells were Bergmann glia and inner granule layer (IGL) astrocytes that were also positive for Aldh1l1, another astroglial marker (Cahoy et al., 2008; Yang et al., 2011) (Fig. 1B,D). No NeuN-positive IGL granule neurons or calbindin-positive Purkinje neurons expressed β-gal at any time point examined in line 77.6 (Fig. 1D). In line 73.12, besides Bergmann glia and IGL astrocytes, there was a very small fraction (1–5%) of granule neurons expressing β-gal, which were possibly derived from postnatal neuronal progenitors that transiently expressed cre (Fig. 1D). No Purkinje neurons expressed reporter protein in line 73.12 at any time point examined. Therefore, in contrast to the commonly used hGFAP-cre line, activation of mGfap-cre was largely restricted to postnatal astrocytes throughout the CNS in both transgenic lines, and in cerebellum, targeting of cre in mGfap-cre line 77.6 was restricted exclusively to postnatal astroglial cells.
Figure 1.
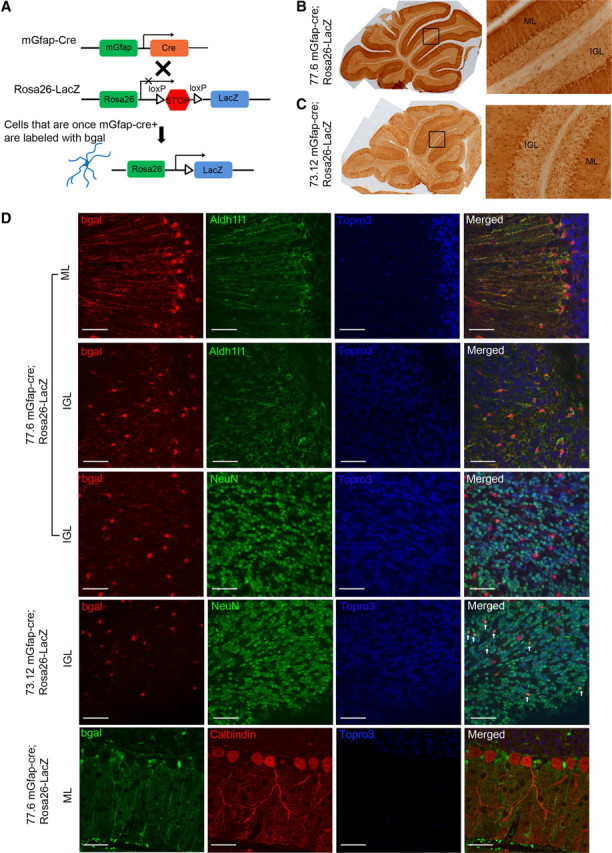
In cerebella, mGfap-cre transgenic lines mainly target postnatal astroglial cells. A, A schematic of lineage tracing using mGfap-cre transgenic mice crossing with Rosa26-LacZ reporter line. B, C, Bright-field immunohistochemistry DAB stainings of the reporter protein β-gal in P35 mGfap-cre; Rosa26-LacZ mice cerebella showed that Bergmann glia and IGL astrocytes were positively labeled (B for line 77.6 and C for line 73.12). ML, Molecular layer. D, Representative confocal images of colabeling of reporter protein β-gal and astroglial marker Aldh1L1, neuronal marker NeuN, or Purkinje cell marker Calbindin in cerebellum. Scale bars, 50 μm. β-gal colocalized well with Aldh1L1 and there was no colocalization of β-gal and Calbindin. There was no colocalization of β-gal and NeuN in line 77.6. The ratio of β-gal+NeuN+/NeuN+ in line 73.12 was calculated to be ∼1–5% from multiple confocal pictures. The white arrows indicate colocalization of β-gal and NeuN in line 73.12.
mGfap-cre; Dicerflox;flox mutant mice developed ataxia, epilepsy, and premature death
To investigate the role of Dicer-dependent pathways in the astroglial lineage, Dicerflox/flox mice (Cobb et al., 2005) were crossbred with mGfap-cre (both 77.6 and 73.12 line) mice (Fig. 2A). mGfap-cre; Dicerflox/+ (hereafter referred to as control) mice were phenotypically indistinguishable from wild-type mice. mGfap-cre; Dicerflox/flox (hereafter referred to as mutant) mice were normal at birth and born with normal Mendelian ratios. Genomic DNA PCR with whole cerebella at presymptomatic/early symptomatic stage showed a small percentage of deleted Dicerflox allele, which was consistent with the fact that the cerebellum harbored numerous granule neurons, and that astroglia were not the major population in the cerebellum (Fig. 2B). Using in vitro cultured neurospheres isolated from postnatal SVZ of mutant mice, where Dicer was looped out in majority of the cells, as a positive control for the genomic PCR, we confirmed that genetic deletion of Dicer floxed alleles did occur in the cerebellum (Fig. 2B). During these experiments, we noted both proliferation and differentiation deficits in Dicer-deficient SVZ adult progenitor cells that will be of interest for future investigations (unpublished observations, J. Tao and Y. E. Sun).
Figure 2.

Dicerflox/flox; mGfap-cre mutant mice developed ataxia. A, A schematic of crossing mGfap-cre transgenic mice with Dicer lox/lox line. B, Genomic DNA PCR showed the ratio of Dicerflox allele (390 bp), Dicerdel allele (309 bp), and wild-type allele (259 bp). DNA samples were extracted either from whole cerebella or in vitro cultured neurospheres derived from SVZ neural progenitor cells. C, Rotarod tests at postnatal day 35 showed a slightly weaker performance of mutant mice compared to control mice and same test repeated at postnatal day 50 showed dramatically worsened performance of mutant mice compared to control mice. N = 7 for control, N = 6 for mutant, **p = 0.004, two-tailed t test. D, Footprint assay at postnatal week 7 showed ataxic gaits in mutant mice: distance between two hindpaws increased and the overlaps between front and hindpaws of the same side were poor. Red paint, Front paws; green paint, hindpaws. N = 2 pairs. p < 0.0001 for hind base distance, p = 0.0004 for overlap, two-tailed t test. E, The Kaplan–Meier survival curves of control and mutant mice. Median survival days are 56 d for 73.12 line (mutant N = 19, control N = 20) and 68 d for 77.6 line (mutant N = 17, control N = 17).
Mutant mice grew normally for the first 5 weeks postnatally, but around postnatal week 7–8, they started to exhibit noticeable wobbly, imbalanced gait. Rotarod training and testing at postnatal 5 week showed a slightly weaker trend of performance by mutant mice as compared to their littermate controls, whereas at postnatal week 7, mutant mice displayed severe impairment in motor function and balance (Fig. 2C). The footprint assay also confirmed an ataxic walking pattern of mutant mice at week 7 with increased hindpaw distances and poor overlaps of front and hindpaws (Fig. 2D). Moreover, the mutant mice exhibited spontaneous epileptic seizures around postnatal week 8 and 9 characterized by uncontrollable movements, Straub tail, and maintained opisthotonos, possibly due to the loss of Dicer in hippocampal subgranular zone neural progenitors, hippocampal, and/or cortical astrocytes. With rapidly aggravating locomotor problems and prolonged seizures, the mutant mice became immobile and became moribund between postnatal weeks 8 and 9. Both 73.12 and 77.6 line mutants exhibited ataxia and epilepsy phenotypes and showed very steep survival curves (Fig. 2E). Line 77.6 mutants survived on average 12 d longer than line 73.12 mutants, possibly due to the fact that fewer hippocampal neurons were targeted in line 77.6 compared to line 73.12, thereby leading to a later onset of epilepsy. Mutant mice typically reached the late symptomatic stage (or end-stage) very quickly with an average of 12 d after the onset of symptoms. End-stage mutant mice also showed slight weight loss and poor grooming.
mGfap-cre; Dicerflox;flox mutant mice had cerebellum degeneration
The brains of end-stage mGfap-cre; Dicerflox;flox mutant mice were grossly normal in size and morphology, but cerebella were severely degenerated with blurred fissures (Fig. 3A). We therefore focused our investigation on the cerebellum for the present study. To discriminate whether the degeneration phenotype that we observed was a cerebellar developmental deficit or a degenerative process that occurred after development had completed, we performed time course Nissl stainings with P7, P15, and P30 control and mutant littermates. It was evident that mGfap-cre; Dicerflox;flox mutant mice had structurally normal cerebellar development and cellular layers were established properly by P30 before the onset of motor symptoms (Fig. 3B). Therefore, in contrast to the early developmental defects of cerebella (e.g., reduction in cerebellar size, loss of granule cells, and disrupted cellular layers) caused by the ablation of Gfap-positive astrocytes during the first postnatal week using hGfap-HSV-TK transgenic mice (Delaney et al., 1996), the degeneration process in mature mGfap-cre; Dicerflox;flox mice was not simply due to a lack of astrocytes at early postnatal developmental stages.
Figure 3.
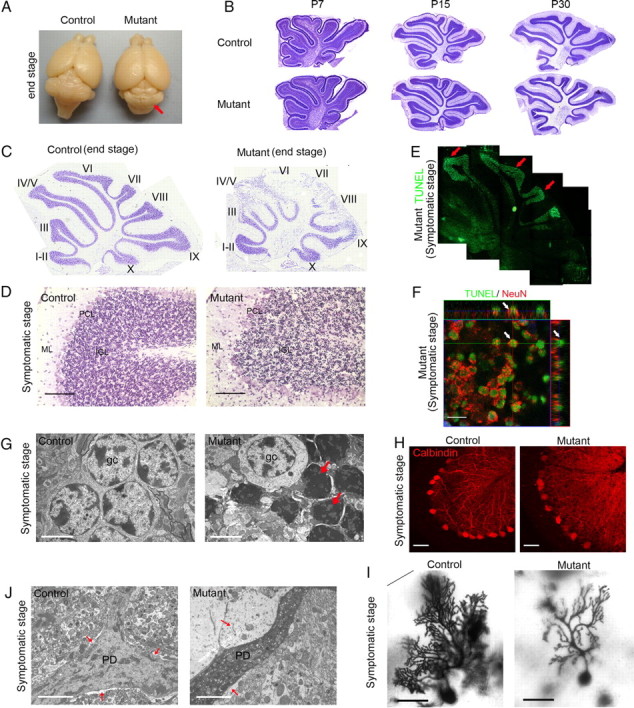
Dicerflox/flox; mGfap-cre mutant mice had normal cerebellar development but around postnatal week 8–9, they had granule cell apoptosis and Purkinje cell dendrite degeneration. A, Compared to control mice, end-stage mutant mice brains showed cerebellar degeneration despite grossly normal morphology of other brain regions (observed with 100% penetrance). B, Nissl stainings of P7, P15, and P30 control and mutant littermate mice cerebella showed normal cerebellar development (N = 3 for each age). C, Nissl stainings of end-stage mutant cerebella showed severe cell loss (N > 10). D, Nissl stainings of late-stage symptomatic mutant mice showed massive condensed nuclei in cerebellar IGL (N = 4). Scale bar, 100 μm. E, TUNEL assays of symptomatic stage mutant mice brains showed massive IGL cells undergoing apoptosis (N = 4). F, A representative orthogonal image showed three-dimensional analysis of individual cells that were positive for both TUNEL and NeuN. Scale bar, 10 μm. G, Electron microscopy pictures showed that in late symptomatic stage mutant cerebella, granule cells became condensed and darkened as marked by arrows. Scale bar, 321 nm. N = 2. H, Calbindin stainings of Purkinje cell bodies and dendrites of symptomatic stage mutant and control cerebella (N = 3). Scale bar, 50 μm. I, Golgi stainings showed markedly reduced dendritic arborization of Purkinje cell in late symptomatic stage mutant cerebellum. Scale bar, 100 μm. N = 2. J, Representative electron microscopy pictures showed that compared to control littermate, late symptomatic stage mutant Purkinje cell dendrites (marked as PD) had increased electron density and surrounding Bergmann glia (marked by arrow) showed swollen cytoplasm with very low electron density. Scale bar, 321 nm. N = 2.
In end-stage (P62 for 73.12 line and P70 for 77.6 line) mutant cerebella, Nissl staining showed that IGL cell density was dramatically decreased, although cerebellar layers and lobes developed normally early on (Fig. 3C). To determine whether there was any cell loss at the time of the onset of neurological symptoms, symptomatic mutant mice (P55–P60) cerebellar sections were stained with cresyl violet. The presence of massive, dark condensed nuclei in IGL suggested acute synchronized cell death (Fig. 3D). Consistent with the Nissl staining results, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay in brain sections of symptomatic stage mice showed abundant positive signals throughout the cerebellum IGL, but nowhere else in the mutant brain (Fig. 3E). Since granule neurons are the most abundant cells in the IGL, we performed double labeling with TUNEL and a neuronal marker, NeuN. The >90% colocalization of the two markers implied that the majority of granule neurons underwent cell death (Fig. 3F). Electron microscopy analyses also demonstrated the presence of a large number of condensed darkened granule cells in symptomatic mutant mice cerebella (Fig. 3G). In addition to massive granule cell death, although Purkinje cell bodies remained largely intact until late symptomatic stage, they showed disintegration of dendritic arborizations as indicated by immunostaining of calbindin, which labeled Purkinje cell dendrites (Fig. 3H). Purkinje dendritic degeneration was further confirmed by Golgi staining (Fig. 3I). Moreover, in Purkinje cell dendrites, we observed prominent vacuole formation, darkened cytoplasm, and rounded mitochondria, which were indicative of dark cell degeneration (Fig. 3J). The observation that major neuronal populations underwent degeneration while the cell type in which Dicer was deleted was Gfap-positive astroglia indicates a non-cell-autonomous effect.
It is noteworthy that Bergmann glia, a major cerebellar astroglial population in which Dicer was deleted, remained alive and exhibited thickened, swollen processes (Fig. 3J). To investigate whether the morphological change of Bergmann glia is a secondary response to neuronal degeneration, a manifestation of primary astrocytic pathology, or both, Gfap immunohistochemical analyses were performed in sagittal cerebellar sections of mice at various developmental stages. While mutant mice seemed to have normal astroglia morphology and Gfap immunointensity during early postnatal cerebellar development (up to P15), mutant Bergmann glia showed increased Gfap immunoreactivity as early as P30 (presymptomatic stage) (Fig. 4A). At early symptomatic stage (P42), mutant Bergmann glia processes were associated with elevated levels of Gfap expression, conspicuously thickened, and disorganized compared with the fine Gfap-positive glial processes oriented perpendicular to the pia in control mice (Fig. 4B,C). In contrast, in IGL astrocytes, both Gfap and S100β staining were decreased (Fig. 4D,E) at early symptomatic stage (P42), while the Aldh1l1 staining pattern did not change. Furthermore, no obvious change in total cell number (by corresponding nuclear staining) was observed in IGL, nor were there any TUNEL-positive cells detected at presymptomatic/early symptomatic stages, suggesting that the decrease of Gfap immunoreactivity in IGL astrocytes did not indicate a loss of IGL astroglia, but more likely reflected changes in glial protein expression in Dicer-deficient astrocytes. These results suggest that the marked morphological abnormality of both IGL astrocytes and Bergmann glia precedes any obvious behavioral impairments or neuronal death, and therefore may represent early or upstream events in the progression of pathogenesis.
Figure 4.
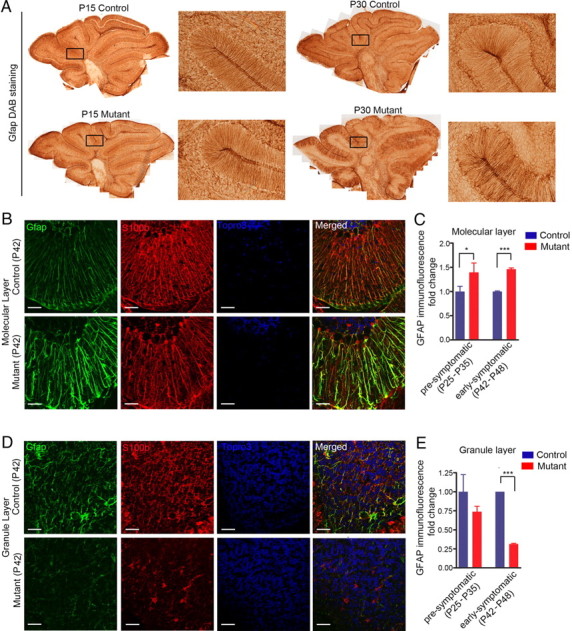
Astroglial morphological changes occurred before neurological symptoms. A, Gfap DAB stainings showed no difference at early postnatal stages (P15) and the earliest morphological changes were observed at ∼P30. B, Bergman glia in mutant cerebella showed increased Gfap staining. C, Quantification of molecular layer Gfap immunofluorescence fold change of presymptomatic (P24–P32) and early symptomatic (P42–P48) mice cerebella. Two-tailed t test, *p < 0.05, ***p < 0.001. D, IGL astrocytes showed decreased Gfap and S100β staining. E, Quantification of granule layer Gfap immunofluorescence fold change of presymptomatic (P24–P32) and early symptomatic (P42–P48) mice cerebella. Two-tailed t test, ***p < 0.001.
Deletion of Dicer altered astrocytic transcriptome in presymptomatic stage cerebella
To reveal the early molecular changes at the presymptomatic stage that might be responsible for more severe neuronal symptoms at the end-stage, we performed whole-genome transcription profiling with four biologically independent pairs of control and mutant cerebella at P30. As shown in Figure 5A, 367 genes were upregulated and 267 genes were downregulated consistently in all four pairs of mutant cerebella compared to their littermate controls (FDR < 0.05). To further dissect relative contributions from different cell types of the cerebellum to the dysregulated gene sets, we cross-referenced our dysregulated gene list to previously annotated databases of cell type-specific genes in various cerebellar glial and neuronal cells (Doyle et al., 2008). This analysis indicated that the majority of dysregulated genes were highly enriched in astrocytic cells (Bergmann glia and astrocytes), while very few neuron-specific genes showed transcriptional changes (Fig. 5B). Although we could not exclude the possibility that some dysregulated genes might arise from neuronal cell types, the majority of the transcriptional changes were associated with astrocyte-specific genes. To further reveal the functions of dysregulated genes in mutant cerebellar astrocytes, we next performed gene ontology (GO) analyses. Among the upregulated genes, which included hallmark genes of reactive astrocytes (Gfap, Vimentin, Tenascin C, etc.), 148 genes encoded for glycoproteins, especially genes which functioned in extracellular matrix, cell adhesion, and calcium ion binding (Fig. 5C). A group of chemotaxis genes were also upregulated, although there was no increase in Iba1+ microglia in mutant cerebella until symptomatic stage, suggesting that these upregulated cytokine/chemokine genes might serve as signals for later microglia activation. In addition, a group of solute carrier family genes was upregulated significantly, although little was known about their functions in the brain. A group of cell cycle-related genes were also upregulated. In contrast, a cohort of important glial-specific functional genes were downregulated, which were mostly involved in oxidation reduction, glutathione metabolism, chemical homeostasis, and glutamate transport (Fig. 5C). Also, a group of synaptic genes were downregulated, which might be due to secondary neuronal transcriptional changes.
Figure 5.
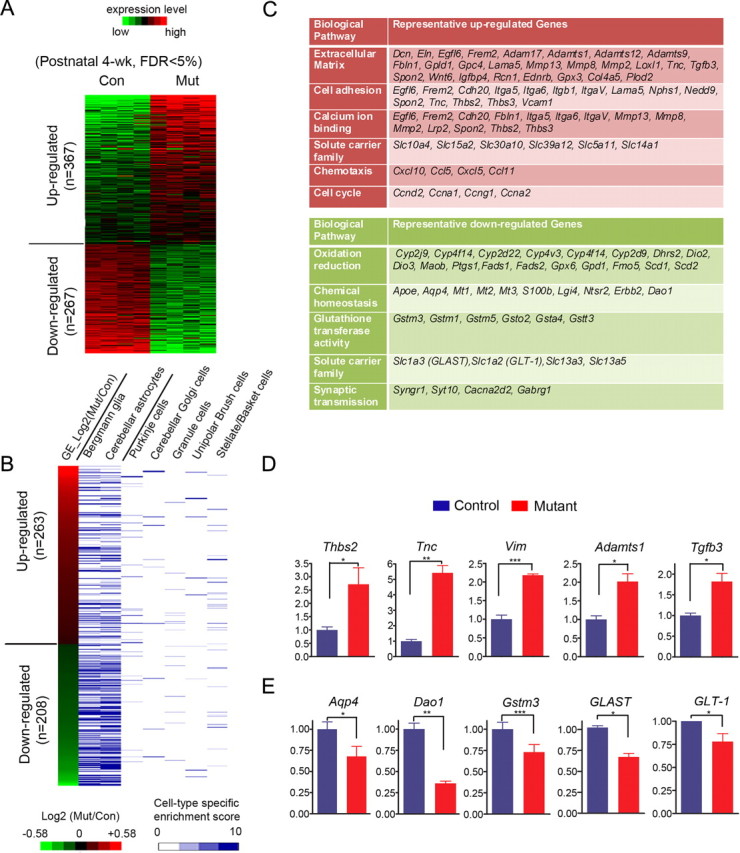
Transcription profiling of control versus mutant mice cerebella at presymptomatic stage (P30) showed a cohort of astrocytic genes dysregulated. A, Heat map representations of relative changes in gene expression (from four pairs of littermate cerebella) between control and mutant cerebella at the presymptomatic stage were shown for differentially expressed genes (FDR < 0.05). B, Heat map representations of cerebellar cell type-specific expression patterns of differentially expressed genes between control and mutant cerebella. The heat map is ranked by changes in expression between control and mutant cerebella. The information on cell type-specific gene expression profiles was derived from a previously published dataset (Doyle et al., 2008). There were 263 upregulated genes and 208 downregulated genes mappable to the reference dataset. The cell type-specific enrichment score represented the measurement of relative expression level of a given gene compared to a universal reference. C, Tables showed representative upregulated (red) and downregulated genes (green) by gene ontology analyses. D, RT-QPCR validation of representative upregulated genes. Two-tailed paired t tests, N = 4. *p < 0.05, **p < 0.01, or ***p < 0.005. E, RT-QPCR validation of representative downregulated genes involved in oxidation reduction and glutamate transport. Two-tailed paired t tests, N = 4, *p < 0.05, **p < 0.01.
The observation that many immature/reactive astrocyte genes were upregulated, while astrocytic genes related to mature astrocyte functions were downregulated in response to Dicer deletion, raised the possibility that Dicer was required for astrocytes to become fully functional during astrocytic maturation process in the first three postnatal weeks. Recent transcriptome analyses of acutely isolated purified astrocytes demonstrate that astrocytes undergo substantial changes in transcriptome from developing (P7–P8) to mature (P17 and after) stages (Cahoy et al., 2008), indicating that astrocytes at different developmental stages are associated with distinct genetic programs that may determine their functional maturity. To examine whether dysregulated genes in Dicer mutant mice (P30) were associated with specific developmental stages of astrocytes, we cross-referenced Dicer-dependent gene expression changes to lists of genes highly expressed in developing/immature and mature postnatal astrocytes, respectively. Interestingly, this analysis indicated that 62 genes were both downregulated in Dicer mutant and specifically enriched in mature astrocytes, which was significantly higher than that expected by chance (p = 7.0 × 10−15, Fisher's exact test) (representative genes listed in Table 1). For example, GLT-1 (also known as Slc1a2), a glial transporter highly expressed only in mature, not developing/immature, astrocytes was reproducibly downregulated in absence of Dicer in astrocytes at P30. BC055107, one of the genes most strongly upregulated during astrocyte development and suggested to maintain cells in a quiescent state (Cahoy et al., 2008), was also markedly downregulated in Dicer mutant. Other genes highly enriched in mature astrocytes and downregulated in Dicer mutant were involved in oxidation reduction and cellular homeostasis pathways. Similarly, 54 genes were both upregulated in Dicer mutant astrocytes and enriched in developing/immature astrocytes, which was also significantly higher than that expected by chance (p = 1.1 × 10−5, Fisher's extract test, representative genes listed in Table 2). The representative genes were functionally related to cell cycle (Cdc2a, Ccna2, Ccnd2, Melk, etc.) and neural precursor cells (Doublecortin, Nedd9, Nestin, etc.). Conversely, 37 genes enriched in developing astrocytes were also further downregulated in Dicer mutant (p = 0.08). Moreover, only 19 genes enriched in mature astrocytes were found to be upregulated in Dicer mutant (p = 0.67). Thus, Dicer-deficient astrocytes at the mature stage seem to acquire a transcription profile partially reminiscent of immature/developing astrocytes.
Table 1.
Representative genes that are downregulated in Dicer mutant and enriched in mature astrocytes
| Gene symbol | Gene title | Biological pathway | Downregulation fold in Dicer Mutant CB | FDR | Enrichment fold (mature over immature astrocytes) |
|---|---|---|---|---|---|
| Cyp2d22 | Cytochrome P450, family 2, subfamily d, polypeptide 22 | Oxidation reduction | 1.547 | 0.0007 | 5.5 |
| Cyp4f14 | Cytochrome P450, family 4, subfamily f, polypeptide 14 | Oxidation reduction | 1.525 | 0.0003 | 2.7 |
| Cyp4v3 | Cytochrome P450, family 4, subfamily v, polypeptide 3 | Oxidation reduction | 1.362 | 0.0207 | 2 |
| Cyp2j9 | Cytochrome P450, family 2, subfamily j, polypeptide 9 | Oxidation reduction | 1.547 | 0 | 1.8 |
| Maob | Monoamine oxidase B | Oxidation reduction | 1.851 | 0 | 3.8 |
| Ptgs1 | Prostaglandin-endoperoxide synthase 1 | Oxidation reduction | 1.504 | 0.0006 | 3.8 |
| Gpd1 | Glycerol-3-phosphate dehydrogenase 1 (soluble) | Oxidation reduction | 1.334 | 0.0008 | 3.3 |
| Dio2 | Deiodinase, iodothyronine, type II | Oxidation reduction | 1.867 | 0 | 2.6 |
| BC055107 | Family with sequence similarity 107, member A | Regulation of cell growth, nuclear factor | 2.557 | 0 | 14.4 |
| Id4 | Inhibitor of DNA binding 4 | Transcription repressor | 1.802 | 0 | 2.4 |
| Lgi4 | Leucine-rich repeat LGI family, member 4 | Cellular homeostasis | 2.327 | 0 | 3.1 |
| Mt1 | Metallothionein 1 | Cellular homeostasis | 1.444 | 0 | 2.2 |
| Mt2 | Metallothionein 2 | cellular homeostasis | 1.478 | 0 | 1.8 |
| S100b | S100 protein, β polypeptide, neural | Cellular homeostasis | 1.474 | 0 | 3.5 |
| Ntsr2 | Neurotensin receptor 2 | Cellular homeostasis | 2.031 | 0 | 2.3 |
| S100a | S100 calcium binding protein A1 | Calcium binding | 1.598 | 0 | 1.6 |
| Fxyd1 | FXYD domain-containing ion transport regulator 1 | Calcium ion homeostasis | 1.46 | 0 | 2.6 |
| Slc1a2 (GLT-1) | Solute carrier family 1 (glial high-affinity glutamate transporter), member 2 | Glutamate transport | 1.509 | 0 | 2.2 |
| Gabrg1 | GABAA receptor, subunit γ-1; similar to GABAA receptor subunit α-2 | Chloride transport, GABA signaling | 1.701 | 0 | 1.6 |
| Gpc5 | Glypican 5 | Extracellular matrix, heparan sulfate proteoglycan binding | 1.602 | 0.0022 | 2.4 |
| Gstm1 | Glutathione S-transferase, μ1 | Glutathione transferase | 1.552 | 0 | 1.6 |
| Gstm5 | Glutathione S-transferase, μ5 | Glutathione transferase | 1.279 | 0.0156 | 1.8 |
| Clu | Clusterin | Anti-apoptosis | 1.707 | 0 | 1.7 |
Table 2.
Representative genes that are upregulated in Dicer mutant and enriched in immature astrocytes
| Gene symbol | Gene title | Biological pathway | Upregulation fold in Dicer mutant CB | FDR | Enrichment fold (immature over mature astrocytes) |
|---|---|---|---|---|---|
| Vim | Vimentin | Intermediate filament-based process | 1.895 | 0 | 3.4 |
| Dcx | Doublecortin | Axon extension, cell differentiation | 1.557 | 0.0004 | 2.8 |
| Nedd9 | Neural precursor cell expressed, developmentally downregulated gene 9 | Cell cycle, cell adhesion | 1.423 | 0.0287 | 2.1 |
| Nes | Nestin | Positive regulation of neural precursor cell proliferation | 1.511 | 0.0013 | 3.6 |
| Frem2 | Fras1-related extracellular matrix protein 2 | Cell adhesion | 5.937 | 0 | 12.9 |
| Melk | Maternal embryonic leucine zipper kinase | Cell cycle | 2.28 | 0.0227 | 9.5 |
| Cdc2a | Cyclin-dependent kinase 1 | Cell cycle | 2.4 | 0 | 12.9 |
| Mki67 | Antigen identified by monoclonal antibody Ki 67 | Cell cycle | 1.837 | 0.0011 | 10.9 |
| Ccna2 | Cyclin A2 | Cell cycle | 1.387 | 0.0435 | 7.5 |
| Cenpf | Centromere protein F | Cell cycle | 1.975 | 0 | 6 |
| Cdca3 | Cell division cycle-associated 3 | Cell cycle | 1.709 | 0.0001 | 4 |
| Ccnb1 | Predicted gene 8416; predicted gene 5593; cyclin B1; similar to cyclin B1; predicted gene 4870 | Cell cycle | 2.494 | 0.0401 | 3.9 |
| Ccnd2 | Cyclin D2 | Cell cycle | 2.039 | 0 | 2.2 |
| Ccnd1 | Cyclin D1 | Cell cycle | 2.016 | 0 | 1.9 |
| Birc5 | Baculoviral IAP repeat-containing 5 | Cell cycle, apoptosis | 1.804 | 0 | 6.5 |
| Ckap2 | Cytoskeleton-associated protein 2 | Cell cycle, apoptosis | 2.133 | 0 | 4.1 |
| Dtl | Denticleless homolog (Drosophila) | Regulation of cell cycle | 2.253 | 0.0007 | 4.3 |
| Hmgb2 | High mobility group box 2 | Nervous system development | 1.575 | 0 | 3.2 |
| Dpysl3 | Dihydropyrimidinase-like 3 | Nervous system development | 1.381 | 0.0015 | 3.1 |
| Adam17 | A disintegrin and metallopeptidase domain 17 | Notch signaling pathway, cell adhesion | 1.427 | 0.014 | 1.6 |
| Cybrd1 | Cytochrome b reductase 1 | Oxidation reduction | 4.758 | 0 | 3 |
| Plod2 | Procollagen lysine, 2-oxoglutarate 5-dioxygenase 2 | Oxidation reduction | 3.531 | 0 | 2.4 |
| Loxl1 | Lysyl oxidase-like 1 | Oxidation reduction | 1.895 | 0 | 2.8 |
| Egln3 | EGL nine homolog 3 (C. elegans) | Oxidation reduction, apoptosis | 1.677 | 0.0001 | 2.6 |
Quantitative RT-PCR analyses further confirmed that typical reactive gliosis-related extracellular matrix genes, such as Thrombospondin2, Tenascin C, Vimentin, and Adamts1 (Fig. 5C), were upregulated in Dicer-deficient cerebella. Tgfb3 is a potential glial-derived factor that is upregulated in reactive astrocytes (Apelt and Schliebs, 2001), and it was also upregulated in mutant cerebella (Fig. 5D). By contrast, genes normally expressed in mature astrocytes were downregulated in Dicer-deficient cerebella. Aqp4, the principal aquaporin in mammalian brain, was expressed in astrocyte foot processes, glia limitans, and ependyma. Downregulation of Aqp4 may alter water transport, K+ kinetics, and neuronal activity in the brain (for review, see Tait et al., 2008). Gstm3, a glutathione S transferase involved in reducing oxidation stress, was also downregulated. The mRNA level of Dao1 (D-amino acid oxidase), an astrocytic protein that catalyzes the degradation of d-serine (Park et al., 2006), was significantly downregulated (Fig. 5E). Glial-derived d-Serine, together with glutamate, activates NMDA receptor. d-Serine level in Dao1 mutant cerebellum is >10-fold higher than that in the wild type (Hamase et al., 2005; Panatier et al., 2006). Moreover, the only two known astrocytic glutamate transporters responsible for taking up excessive glutamate, GLT-1 (Slc1a2) and GLAST (Slc1a3), were both significantly downregulated (Fig. 5E). Downregulation of Dao1, GLT-1, and GLAST indicated the possibility of excitotoxicity. In addition, Glul (glutamine synthetase), which converts glutamate to glutamine in astrocytes and has been implicated as molecule potentially involved in epilepsy (Ortinski et al., 2010), exhibited a modest decrease (fold change = 0.79, FDR = 0.14) in mGfap-cre; Dicerflox/flox mice at P30 (presymptomatic) compared to wild-type littermates.
Reduced expressions of glutamate transporters and impaired glutamate uptake function
To further test the functional outcome of specific Dicer-deficient astrocytic transcriptome alterations, we focused on examining glutamate transport pathways. Glutamate is the predominant excitatory neurotransmitter in the mammalian CNS that activates cells through AMPA, kainate, and/or NMDA receptors. The extracellular concentration of glutamate needs to be kept low to limit tonic activation of the receptors, as excessive glutamate receptor activation can easily damage neurons (for review, see Sheldon and Robinson, 2007). In the cerebellum, where glutamate is a major neurotransmitter, astrocytes provide effective protection of neurons against glutamate excitotoxicity as they clear up excessive extracellular glutamate by active uptake via glutamate transporters GLT-1 and GLAST (Chaudhry et al., 1995; Lehre et al., 1995; Rothstein et al., 1996). In the cerebellum, GLAST is highly expressed in Bergmann glia and GLT-1 is expressed in both Bergmann glia and IGL astrocytes. We thus examined protein levels of GLT-1 and GLAST in the cerebella of mice at both presymptomatic and symptomatic stages. Western blot analyses showed ∼70% decrease of GLT-1 and ∼40% decrease of GLAST at P30 (presymptomatic stage). With progression of neurological symptoms, protein levels of both GLT-1 and GLAST continued to decrease and became barely detectable at P60 (Fig. 6A–D).
Figure 6.

Astroglial glutamate uptake pathway is severely compromised in mutant cerebella. A, B, Western blots and quantifications demonstrated that GLT-1 protein level was markedly downregulated at presymptomatic stage and continued to decrease until end-stage. C, D, Similarly, Western blots and quantifications showed that GLAST protein level was markedly downregulated at presymptomatic stage and continued to decrease until end-stage. E, Glutamate uptake assays with crude cerebellar synaptosomes at early symptomatic stage (P40) and late symptomatic stage (P60) showed that mutant cerebellar glutamate uptake functions were severely compromised.
To assess the in vivo glutamate transporter function, we quantified the uptake of radioactively labeled glutamate in synaptosomes prepared from control and mutant mice cerebella at early symptomatic stage (P40) and at late symptomatic stage (P60). We found that glutamate uptake capacity was decreased by ∼25% in synaptosomes isolated from P40 mutant cerebella and it was further reduced at P60 (Fig. 6E). Collectively, this series of experiments indicate that Dicer deletion in cerebellar astroglial cells impairs astrocytic glutamate transport already at the presymptomatic stage, which may directly lead to excitotoxicity of neurons at the late symptomatic stage.
NMDA receptor-mediated tonic conductance significantly increased in mutant cerebellar granule neurons
To directly test the hypothesis that the decrease of glial glutamate transporter expression in mutant mice cerebella may cause a tonic increase in the concentration of glutamate in the extracellular space and affect neuronal physiology, we recorded cerebellar granule cells in acute slices of presymptomatic or early symptomatic stage (P35–P40) mutant mice and their control littermates before the onset of granule cell apoptosis. At a holding potential of +30 mV and in the presence of extracellular Mg2+, application of the NMDA receptor antagonist d-AP5 at a saturating concentration (50 μm) blocked a tonic current that had a mean conductance of 262 ± 31 pS/pF in mutant granule cells (N = 7) compared to a mean conductance of 51 ± 9 pS/pF in littermate control granule cells (N = 7) (Fig. 7A). Application of TBOA (100 μm), a broad blocker of both glial and neuronal glutamate transporters, further increased the tonic conductance of both control and mutant granule cells to the same level (776 ± 68 pS/pF for control cells, N = 7; 633 ± 71 pS/pF for mutant cells, N = 8) (Fig. 7B). A bootstrap statistical analysis of the TBOA treatment-induced potentiation for the NMDA receptor-mediated conductance calculated showed a significant difference between control and mutant (potentiation fold is ∼20 for control and 4 for mutant) (Fig. 7C). Collectively, these results suggest that at presymptomatic or early symptomatic stage, although the extracellular glutamate concentration has not reached the ceiling level when all glutamate transporters were blocked by TBOA, the increased ambient glutamate in mutant cerebella has already altered the basal physiology of granule cells with more than a fivefold increase in NMDA receptor-mediated tonic conductance (N = 7 for wild type and mutant, respectively, p < 0.001, two-tailed t test).
Figure 7.

NMDA receptor-mediated tonic conductance significantly increased in mutant cerebellar granule neurons. A, Two representative traces showed the different amplitudes of NMDA receptor-mediated tonic currents. Horizontal bars indicated the application of the NMDA receptor antagonist d-AP5 (final concentration ≥ 50 μm). The dotted lines were the mean currents after complete blockade of NMDA receptors for calculating the magnitude of the NMDAR-mediated conductance. B, Representative traces showed that the application of 100 μm TBOA in bath induced a large increase of tonic conductance. The dotted lines indicated the basic conductance level held at +30 mV. C, Simulated distribution map of TBOA treatment potentiation fold for NMDA receptor-dependent conductance calculated by bootstrap statistics. Mean value of control cells was 20.0 with lower bound of 14.0 and higher bound of 26.0 with 95% confidence. Mean value of mutant cells was 3.9 with lower bound of 2.8 and higher bound of 5.2 with 95% confidence.
Discussion
Astrocytic Dicer is essential for normal mature cerebellar neuronal functions
While it is increasingly evident that CNS astrocytes are indispensable for normal brain development, functional differences between immature, mature, and reactive astrocytes are not well understood. Using two independent transgenic lines with Dicer deleted in postnatal astroglia, our study provides in vivo evidence that in mature mouse cerebella, normal neuronal functions are highly dependent on astrocytic Dicer. We found that while mGfap-cre; Dicerflox;flox mutant mice had normal early postnatal cerebellar development, they developed ataxia and cerebellar degeneration at a later postnatal stage (P55–P65) with massive granule neuron apoptosis and Purkinje cell dendrite degeneration. Before the onset of such neuronal dysfunctions, mutant cerebellar astroglia underwent morphological changes with an altered transcriptome partially reminiscent of a more immature or reactive-like status (Cahoy et al., 2008). It is worth pointing out that this comparison is only in proximity to the precise cerebellar astrocyte development, as the reference transcriptome datasets were based on forebrain astrocytes at distinct developmental stages and there are no similar comprehensive transcriptome databases on cerebellar astrocytes yet available. Moreover, we also cross-referenced Dicer-dependent gene expression changes to lists of genes highly expressed in P6 and P30 Bergmann glia, respectively (Koirala and Corfas, 2010). This analysis revealed a consistent trend, as many P30 Bergmann glia-enriched genes were downregulated in Dicer mutant, while some P6 Bergmann glia-enriched genes were upregulated in Dicer mutant. Importantly, while these transcriptional changes do not cause immediate cell death of astrocytes themselves, they lead to neuronal dysfunction and degeneration over time. These results indicate that normal mature brain functions requires functionally mature astrocytes and Dicer-dependent pathways may in part regulate such astrocytic maturation processes (Fig. 8). Furthermore, the published transcriptome datasets also suggest that transcription profile of in vitro cultured primary astroglia isolated from neonatal rodent brain is more similar to that of immature astrocytes (Cahoy et al., 2008), suggesting that in vitro cultured astrocytes are not functionally equivalent to in vivo mature astroglia. This poses a challenge to dissecting out downstream pathways affected by Dicer deletion using in vitro cultured neonatal astroglia.
Figure 8.

A model showed the essential roles of Dicer in regulating astrocytes maturation and functions. In the maturation process of astrocytes, Dicer-dependent pathway repressed genes that were involved in neural progenitor cell lineage, reactive astrocytes, and cell cycles while promoting transcription of mature astrocyte function genes in oxidation reduction, chemical homeostasis, etc. Deletion of Dicer in this process resulted in immature or reactive-like astrocytes that failed to support normal mature brain circuits.
The importance of neuron–glia interaction and astrocytic contribution in neurodegenerative disorders has been well demonstrated in spinocerebellar ataxia, ALS, and other diseases (as reviewed by Ilieva et al., 2009). Here we show that deletion of Dicer in astrocytes is sufficient to cause impairment of glutamate transporter pathways in mature astrocytes and cerebellar neuronal degeneration. A previous study showed that expressing mutant ataxin 7 in Bergmann glia caused non-cell-autonomous toxicity to Purkinje neurons via downregulation of GLAST, which provided independent evidence of the importance of such mechanisms in the cerebellum (Custer et al., 2006). In addition, deletion of the member of the Wnt signaling pathway, adenomatous polyposis coli (APC) in Bergmann glia also leads to non-cell-autonomous Purkinje neuron degeneration (Wang et al., 2011). Our results have thus identified a new regulatory component, the astrocytic Dicer-dependent pathway, of the non-cell-autonomous pathogenesis of neurodegeneration. Whether astrocytic miRNA dysregulation might be involved in human neurodegenerative disorders and which specific miRNAs may play major roles await further investigation. The potential contributions of astrocytic Dicer-dependent pathways to the progression of spontaneous and severe epilepsy, which are found in the mutant mice, are also of great interest and warrant future investigations.
Distinct neuronal responses to loss of astrocytic Dicer
Upon deletion of Dicer in postnatal astrocytes, neurons in different brain regions seem to have distinct responses. The cerebellum is particularly susceptible to such insults: cerebellar granule cells undergo massive apoptosis, whereas Purkinje cell dendrites degenerate. Although there are no detectable neuronal apoptosis/death signals in other parts of the brain, it is likely that cortical or hippocampal neurons also have become dysfunctional as indicated by the progression of epilepsy and the large number of reactive astrocytes in cortex and disorganized astrocytes in hippocampus in mutant mice. The difference between the cerebellum and other brain regions may, at least in part, be due to the intrinsic differences between neuronal subtypes. For example, cerebellar granule cells are known to have selective vulnerability to glutamate excitotoxicity. Their axon terminals are predominantly excitatory with glutamate as the major neurotransmitter. Thus, without functional astrocytic glutamate transporters that recycle extracellular glutamate, the accumulation of excessive glutamate further leads to release of additional glutamate from granule cells, rapidly eliciting a cascade of excitotoxic signals. As previously reviewed (Fonnum and Lock, 2004), other contributing factors may include the small cell size, unique NMDA receptor expressions, low level of calcium-binding proteins, and glutathione pathways. Since the astrocytic glutathione metabolism pathway is also affected in Dicer-depleted cerebellar astrocytes, it is possible that the lack of antioxidant pathway may also contribute to the vulnerability of cerebellar granule cells. Furthermore, since glutamate activates glial release of d-serine, which is a coagonist of NMDA receptor, the sensitivity of cerebellar d-serine level to Dao1 decrease may further contribute to cerebellar susceptibility to excitotoxicity. Another interesting observation is that the central zone (lobules VI–VII) particularly and, to a lesser extent, the posterior zone (lobules VIII–dorsal IX) of the cerebellum undergo degeneration first; the anterior zone (lobules I–V) and nodular zone (ventral lobule IX and lobule X) are slower in degeneration progression. The underlying mechanisms will need further investigation.
Heterogeneity of astrocytes
The deletion of Dicer in astrocytes caused different astroglial morphological changes in different brain regions: cerebellar Bergmann glia and cortical astrocytes showed reactive gliosis-like morphology, hippocampal astrocytes were disorganized, and cerebellar IGL astrocytes showed downregulated Gfap expression (unpublished observations, J. Tao and Y. E. Sun). These observations are consistent with the notion that astrocytes are heterogeneous and may have different subtypes (as reviewed by Zhang and Barres, 2010). It is noteworthy that similar to Bergmann glia and IGL astrocytes, hippocampal and cortical astrocytes show downregulation of GLT-1 in mutant mice (unpublished observations, J. Tao and Y. E. Sun). This is consistent with a previous report that homozygous GLT-1 null mice have lethal spontaneous seizures (Tanaka et al., 1997). Therefore, it is possible that the morphological changes may be different in distinct populations of astrocytes, but the underlying glutamate transporter dysregulation remains conserved. A recent study reported that using lentiviral vectors expressing mutant huntingtin protein in mouse striatal astrocytes led to progressive reactive astrogliosis with marked decrease of both GLT-1 and GLAST (Faideau et al., 2010). In conjunction with our findings, it is tempting to speculate that multiple mutations in protein coding genes or Dicer-dependent miRNA pathways in astrocytes may all contribute to altering glial glutamate transport and gliosis, which further lead to neuronal pathogenesis. Therefore, discovering the Dicer-dependent miRNAs and their downstream mRNA targets in mature astrocytes will have important implications in basic and clinical research.
Footnotes
This work was supported by National Institutes of Health Grant EUREKARO1 MH048095 and the National Basic Research Program of China (2011CBA01106) to Y.E.S. and NIH/NINDS Grants NS049501 to X.W.Y. and NS057624 to M.V.S. We thank all the Sun laboratory members, especially Dr. Volkan Coskun for helpful discussions and suggestions. We thank Dr. Sergey A. Krupenko for kindly providing Aldh1L1 antibody, Dr. Thomas Otis for technical advice, Marianne Cilluffo and Brain Research Institute imaging core facility for help with EM, and Dr. Guodong Li, Dr. Bingbing Song, Dr. Shaun Fouse, and Rose Korsak for technical supports.
References
- Apelt J, Schliebs R. Beta-amyloid-induced glial expression of both pro- and anti-inflammatory cytokines in cerebral cortex of aged transgenic Tg2576 mice with Alzheimer plaque pathology. Brain Res. 2001;894:21–30. doi: 10.1016/s0006-8993(00)03176-0. [DOI] [PubMed] [Google Scholar]
- Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- Bartel DP. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- Bushong EA, Martone ME, Jones YZ, Ellisman MH. Protoplasmic astrocytes in CA1 stratum radiatum occupy separate anatomical domains. J Neurosci. 2002;22:183–192. doi: 10.1523/JNEUROSCI.22-01-00183.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, Thompson WJ, Barres BA. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry FA, Lehre KP, van Lookeren Campagne M, Ottersen OP, Danbolt NC, Storm-Mathisen J. Glutamate transporters in glial plasma membranes: highly differentiated localizations revealed by quantitative ultrastructural immunocytochemistry. Neuron. 1995;15:711–720. doi: 10.1016/0896-6273(95)90158-2. [DOI] [PubMed] [Google Scholar]
- Christopherson KS, Ullian EM, Stokes CC, Mullowney CE, Hell JW, Agah A, Lawler J, Mosher DF, Bornstein P, Barres BA. Thrombospondins are astrocyte-secreted proteins that promote CNS synaptogenesis. Cell. 2005;120:421–433. doi: 10.1016/j.cell.2004.12.020. [DOI] [PubMed] [Google Scholar]
- Cobb BS, Nesterova TB, Thompson E, Hertweck A, O'Connor E, Godwin J, Wilson CB, Brockdorff N, Fisher AG, Smale ST, Merkenschlager M. T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med. 2005;201:1367–1373. doi: 10.1084/jem.20050572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Custer SK, Garden GA, Gill N, Rueb U, Libby RT, Schultz C, Guyenet SJ, Deller T, Westrum LE, Sopher BL, La Spada AR. Bergmann glia expression of polyglutamine-expanded ataxin-7 produces neurodegeneration by impairing glutamate transport. Nat Neurosci. 2006;9:1302–1311. doi: 10.1038/nn1750. [DOI] [PubMed] [Google Scholar]
- Davis TH, Cuellar TL, Koch SM, Barker AJ, Harfe BD, McManus MT, Ullian EM. Conditional loss of Dicer disrupts cellular and tissue morphogenesis in the cortex and hippocampus. J Neurosci. 2008;28:4322–4330. doi: 10.1523/JNEUROSCI.4815-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Pietri Tonelli D, Pulvers JN, Haffner C, Murchison EP, Hannon GJ, Huttner WB. miRNAs are essential for survival and differentiation of newborn neurons but not for expansion of neural progenitors during early neurogenesis in the mouse embryonic neocortex. Development. 2008;135:3911–3921. doi: 10.1242/dev.025080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney CL, Brenner M, Messing A. Conditional ablation of cerebellar astrocytes in postnatal transgenic mice. J Neurosci. 1996;16:6908–6918. doi: 10.1523/JNEUROSCI.16-21-06908.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle JP, Dougherty JD, Heiman M, Schmidt EF, Stevens TR, Ma G, Bupp S, Shrestha P, Shah RD, Doughty ML, Gong S, Greengard P, Heintz N. Application of a translational profiling approach for the comparative analysis of CNS cell types. Cell. 2008;135:749–762. doi: 10.1016/j.cell.2008.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugas JC, Cuellar TL, Scholze A, Ason B, Ibrahim A, Emery B, Zamanian JL, Foo LC, McManus MT, Barres BA. Dicer1 and miR-219 Are required for normal oligodendrocyte differentiation and myelination. Neuron. 2010;65:597–611. doi: 10.1016/j.neuron.2010.01.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eroglu C, Allen NJ, Susman MW, O'Rourke NA, Park CY, Ozkan E, Chakraborty C, Mulinyawe SB, Annis DS, Huberman AD, Green EM, Lawler J, Dolmetsch R, Garcia KC, Smith SJ, Luo ZD, Rosenthal A, Mosher DF, Barres BA. Gabapentin receptor alpha2delta-1 is a neuronal thrombospondin receptor responsible for excitatory CNS synaptogenesis. Cell. 2009;139:380–392. doi: 10.1016/j.cell.2009.09.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Déglon N, Ferrante RJ, Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington's disease subjects. Hum Mol Genet. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fonnum F, Lock EA. The contributions of excitotoxicity, glutathione depletion and DNA repair in chemically induced injury to neurones: exemplified with toxic effects on cerebellar granule cells. J Neurochem. 2004;88:513–531. doi: 10.1046/j.1471-4159.2003.02211.x. [DOI] [PubMed] [Google Scholar]
- Freeman MR. Specification and morphogenesis of astrocytes. Science. 2010;330:774–778. doi: 10.1126/science.1190928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia AD, Doan NB, Imura T, Bush TG, Sofroniew MV. GFAP-expressing progenitors are the principal source of constitutive neurogenesis in adult mouse forebrain. Nat Neurosci. 2004;7:1233–1241. doi: 10.1038/nn1340. [DOI] [PubMed] [Google Scholar]
- Glykys J, Mody I. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J Physiol. 2007;582:1163–1178. doi: 10.1113/jphysiol.2007.134460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregorian C, Nakashima J, Dry SM, Nghiemphu PL, Smith KB, Ao Y, Dang J, Lawson G, Mellinghoff IK, Mischel PS, Phelps M, Parada LF, Liu X, Sofroniew MV, Eilber FC, Wu H. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc Natl Acad Sci U S A. 2009;106:19479–19484. doi: 10.1073/pnas.0910398106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halassa MM, Haydon PG. Integrated brain circuits: astrocytic networks modulate neuronal activity and behavior. Annu Rev Physiol. 2010;72:335–355. doi: 10.1146/annurev-physiol-021909-135843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamase K, Konno R, Morikawa A, Zaitsu K. Sensitive determination of D-amino acids in mammals and the effect of D-amino-acid oxidase activity on their amounts. Biol Pharm Bull. 2005;28:1578–1584. doi: 10.1248/bpb.28.1578. [DOI] [PubMed] [Google Scholar]
- He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–531. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- Herrmann JE, Imura T, Song B, Qi J, Ao Y, Nguyen TK, Korsak RA, Takeda K, Akira S, Sofroniew MV. STAT3 is a critical regulator of astrogliosis and scar formation after spinal cord injury. J Neurosci. 2008;28:7231–7243. doi: 10.1523/JNEUROSCI.1709-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutvágner G, McLachlan J, Pasquinelli AE, Bálint E, Tuschl T, Zamore PD. A cellular function for the RNA-interference enzyme Dicer in the maturation of the let-7 small temporal RNA. Science. 2001;293:834–838. doi: 10.1126/science.1062961. [DOI] [PubMed] [Google Scholar]
- Ilieva H, Polymenidou M, Cleveland DW. Non-cell autonomous toxicity in neurodegenerative disorders: ALS and beyond. J Cell Biol. 2009;187:761–772. doi: 10.1083/jcb.200908164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koirala S, Corfas G. Identification of novel glial genes by single-cell transcriptional profiling of Bergmann glial cells from mouse cerebellum. PLoS One. 2010;5:e9198. doi: 10.1371/journal.pone.0009198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Ahn C, Han J, Choi H, Kim J, Yim J, Lee J, Provost P, Rådmark O, Kim S, Kim VN. The nuclear RNase III Drosha initiates microRNA processing. Nature. 2003;425:415–419. doi: 10.1038/nature01957. [DOI] [PubMed] [Google Scholar]
- Lehre KP, Levy LM, Ottersen OP, Storm-Mathisen J, Danbolt NC. Differential expression of two glial glutamate transporters in the rat brain: quantitative and immunocytochemical observations. J Neurosci. 1995;15:1835–1853. doi: 10.1523/JNEUROSCI.15-03-01835.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington's disease transgenic mice. Neurobiol Dis. 2001;8:807–821. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Oliet SH, Piet R, Poulain DA. Control of glutamate clearance and synaptic efficacy by glial coverage of neurons. Science. 2001;292:923–926. doi: 10.1126/science.1059162. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Dong J, Mungenast A, Yue C, Takano H, Watson DJ, Haydon PG, Coulter DA. Selective induction of astrocytic gliosis generates deficits in neuronal inhibition. Nat Neurosci. 2010;13:584–591. doi: 10.1038/nn.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatier A, Theodosis DT, Mothet JP, Touquet B, Pollegioni L, Poulain DA, Oliet SH. Glia-derived D-serine controls NMDA receptor activity and synaptic memory. Cell. 2006;125:775–784. doi: 10.1016/j.cell.2006.02.051. [DOI] [PubMed] [Google Scholar]
- Park HK, Shishido Y, Ichise-Shishido S, Kawazoe T, Ono K, Iwana S, Tomita Y, Yorita K, Sakai T, Fukui K. Potential role for astroglial D-amino acid oxidase in extracellular D-serine metabolism and cytotoxicity. J Biochem. 2006;139:295–304. doi: 10.1093/jb/mvj036. [DOI] [PubMed] [Google Scholar]
- Rana TM. Illuminating the silence: understanding the structure and function of small RNAs. Nat Rev Mol Cell Biol. 2007;8:23–36. doi: 10.1038/nrm2085. [DOI] [PubMed] [Google Scholar]
- Rothstein JD, Dykes-Hoberg M, Pardo CA, Bristol LA, Jin L, Kuncl RW, Kanai Y, Hediger MA, Wang Y, Schielke JP, Welty DF. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron. 1996;16:675–686. doi: 10.1016/s0896-6273(00)80086-0. [DOI] [PubMed] [Google Scholar]
- Saba R, Schratt GM. MicroRNAs in neuronal development, function and dysfunction. Brain Res. 2010;1338:3–13. doi: 10.1016/j.brainres.2010.03.107. [DOI] [PubMed] [Google Scholar]
- Schaefer A, O'Carroll D, Tan CL, Hillman D, Sugimori M, Llinas R, Greengard P. Cerebellar neurodegeneration in the absence of microRNAs. J Exp Med. 2007;204:1553–1558. doi: 10.1084/jem.20070823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheldon AL, Robinson MB. The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochem Int. 2007;51:333–355. doi: 10.1016/j.neuint.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D, Shin JY, McManus MT, Ptácek LJ, Fu YH. Dicer ablation in oligodendrocytes provokes neuronal impairment in mice. Ann Neurol. 2009;66:843–857. doi: 10.1002/ana.21927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009;32:638–647. doi: 10.1016/j.tins.2009.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV. Astrocytes: biology and pathology. Acta Neuropathol. 2010;119:7–35. doi: 10.1007/s00401-009-0619-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tait MJ, Saadoun S, Bell BA, Papadopoulos MC. Water movements in the brain: role of aquaporins. Trends Neurosci. 2008;31:37–43. doi: 10.1016/j.tins.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Tanaka K, Watase K, Manabe T, Yamada K, Watanabe M, Takahashi K, Iwama H, Nishikawa T, Ichihara N, Kikuchi T, Okuyama S, Kawashima N, Hori S, Takimoto M, Wada K. Epilepsy and exacerbation of brain injury in mice lacking the glutamate transporter GLT-1. Science. 1997;276:1699–1702. doi: 10.1126/science.276.5319.1699. [DOI] [PubMed] [Google Scholar]
- Volterra A, Meldolesi J. Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci. 2005;6:626–640. doi: 10.1038/nrn1722. [DOI] [PubMed] [Google Scholar]
- Wang X, Imura T, Sofroniew MV, Fushiki S. Loss of adenomatous polyposis coli in Bergmann glia disrupts their unique architecture and leads to cell nonautonomous neurodegeneration of cerebellar Purkinje neurons. Glia. 2011;59:857–868. doi: 10.1002/glia.21154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Tao J, Chen PJ, Shahab A, Ge W, Hart RP, Ruan X, Ruan Y, Sun YE. Genome-wide analysis reveals methyl-CpG-binding protein 2-dependent regulation of microRNAs in a mouse model of Rett syndrome. Proc Natl Acad Sci U S A. 2010;107:18161–18166. doi: 10.1073/pnas.1005595107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Vidensky S, Jin L, Jie C, Lorenzini I, Frankl M, Rothstein JD. Molecular comparison of GLT1+ and ALDH1L1+ astrocytes in vivo in astroglial reporter mice. Glia. 2011;59:200–207. doi: 10.1002/glia.21089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Barres BA. Astrocyte heterogeneity: an underappreciated topic in neurobiology. Curr Opin Neurobiol. 2010;20:588–594. doi: 10.1016/j.conb.2010.06.005. [DOI] [PubMed] [Google Scholar]
- Zhao X, He X, Han X, Yu Y, Ye F, Chen Y, Hoang T, Xu X, Mi QS, Xin M, Wang F, Appel B, Lu QR. MicroRNA-mediated control of oligodendrocyte differentiation. Neuron. 2010;65:612–626. doi: 10.1016/j.neuron.2010.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo L, Theis M, Alvarez-Maya I, Brenner M, Willecke K, Messing A. hGFAP-cre transgenic mice for manipulation of glial and neuronal function in vivo. Genesis. 2001;31:85–94. doi: 10.1002/gene.10008. [DOI] [PubMed] [Google Scholar]