

Abstract
Cowpox virus, a zoonotic poxvirus endemic to Eurasia, infects a large number of host species which makes its eradication impossible. The elimination of world-wide smallpox vaccination programs renders the human population increasingly susceptible to infection by orthopoxviruses resulting in a growing number of zoonotic infections including CPXV transmitted from domestic animals to humans. The ability of CPXV to infect a wide range of mammalian host is likely due to the fact that, among the orthopoxviruses, CPXV encodes the most complete set of open reading frames expected to encode immunomodulatory proteins. This renders CPXV particularly interesting for studying poxviral strategies to evade and counteract the host immune responses.
Keywords: Poxvirus, Orthopoxvirus, Cowpox, Vaccinia, Ectromelia, Monkeypox, Variola
1. Introduction
To successfully infect their hosts and produce infectious progeny capable of spreading through the host population, poxviruses have to limit recognition and destruction by the host’s immune system. In order to escape immune surveillance, poxviruses employ a number of different strategies. These escape mechanisms include interference with processing and presentation of the viral antigens, expression of decoy receptors/ligands and inactivation of key immune components of the host defense response.
Poxviruses comprise a large family of linear dsDNA viruses. Cowpox virus (CPXV), a zoonotic poxvirus belongs to the most studied genus of Orthopoxviridae (OPXV) that includes viruses isolated from mammals such as the human-specific pathogen variola virus (VARV), the causative agent of smallpox. This genus also includes the murine-specific ectromelia virus (mousepox; ECTV), as well as other viruses with a broad host range such as monkeypox (MPXV). Vaccinia virus (VACV), the most widely studied OPXV due to its successful use as smallpox vaccine, was widely considered to be derived from CPXV [1]. However, sequence comparisons suggest that VACV might have originated from horsepox virus [2]. Among the OPXVs, CPXV possesses the largest genome (∼224–228 kbp) [3] and possibly infects the widest range of host species. Rodents are thought to be its natural host, but the virus can occasionally infect domestic animals and be transmitted to humans [4]. With the eradication of VARV and the diminished number of smallpox vaccinations, CPXV and MPXV are considered to be the main source for potential OPXV infections in humans. In healthy individuals CPXV infection usually causes localized skin lesions, however in immunocompromized patients the infection may lead to severe generalized skin infection and lethal outcome [5]. Recent outbreaks of CPXV infection in humans have been reported from Germany and France in 2009 [6], [7]. In addition, CPXV infection outbreaks occur periodically in zoo animals [8]. The largest fatal outbreak happened in a colony of new world monkeys in Germany in 2002 [9].
In contrast to other dsDNA viruses, poxviruses encode their own DNA replication and transcription machinery and they are able to replicate in the cytoplasm, forming so-called virus factories. The poxvirus replication cycle within the cell is depicted in Fig. 1 and has been previously reviewed [10], [11]. Upon virus entry into the cell, virions are uncoated, the DNA is replicated, and viral genes are expressed in a coordinated fashion followed by virion assembly and exit from the cell. Poxviruses assemble in a complex process of viral morphogenesis that includes four infectious virion forms (Fig. 1): (i) the most abundant, intracellular mature virus (IMV or MV), is released by the cell during its lysis; (ii) intracellular enveloped virus (IEM or WV), an intermediate form of virus is produced by budding of IMV particle through the trans-Golgi network (TGN) membrane; (iii) cell-associated enveloped virus (CEV) is responsible for cell-to-cell spread of the virus, and (iv) extracellular virus (EEV or EV) is critical for dissemination of the virus within the host.
Fig. 1.
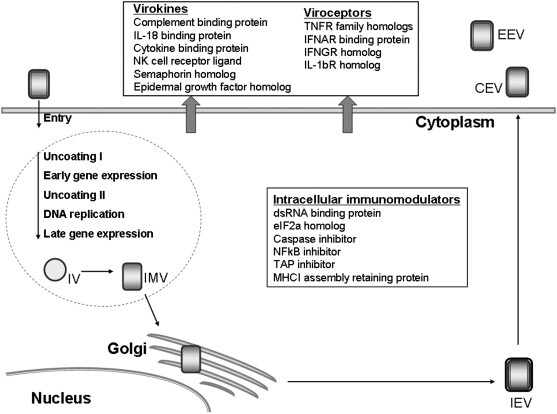
CPXV replication cycle and immunomodulatory proteins.
A large number of poxvirus genomes have been sequenced, including the three CPXV strains Brighton Red (BR), GRI-90 (GRI), and Germany-91 (Ger91) [12]. The involvement of a number of viral proteins in modulating the immune response was discovered based on sequence similarities to cellular immune proteins [13]. Sequence analysis also revealed that the poxviral genes essential for virus replication are highly conserved and tend to localize in the middle of the genome whereas the species-specific and immunomodulating genes are generally found at the termini [14]. The clustering of genes non-essential for replication possibly allows these viruses to rearrange host-modulatory gene sequences or acquire new genes. As a result, the sequence, structure, and function of these terminal sequences are highly diverse. Comparative analysis of CPXV and other OPXV genomes strongly suggests that these viruses had a common ancestor. Because CPXV possesses the largest genome which contains many ORFs found in other OPXV, as well as some unique ORFs, CPXV was suggested to be the most ancient and closest to the common ancestor virus [15]. Interestingly, VARV lacks many of these putative ancestral genes which likely restricted its host range to a single group, humans, which facilitated the eradication of this plague [16], [17]. CPXV encodes a large number of immune evasion proteins that collectively target a wide range of anti-viral host responses (Fig. 1; Table 1 ) [15]. In the current review we will focus on these immunomodulatory genes, with particular emphasis on those genes that are CPXV-specific. In Table 1, we give a list of all immunomodulatory proteins of CPXV identified to date. Several of these proteins will be discussed in more detail below.
Table 1.
Cowpox virus encoded immunomodulating proteins and their orthopoxviral orthologs (the Poxvirus Bioinformatics Resource Center, http://www.poxvirus.org).
| Immunomodulating proteins | CPXV |
VARV GAR-66 |
MPXV ZAR-1979 |
ECTV MOS |
VACV COP |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BR |
GR I |
Ger91 |
||||||||||||
| ORF | Size a.a. | ORF | Size a.a. | ORF | Size a.a. | ORF | Size a.a. | ORF | Size a.a. | ORF | Size a.a. | ORF | Size a.a. | |
| Complement control protein IMP Late, Secreted |
034 | 263 | 031 | 259 | 032 | 263 | SPICE 018 |
263 | MOPICE 019 |
216 | 018 | 262 | VCP C3L, 028 |
263 |
| Ankyrin repeat NFkb inhibitor, CPXV 006 early/late | 006/231 | 619 | 003/212 | 586 | 003/217 | 591 | 206 G1R |
585 | 003/200 | 587 | 002/176 | 587 | – | – |
| Ankyrin repeat NFkb inhibitor, CP77 early |
025 | 668 | 023 | 668 | 023 | 668 | 008 | 354 | 012 | 660 | – | – | – | – |
| Ankyrin repeat NFkb inhibitor, K1L ortholog | 041 | 284 | 038 | 284 | 039 | 285 | 027 | 66 | 028 | 284 | 023 | 285 | 036 K1L |
284 |
| TNFR homolog CrmB Early, secreted |
005/232 014 |
355 202 |
002/213 012 |
351 202 |
002/218 012 |
348 202 |
207 G2R |
349 | 002, J2L/201,J2R |
348 | – | – | 004, C22L/264, B28R |
122 |
| TNFR homolog CrmC Late, secreted |
197 | 186 | 180 | 186 | 179 | 186 | – | – | – | – | – | – | 224, A53R |
103 |
| TNFR homolog CrmD Late, secreted |
227 | 320 | 207 | 322 | 209 | 81 | – | – | – | – | 003/175 | 320 | – | – |
| TNFR homolog CrmE Secreted |
– | – | 208 | 167 | 211 | 167 | – | – | – | – | – | – | – | – |
| CD30 receptor homolog, vCD30 Late, secreted |
015 | 110 | 013 | 111 | 013 | 109 | – | – | – | – | 009 | 111 | – | – |
| Caspase inhibitor CrmA (SPI-2) Early, secreted |
213 | 341 | 203 | 375 | 195 | 346 | 192 | 344 | 181 | 344 | 166 | 344 | 246, B13R |
116 |
| IFN-a/b binding protein Early, secreted |
218 | 366 | 200 | 351 | 200 | 355 | 199/ D9R |
355 | 187 | 352 | 171 | 358 | 254/B19R | 353 |
| IFNGR homolog Early, secreted |
208 | 266 | 190 | 271 | 189 | 266 | 187 | 266 | 177 | 267 | 163 | 266 | 241, B8R |
272 |
| dsRNA-binding protein Early |
071 | 190 | 064 | 190 | 064 | 194 | 051 | 192 | 052 | 153 | 045 | 190 | 072, E3L |
190 |
| PKR inhibitor Early |
043 | 88 | 040 | 88 | 041 | 88 | 29 | 88 | ZAR-029 | 43 | – | – | 049, K3L |
88 |
| IL-1b receptor Early, secreted |
215 | 326 | 197 | 326 | 197 | 325 | 196 | 69 | 183 | 210 | 168 | 328 | 250, B16R |
290 |
| IL-18-receptor Secreted |
223 | 372 | 203 | 375 | 203 | 372 | 204 | 372 | 191 | 375 | 173 | 370 | 015/ C12L |
|
| Chemokine binding protein vCCI Early/late, secreted |
003/233 | 246 | 001/214 | 225 | 001/219 | 252 | 208 | 253 | 001/202 | 246 | 001/177 | 247 | 001, H5R/265, C23L |
244 |
| NKG2D binding protein (OMCP) Secreted |
018 | 171 | 016 | 178 | 016 | 174 | – | – | 198,N3R | 176 | – | – | – | – |
| TAP-inhibitor, early |
012 | 69 | 010 NK cell evasion? |
96 | 010 NK cell evasion? |
160 | – | – | – | – | 007 NK cell evasion? |
103 | – | – |
| MHC I binding protein, CPXV 203, early |
209 | 225 | 191 | 225 | 191 | 221 | – | – | 178 | 221 | 242 | 77 | – | – |
| Semaphorin Late, Secreted |
182 | 409 | 165 | 402 | 161 | 404 | 157 | 74 | – | – | 144 | 399 | 205, A39R |
403 |
| 3b-hydroxysteroid dehydrogenase Early |
188 | 345 | 171 | 346 | 170 | 346 | 164 | 101 | 161 | 346 | 148 | 346 | 211, A44L | 346 |
| Cowpox growth factor, Early, secreted |
021 | 139 | 019 | 138 | 019 | 138 | 003 | 140 | 008 | 142 | 011 | 83 | 016, C11R |
142 |
2. Complement evasion
Complement activation is one of the first responses of the innate immune system to an invading pathogen. The complement system employs a complex cascade of proteolytic cleavages of more than 30 plasma and cell membrane proteins and leads to induction of an inflammatory response, phagocyte and neutrophil chemotaxis, pathogen neutralization and subsequent opsonization, and lysis of the infected cells (reviewed in Refs. [18], [19]). The activation can be initiated via three independent pathways: (i) classical, binding of the first component in the cascade C1q to an antibody–antigen complex; (ii) alternative, a spontaneous hydrolysis of the downstream complement component 3 (C3) convertase and its interaction with pathogen surface; (iii) the mannose-binding lectin (MBL) pathway triggered by MBL binding to mannose residues on the pathogen surface. All three pathways converge at the stage of cleavage of C3 into C3a, antimicrobial peptide and C3b, an opsonin that binds to the pathogen and labels it for degradation. Because the effector compounds generated in the complement cascade can be delivered to any surface including host membranes, intact host cells protect themselves by expressing multiple complement regulatory proteins, including complement receptor 1 (CR1), decay accelerating factor (DAF), and membrane cofactor of proteolysis (MCP) located in the plasma membrane [18].
Like many other enveloped viruses, poxviruses use host complement control proteins to avoid activating the complement system [20], [21], [22]. It was demonstrated that in contrast to IMV proteins, EEV carrying cellular complement control proteins on their surface were resistant to complement activation [23]. In addition, CPXV and other OPXV encode proteins with structural and amino acid similarity to host complement control proteins with the largest sequence homology to the C4b-binding protein [24], [25]. The complement control protein of CPXV, called inflammation modulatory protein (IMP), is highly similar to its OPXV orthologs. These proteins were shown to inhibit both classical and alternative pathways via binding C3 and C4 and acting as a cofactor for factor I, a host complement control protein that cleaves and inactivates C3b and C4b [13]. CPXV IMP functions were tested using an IMP knockout virus. The protein was shown to block complement-mediated hemolysis and limit the inflammatory responses in infected mice [24].
3. Inhibition of TNF-induced responses
Tumor necrosis factor (TNF; TNF-α) is a pro-inflammatory cytokine that activates the innate immune responses to infection, promotes an anti-viral state in cells, controls apoptosis/survival of cells, and cell differentiation. TNF is secreted by a wide variety of cells including macrophages and binds to two surface receptors: TNFR1, expressed by most cell types and TNFR2, found on immune and endothelial cells [26]. The same set of receptors can also bind a soluble form of another member of the TNF superfamily, lymphotoxin (LT)-α (TNF-β). LT-α is produced by activated T-cells and NK cells and along with TNF it forms a signaling network that is essential for efficient induction of innate and adaptive immunity [27]. TNF expression is initiated via recognition of a pathogen by multiple host cell pattern recognition receptors (PRRs) including toll-like receptors (TLRs) and cytoplasmic nucleic acid sensors [28]. Five TLRs have been demonstrated to sense viral infection. TLR2 and TLR4 were shown to detect viral particles [28]. TLR3, TLR7 and TLR8, and TLR9 located in the endosomes are suggested to recognize viral nucleic acids, dsRNA (a viral transcription by-product), ssRNA, and unmethylated CpG motifs of dsDNA, respectively [28]. In addition, virus produced nucleic acids can be recognized by cytoplasmic RNA sensors, retinoic acid inducible gene I (RIG-I) and melanoma differentiation associated gene 5 (MDA5) and a cytoplasmic DNA sensor, DNA-dependent activator of interferon (IFN)-regulatory factors (DAI) [29], [30]. In particular, the RIG-I-mediated pathway was demonstrated to be important for induction of both TNF and type I IFN (discussed below) during myxoma virus infection in non-permissive cell lines [31]. Induction of the signaling pathways results in activation of NFκB, a key transcription factor for pro-inflammatory genes including TNF and type I IFN (discussed below). NFκB inhibitory proteins that sequester NFκB in the cytoplasm become phosphorylated and inactivated by IκB kinase (IKK) which triggers release and translocation of NFκB subunits to the nucleus. In addition to pro-inflammatory functions, TNF exerts an anti-inflammatory effect by amplifying NFκB-dependent expression of anti-apoptotic factors and activation of the mitogen-activated protein kinase (MAPK) signaling network. This leads to the assembly of complex I containing NFκB controlled anti-apoptotic gene products and pro-apoptotic complex II containing caspase-8, respectively. The outcome, cell survival or caspase-mediated apoptosis depends on the efficiency of formation of each of the complexes [32].
CPXV and other OPXV counteract TNF-mediated responses at several different stages: they inhibit NFκB activation to prevent TNF expression in the first place; intercept TNF and LT-α to subvert TNF-signal transduction; and inhibit caspase-8 and granzyme B to overturn the induction of apoptosis in infected cells [13], [33].
CPXV encodes at least three NFκB inhibiting proteins: CP77 and CPXV 006 expressed by pathogenic OPXV and an ortholog of VACV K1L common to all OPXV (Table 1) [33]. All three proteins have structural similarities and possess multiple predicted ankyrin repeats (ANK), protein–protein interaction motifs found in many cellular proteins including NFκB binding proteins. However they have low sequence identity and seem to function using distinct mechanisms and inhibit different stages of NFκB activation pathway. CP77 contains nine N-terminal ANK and an F-box like C-terminal domain that facilitate binding of the protein to the NFκB/p65 and the SCF, E3 ubiquitin ligase complex, respectively. Both of these functions are essential for prevention of NFκB migration into the nucleus suggesting that CP77 functions as a bridging molecule between NFκB and SCF and, possibly, targets it for ubiquitination and subsequent degradation by proteasome [34]. Alternatively, the protein may play a role as “surrogate” IkB-like domain and interfere with NFκB translocation into the nucleus. CP77 was also shown to play the role of a host range (hr) factor and rescue replication of VACV hr-mutants lacking either C7L or K1L gene in non-permissive cell lines [33]. The second NFκB inhibitor, CPXV 006 is encoded by all pathogenic OPXV. The protein contains six N-terminal ANKs. It directly interacts and interferes with degradation of the NFκB/p105 precursor protein and as a result prevents release and nuclear migration of NFκB. Using a CPXV 006 knockout virus, the protein was demonstrated to function upstream of IKK [35]. Deletion of CPXV 006, restored phosphorylation of IKK, activation and nuclear localization of NFkB, and expression of NFκB-controlled pro-inflammatory cytokines in infected cells [35]. In-vivo experiments demonstrated that in the absence of CPXV 006, CPXV was attenuated with elevated inflammatory responses at the sites of virus replication [35]. The third protein, an ortholog of VACV K1L (96% identity) possesses six predicted ANKs. K1L was shown to interfere with NFκB activation by preventing degradation of IκBα, a cellular NFκB inhibitor [33]. In addition to these three proteins, CPXV encodes several other ANK proteins [12] for which the functions have yet to be determined. However considering that several poxviral ANK proteins were shown to play a role as host range determinants and immunomodulators, these proteins may also be important for CPXV immune evasion.
Four different TNF and/or LT-α binding proteins named cytokine response modifier (Crm) B, CrmC, CrmD, and CrmE were found in different poxviruses, but only CPXV was shown to express all four of them (Table 1) [13], [16], [36]. These proteins display a variable number of cysteine-rich domains (CRD) characteristic of TNFR families and represent a family of poxviral viroceptors. CPXV-encoded Crm proteins have different temporal expression and show diverse species specificities which contribute to its capability to infect a wide variety of hosts. CrmB protein, expressed by triplicated ORFs in CPXV, is found in all other OPXV [16]. It is a secreted protein that closely resembles TNFRII protein and shares ∼42% identity with its ligand-binding domain. CrmB was shown to specifically bind both mouse TNF and human LT-α [37]. In-vitro, recombinant CPXV CrmB efficiently protected cells from the cytolytic effect of mouse TNF, but unlike the VARV ortholog, it was less potent against human or rabbit TNF [16]. Similar to CrmB, CrmC is a soluble secreted protein and is homologous to TNFR2. However its sequence lacks 150 conserved amino acid residues found at the C-terminus of CrmB protein and, unlike CrmB, it specifically binds to and protects from TNF but not LT-α [38]. CrmD is expressed only by CPXV and ECTV (97% identity [39]). It also shares homology to the ligand-binding domain of TNFRII. Both CPXV CrmD and its mouse ortholog competitively bind and block the cytolytic activity of human, rat, and mouse TNF and human LT-α [39]. The fourth TNFR-like protein, CrmE is expressed uniquely by CPXV [40]. Albeit its truncated orthologs are also found in VACV and MPXV their functions are unknown [12]. The CrmE amino acid sequence has limited homology to other CPXV Crm proteins (∼33–43%) as well as cellular TNFRI (∼28%) and TNFRII (31%) [12], [40], [41]. Although CPXV CrmE can specifically bind human, rat, and mouse TNF, it protects only against human TNF cytolysis [12], [40], [41]. It did not bind LT-α or any other ligands of TNF superfamily [40].
CPXV vCD30, a soluble homolog of cellular CD30 receptor is considered the fifth poxviral TNFR family member [42]. Similarly to other TNFR proteins, vCD30 possesses two cysteine-rich domains and closely resembles the mouse CD30 protein. CD30, a TNFR family member is expressed at low levels on resting lymphocytes, NK cells, and macrophages, but its expression is induced on activated or virally transformed cells [43]. The only known ligand for CD30 is CD153 (CD30L) expressed on activated T-cells, B-cells, monocytes, macrophages and other hematopoietic cells. Interactions between CD30 and CD153 were shown to be important for T-cell and B-cell co-stimulation and proliferation [43]. vCD30 was shown to specifically and competitively bind to CD30L and is suggested to interfere with the CD30–CD153 interaction [42]. vCD30 orthologs were also found in ECTV (91% identity) and in two recently sequenced genomes of horsepox and deerpox, but not in other poxviruses [12], [44]. Recombinant ECTV vCD30 inhibits T-cell activation and the induction of type 1 cytokine-mediated inflammatory responses [44]. However deletion of vCD30 did not have a significant effect on viral virulence in infected mouse models [45].
CPXV cytokine response modifier A (CrmA), the first discovered poxviral caspase inhibitor [46] was also found in other poxviruses including VACV (Table 1; reviewed by Ref. [13]). CrmA proteins belong to the serine protease inhibitor superfamily (serpins or SPI) and are thought to act as suicide substrates. CPXV CrmA is the most potent inhibitor compared to its orthologs and can efficiently inhibit both caspase-8 and granzyme B and consequently protect infected cells from TNF-induced apoptosis and T-cell-mediated cytotoxicity. In addition, CrmA was shown to inhibit caspase-1, required for proteolytic maturation of IL-1β and IL-18 suggesting that it also plays a role in downregulation of cytokine signaling [13].
4. Blockade of interferon response
Interferons have several functions in activating the innate immune response to viral infection [47]. In particular, type I interferons block viral spread to uninfected cells by inducing expression of multiple anti-viral proteins that can interfere with every step of the viral life cycle. IFN-α and IFN-β are produced upon viral infection by almost all cell types including fibroblasts, dendritic cells, hepatocytes. As mentioned earlier, detection of viral components by TLRs and cytoplasmic sensors activates the transcription factor NFκB which is essential for IFN-β expression. Another transcription factor important for IFN-β gene expression, interferon regulatory factor 3 (IRF3), was shown to be activated via TLR3 and RIG-I-mediated signal transduction pathways [31], [47]. In addition to IFN-β induction, IRF3 activates several interferon-stimulated genes (ISGs) directly. This initial response is further amplified when secreted IFN-β binds to its receptor on the surface of infected and uninfected cells and induces expression of IRF7 and multiple ISGs. Both IFN-α and INF-β bind to a single receptor, IFNAR, which is coupled to the Janus-family protein tyrosine kinases (JAK) that phosphorylate signal transducing activators of transcription (STAT) 1 and 2. Upon phosphorylation, STAT proteins translocate into the nucleus where they complex with interferon regulatory factor 9 (IRF9) and initiate transcription of ISGs via binding to IFN-stimulated response elements (ISRE) in the promoter sequences. Among ISGs are 2′–5′-oligoadenylate synthetase (OAS) activating the latent endoribonuclease (RNase L) and protein kinase R (PKR) inactivating a translation initiation factor eIF-2 by phosphorylation which results in degradation of viral RNA and shut-down of protein synthesis. IFN-dependent signal transduction is tightly regulated by various mechanisms, including IFNAR degradation/internalization, dephosphorylation/degradation of JAK-STAT pathway components, and blocking transcriptional activation by protein inhibitors of activated STAT (PIAS).
CPXV and other OPXVs use several strategies to counteract the interferon response. They sequester dsRNA and interfere with PKR signaling to prevent initiation of interferon response; express decoy receptors for type I and II IFNs and interferon induced cytokines, IL-18 and IL-1b [13], [48].
Type I IFN binding proteins are encoded by all OPXVs including CPXV [12]. However, these proteins have limited homology to INFAR. They are structurally more closely related to the IL-1 receptor and belong to the immunoglobulin superfamily [49]. The VACV ortholog, B18R was shown to competitively bind to and inhibit a broad range of type I IFN species, block induction of the anti-viral response in infected and uninfected cells, and prevent IFN-α response in infected mice [13], [50]. Homologs of cellular IFNGR are encoded by all OPXV [13], [51]. In contrast to species-specific cellular IFNGR proteins, all OPXV homologs bind and inhibit wide range of IFN-γ species. The biological activity of the IFNGR homolog was demonstrated in a rabbit model using recombinant VACV with deleted IFNGR homolog (B8R) ORF. Consistent with high affinity of B8R for rabbit IFN-γ, the knockout virus had an attenuated phenotype [52].
E3L, a dsRNA-binding and sequestering protein and K3L, an eIF2α homolog that functions as a non-phosphorylatable pseudosubstrate for PKR, are expressed by CPXV, VACV, and other OPXVs (Table 1). These proteins function as direct inhibitors of PKR and OAS signaling pathways. In addition, E3L was shown to inhibit IRF3 activation and consequently upregulation of INF-β expression (reviewed in Refs. [13], [48]).
5. Suppression of cytokine signaling
Cytokines and chemokines are produced in response to viral infection and orchestrate migration of the immune cells to the sites of infection and induction of an anti-viral defense. Expression of the cytokines is controlled by all poxviruses on many levels. OPXVs in particular were shown to interfere with activation of NFkB required for cytokine expression, TNF and IFN signaling, and inactivate caspase-1, required for processing of IL-1β and IL-18 cytokines (discussed above). In addition to this, these viruses encode secreted decoy receptors that intercept IL-1β, IL-18 cytokines and CC-chemokines (Table 1) [13]. Viral IL-1β receptors produced by VACV, CPXV, and ECTV display limited homology to the cellular receptor, however they were found to specifically bind IL-1b thereby preventing its interaction with the cellular receptor and blocking proliferation of B- and T-cells [13]. Similarly, amino acid sequences of CPXV, VACV, and ECTV IL-18 binding proteins (IL-18BP) are not related to the cellular IL-18-receptor, yet IL-18BP was demonstrated to efficiently block IL-18 interactions with the cellular receptor and interfere with NFκB activation and IFN-γ induction [53]. Interestingly, although IL-1β and IL-18 and their cellular receptors are related proteins, IL-18BP and IL-1βR displayed high specificity towards their targets: IL-18BP was not able to bind IL-1β and vice versa IL-1βR did not bind IL-18 [53]. Viral chemokine inhibitor (vCCI) expressed by all OPXVs (Table 1) specifically binds to and inhibits a particular subfamily of chemokines, CC-chemokines that were shown to attract macrophages and T-cells [13]. Again, vCCI did not exhibit amino acid sequence homology to any know chemokine receptor yet it was able to efficiently block binding of all tested CC-chemokines to their cellular receptors and inhibit monocyte chemotaxis [54].
6. Inhibition of NK cell activation
Although NK cells derive from a common lymphoid progenitor cells and kill the infected target cells by cytolysis, they are distinct from T-cell lymphocytes and function as a part of innate immunity. NK cell activation is tightly regulated and depends on the balance between the signals transduced by activating and inhibitory receptors many of which use MHC I or MHC I-like molecules as their ligands. NK cells directly survey target cells for the appropriate MHC I expression. Recognition of MHC I molecules by inhibitory NK cell receptors prevents NK cells from attacking “self”. Conversely, downregulation of MHC I by viruses (“missing self”) will result in the loss of inhibitory signaling and will lead to activation of the NK cells when they are additionally stimulated via activating receptors [55]. Among activating receptors are NKG2D and natural cytotoxicity receptors (NCR) located on the NK cell surface. NKG2D binds infection-induced MHC I-like ligands and can cause NK cell activation even if the target cell has normal MHC I expression [55], [56]. In addition, NKG2D is expressed by CD8+ T-cells and suggested to function as co-stimulatory receptor for these cells. In order to avoid NK cell activation many viruses, in particular herpesvirses, were shown to downmodulate NKG2D ligands [55].
The role of NK cells in immune response to OPXV infection have been studied in-vivo using ECTV infected mice [57]. It was shown that an NK cell response, mounted within 48 h post infection and several days before the CD8+ T-cell response was detected, was important for control of early virus dissemination and efficient induction of the adaptive response. Moreover upregulation of NKG2D and its ligands during infection was required for optimal NK cell activation and cytotoxicity [57]. Sequence analysis of OPXV genomes revealed that CPXV and MPXV encode a secreted protein termed as OPXV MHC I-like protein (OMCP). It was further shown that the OMCP protein binds with high affinity to both human and mouse NKG2D receptors (Table 1) [58]. In in-vitro assays, OMPC competitively blocked NKG2D interaction with the cellular ligands and inhibited NK cell-mediated cytotoxicity [58]. Considering that NKG2D is important for activation of both NK and T-cells, expression of NKG2D inhibitory ligand by CPXV and MPXV seems to be less selective and more advantageous strategy compared with downregulation of the cellular NKG2D ligands employed by herpesviruses. Recently, a new cellular receptor for OMPC, FcR-like 5 (FCRL5)/immunoglobulin receptor translocation-associated protein 2 (IRTA2) has been described [59]. This receptor is expressed by naive and memory B-cells and plasma cells [60], [61]. No cellular ligand for FCRL5 has been identified to date, but the receptor is suggested to be involved in B-cell differentiation and, potentially, be an inhibitory co-receptor for B-cell receptor signaling [62].
7. T-cell evasion by downregulation of MHC I expression
CD8+ T-cell-mediated responses play an important role in the control of intracellular pathogens, particularly viral infection. T-cells scan MHC I complexes on the cell surface and become activated once their T-cell receptor recognizes an antigen (Ag)-derived peptide presented by MHC I. When viral as well as cellular proteins are degraded by proteasomes, short peptides are translocated into the endoplasmic reticulum (ER) lumen by transporter associated with antigen processing (TAP). In the ER, peptides are trimmed to 8–10 amino acids and assemble with MHC I molecules. This complex travels to the cell surface to present the peptide to CD8+ T-cells. The process of MHC I-peptide assembly is tightly regulated by a cellular quality control system: misfolded or “empty” MHC I molecules do not leave the ER and undergo ER-associated degradation (ERAD) [63]. In order to avoid presentation of viral peptides and prevent CD8+ cell-mediated killing of the infected cells many viruses interfere with the MHC I presentation pathway at different stages such as MHC I expression, proteasomal degradation of the proteins, TAP-mediated transport of the peptides, assembly of the MHC I peptide complex, or trafficking of this complex to the plasma membrane [64].
Results obtained with VACV and ECTV suggested that OPXV do not inhibit MHC I-dependent peptide presentation. However, we recently observed that, unlike VACV or ECTV, CPXV infection blocks MHC I cell surface expression which also correlated with inhibition of CD8+ T-cell stimulation by the infected Ag presenting cells [65]. It was now demonstrated that CPXV encodes two ORFs, 203 and BR-012 which interfere with expression of mouse and human species of MHC I by two distinct mechanisms [66], [67], [68]. The 203 protein is encoded by all three CPXV strains (BR, GRI, and Ger91), but it is truncated in VACV and has more diverse sequence in MPXV (64% identity) (Table 1). CPXV 203 binds to and retains MHC I within the ER by means of the ER retention motif KTEL [67]. CPXV12 orthologs are present in all three CPXV strains (Table 1), however only BR-012 downregulates MHC I expression [68]. BR-012 is a truncated version of its GRI and Ger91orthologs. In the latter, this ORF encodes a C-type lectin domain that is a putative ligand for the NK-inhibitory receptor NKR-P1B, thus potentially contributing to NK cell evasion. In contrast, BR-012 is missing most of the C-type lectin domain and instead it interferes with MHC I presentation. BR-012 is integral to the ER-membrane with the C-terminus protruding into the ER lumen. BR-012 was shown to inhibit TAP-mediated peptide transport and dissociation of MHC I from peptide-loading complex eventually leading to MHC I degradation [66], [68]. BR-012 is so far the only poxviral TAP-inhibitor known to date. Deletion of both ORFs, 203 or BR-12 was required for full rescue of MHC I surface expression in infected cells and resulted in complete restoration of CD8+ T-cell activation [66], [67], [68]. These data suggested that the two unrelated and independently functioning proteins act in concert in order to prevent antigen presentation and CD8+ T-cell-mediated killing of the infected cells. The role of both 203 and BR-12 as virulence factors was tested in-vivo using a mouse model. The mutant virus with deleted ORFs 203 and 012 exhibited a significantly attenuated phenotype compared with the wild type virus [66]. Importantly, virulence was restored upon CD8+ T-cell depletion suggesting that CD8+ T-cells are unable to control CPXV due to MHC I downregulation by 203 and BR-012.
8. Other immunomodulating proteins
Additional CPXV-encoded proteins with putative immunomodulatory functions which have yet to be characterized include orthologs of VACV growth factor (VGF), a secreted homolog of epidermal growth factor [69] that induces proliferation in quiescent cells required for efficient virus replication; 3β-hydroxysteroid dehydrogenase, a steroid hormone and a virulence factor; and a homolog of cellular regulatory proteins termed semaphorins that is likely to play a role in mediating an inflammation response (reviewed in Ref. [13]).
9. Conclusion
CPXV immunomodulating strategies discussed in this review affect many innate and adaptive immune response pathways. These strategies are thought to allow the virus to control and manipulate such critical aspects as detection of the pathogen by complement, induction of the cytokine responses, establishment of the anti-viral state, and activation of NK and T cell cytotoxicity. Importantly, CPXV infects small animals and it is thus possible to experimentally test the role of these various immunomodulatory strategies for viral pathogenesis. The function of many of these “non-essential” ORFs in the CPXV genome is unknown due to the fact that CPXV possesses the largest OPXV genome and it has not been studied extensively in the past. However, most of these non-essential genes are found in at least one other OPXV family member suggesting that by studying CPXV-“specific” immunomodulators we will gain insights that are likely relevant for this entire viral family of pathogens.
Acknowledgements
We thank you Dr. Matthew A. Fischer for critical reading of the manuscript.
References
- 1.Baxby D. The origins of vaccinia virus. J. Infect. Dis. 1977;136:453–455. doi: 10.1093/infdis/136.3.453. [DOI] [PubMed] [Google Scholar]
- 2.Tulman E.R., Delhon G., Afonso C.L., Lu Z., Zsak L., Sandybaev N.T., Kerembekova U.Z., Zaitsev V.L., Kutish G.F., Rock D.L. Genome of horsepox virus. J. Virol. 2006;80:9244–9258. doi: 10.1128/JVI.00945-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Esposito J.J., Knight J.C. Orthopoxvirus DNA: a comparison of restriction profiles and maps. Virology. 1985;143:230–251. doi: 10.1016/0042-6822(85)90111-4. [DOI] [PubMed] [Google Scholar]
- 4.Baxby D., Bennett M. Poxvirus zoonoses. J. Med. Microbiol. 1997;46:17–20. 28–33. [PubMed] [Google Scholar]
- 5.Eis-Hubinger A.M., Gerritzen A., Schneweis K.E., Pfeiff B., Pullmann H., Mayr A., Czerny C.P. Fatal cowpox-like virus infection transmitted by cat. Lancet. 1990;336:880. doi: 10.1016/0140-6736(90)92387-w. [DOI] [PubMed] [Google Scholar]
- 6.Campe H., Zimmermann P., Glos K., Bayer M., Bergemann H., Dreweck C., Graf P., Weber B.K., Meyer H., Buttner M., Busch U., Sing A. Cowpox virus transmission from pet rats to humans, Germany. Emerg. Infect. Dis. 2009;15:777–780. doi: 10.3201/eid1505.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ninove L., Domart Y., Vervel C., Voinot C., Salez N., Raoult D., Meyer H., Capek I., Zandotti C., Charrel R.N. Cowpox virus transmission from pet rats to humans, France. Emerg. Infect. Dis. 2009;15:781–784. doi: 10.3201/eid1505.090235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kurth A., Straube M., Kuczka A., Dunsche A.J., Meyer H., Nitsche A. Cowpox virus outbreak in banded mongooses (Mungos mungo) and jaguarundis (Herpailurus yagouaroundi) with a time-delayed infection to humans. PloS One. 2009;4:e6883. doi: 10.1371/journal.pone.0006883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Matz-Rensing K., Ellerbrok H., Ehlers B., Pauli G., Floto A., Alex M., Czerny C.P., Kaup F.J. Fatal poxvirus outbreak in a colony of new world monkeys. Vet. Pathol. 2006;43:212–218. doi: 10.1354/vp.43-2-212. [DOI] [PubMed] [Google Scholar]
- 10.Condit R.C., Moussatche N., Traktman P. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 2006;66:31–124. doi: 10.1016/S0065-3527(06)66002-8. [DOI] [PubMed] [Google Scholar]
- 11.Moss B. Lippincott Williams & Wilkins; 2007. Poxviridae: The Viruses and Their Replication. [Google Scholar]
- 12.Lefkowitz E.J., Upton C., Changayil S.S., Buck C., Traktman P., Buller R.M. Poxvirus Bioinformatics Resource Center: a comprehensive Poxviridae informational and analytical resource. Nucleic Acids Res. 2005;33:D311–D316. doi: 10.1093/nar/gki110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Seet B.T., Johnston J.B., Brunetti C.R., Barrett J.W., Everett H., Cameron C., Sypula J., Nazarian S.H., Lucas A., McFadden G. Poxviruses and immune evasion. Annu. Rev. Immunol. 2003;21:377–423. doi: 10.1146/annurev.immunol.21.120601.141049. [DOI] [PubMed] [Google Scholar]
- 14.Gubser C., Hue S., Kellam P., Smith G.L. Poxvirus genomes: a phylogenetic analysis. J. Gen. Virol. 2004;85:105–117. doi: 10.1099/vir.0.19565-0. [DOI] [PubMed] [Google Scholar]
- 15.Shchelkunov S.N., Safronov P.F., Totmenin A.V., Petrov N.A., Ryazankina O.I., Gutorov V.V., Kotwal G.J. The genomic sequence analysis of the left and right species-specific terminal region of a cowpox virus strain reveals unique sequences and a cluster of intact ORFs for immunomodulatory and host range proteins. Virology. 1998;243:432–460. doi: 10.1006/viro.1998.9039. [DOI] [PubMed] [Google Scholar]
- 16.Gileva I.P., Nepomnyashchikh T.S., Antonets D.V., Lebedev L.R., Kochneva G.V., Grazhdantseva A.V., Shchelkunov S.N. Properties of the recombinant TNF-binding proteins from variola, monkeypox, and cowpox viruses are different. Biochim. Biophys. Acta. 2006;1764:1710–1718. doi: 10.1016/j.bbapap.2006.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liszewski M.K., Leung M.K., Hauhart R., Buller R.M., Bertram P., Wang X., Rosengard A.M., Kotwal G.J., Atkinson J.P. Structure and regulatory profile of the monkeypox inhibitor of complement: comparison to homologs in vaccinia and variola and evidence for dimer formation. J. Immunol. 2006;176:3725–3734. doi: 10.4049/jimmunol.176.6.3725. [DOI] [PubMed] [Google Scholar]
- 18.Zipfel P.F., Skerka C. Complement regulators and inhibitory proteins. Nat. Rev. Immunol. 2009;9:729–740. doi: 10.1038/nri2620. [DOI] [PubMed] [Google Scholar]
- 19.Dunkelberger J.R., Song W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010;20:34–50. doi: 10.1038/cr.2009.139. [DOI] [PubMed] [Google Scholar]
- 20.Saifuddin M., Parker C.J., Peeples M.E., Gorny M.K., Zolla-Pazner S., Ghassemi M., Rooney I.A., Atkinson J.P., Spear G.T. Role of virion-associated glycosylphosphatidylinositol-linked proteins CD55 and CD59 in complement resistance of cell line-derived and primary isolates of HIV-1. J. Exp. Med. 1995;182:501–509. doi: 10.1084/jem.182.2.501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Spear G.T., Lurain N.S., Parker C.J., Ghassemi M., Payne G.H., Saifuddin M. Host cell-derived complement control proteins CD55 and CD59 are incorporated into the virions of two unrelated enveloped viruses. Human T cell leukemia/lymphoma virus type I (HTLV-I) and human cytomegalovirus (HCMV) J. Immunol. 1995;155:4376–4381. [PubMed] [Google Scholar]
- 22.Shaw M.L., Stone K.L., Colangelo C.M., Gulcicek E.E., Palese P. Cellular proteins in influenza virus particles. PLoS Pathog. 2008;4:e1000085. doi: 10.1371/journal.ppat.1000085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vanderplasschen A., Mathew E., Hollinshead M., Sim R.B., Smith G.L. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. U S A. 1998;95:7544–7549. doi: 10.1073/pnas.95.13.7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Miller C.G., Shchelkunov S.N., Kotwal G.J. The cowpox virus-encoded homolog of the vaccinia virus complement control protein is an inflammation modulatory protein. Virology. 1997;229:126–133. doi: 10.1006/viro.1996.8396. [DOI] [PubMed] [Google Scholar]
- 25.Kotwal G.J., Moss B. Vaccinia virus encodes a secretory polypeptide structurally related to complement control proteins. Nature. 1988;335:176–178. doi: 10.1038/335176a0. [DOI] [PubMed] [Google Scholar]
- 26.Pennica D., Nedwin G.E., Hayflick J.S., Seeburg P.H., Derynck R., Palladino M.A., Kohr W.J., Aggarwal B.B., Goeddel D.V. Human tumour necrosis factor: precursor structure, expression and homology to lymphotoxin. Nature. 1984;312:724–729. doi: 10.1038/312724a0. [DOI] [PubMed] [Google Scholar]
- 27.Ware C.F. Network communications: lymphotoxins, LIGHT, and TNF. Annu. Rev. Immunol. 2005;23:787–819. doi: 10.1146/annurev.immunol.23.021704.115719. [DOI] [PubMed] [Google Scholar]
- 28.Akira S., Uematsu S., Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 29.Kato H., Takeuchi O., Sato S., Yoneyama M., Yamamoto M., Matsui K., Uematsu S., Jung A., Kawai T., Ishii K.J., Yamaguchi O., Otsu K., Tsujimura T., Koh C.S., Reis e Sousa C., Matsuura Y., Fujita T., Akira S. Differential roles of MDA5 and RIG-I helicases in the recognition of RNA viruses. Nature. 2006;441:101–105. doi: 10.1038/nature04734. [DOI] [PubMed] [Google Scholar]
- 30.Takaoka A., Wang Z., Choi M.K., Yanai H., Negishi H., Ban T., Lu Y., Miyagishi M., Kodama T., Honda K., Ohba Y., Taniguchi T. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature. 2007;448:501–505. doi: 10.1038/nature06013. [DOI] [PubMed] [Google Scholar]
- 31.Wang F., Gao X., Barrett J.W., Shao Q., Bartee E., Mohamed M.R., Rahman M., Werden S., Irvine T., Cao J., Dekaban G.A., McFadden G. RIG-I mediates the co-induction of tumor necrosis factor and type I interferon elicited by myxoma virus in primary human macrophages. PLoS Pathog. 2008;4:e1000099. doi: 10.1371/journal.ppat.1000099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Van Herreweghe F., Festjens N., Declercq W., Vandenabeele P. Tumor necrosis factor-mediated cell death: to break or to burst, that’s the question. Cell Mol. Life Sci. 2010;67:1567–1579. doi: 10.1007/s00018-010-0283-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mohamed M.R., McFadden G. NFkB inhibitors: strategies from poxviruses. Cell Cycle. 2009;8:3125–3132. doi: 10.4161/cc.8.19.9683. [DOI] [PubMed] [Google Scholar]
- 34.Chang S.J., Hsiao J.C., Sonnberg S., Chiang C.T., Yang M.H., Tzou D.L., Mercer A.A., Chang W. Poxvirus host range protein CP77 contains an F-box-like domain that is necessary to suppress NF-kappaB activation by tumor necrosis factor alpha but is independent of its host range function. J Virol. 2009;83:4140–4152. doi: 10.1128/JVI.01835-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mohamed M.R., Rahman M.M., Rice A., Moyer R.W., Werden S.J., McFadden G. Cowpox virus expresses a novel ankyrin repeat NF-kappaB inhibitor that controls inflammatory cell influx into virus-infected tissues and is critical for virus pathogenesis. J. Virology. 2009;83:9223–9236. doi: 10.1128/JVI.00861-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Shchelkunov S.N. Immunomodulatory proteins of orthopoxviruses. Mol. Biol. (Mosk) 2003;37:41–53. [PubMed] [Google Scholar]
- 37.Hu F.Q., Smith C.A., Pickup D.J. Cowpox virus contains two copies of an early gene encoding a soluble secreted form of the type II TNF receptor. Virology. 1994;204:343–356. doi: 10.1006/viro.1994.1539. [DOI] [PubMed] [Google Scholar]
- 38.Smith C.A., Hu F.Q., Smith T.D., Richards C.L., Smolak P., Goodwin R.G., Pickup D.J. Cowpox virus genome encodes a second soluble homologue of cellular TNF receptors, distinct from CrmB, that binds TNF but not LT alpha. Virology. 1996;223:132–147. doi: 10.1006/viro.1996.0462. [DOI] [PubMed] [Google Scholar]
- 39.Loparev V.N., Parsons J.M., Knight J.C., Panus J.F., Ray C.A., Buller R.M., Pickup D.J., Esposito J.J. A third distinct tumor necrosis factor receptor of orthopoxviruses. Proc. Natl. Acad. Sci. U S A. 1998;95:3786–3791. doi: 10.1073/pnas.95.7.3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Saraiva M., Alcami A. CrmE, a novel soluble tumor necrosis factor receptor encoded by poxviruses. J. Virology. 2001;75:226–233. doi: 10.1128/JVI.75.1.226-233.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Graham S.C., Bahar M.W., Abrescia N.G., Smith G.L., Stuart D.I., Grimes J.M. Structure of CrmE, a virus-encoded tumour necrosis factor receptor. J. Mol. Biol. 2007;372:660–671. doi: 10.1016/j.jmb.2007.06.082. [DOI] [PubMed] [Google Scholar]
- 42.Panus J.F., Smith C.A., Ray C.A., Smith T.D., Patel D.D., Pickup D.J. Cowpox virus encodes a fifth member of the tumor necrosis factor receptor family: a soluble, secreted CD30 homologue. Proc. Natl. Acad. Sci. U S A. 2002;99:8348–8353. doi: 10.1073/pnas.122238599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kennedy M.K., Willis C.R., Armitage R.J. Deciphering CD30 ligand biology and its role in humoral immunity. Immunology. 2006;118:143–152. doi: 10.1111/j.1365-2567.2006.02354.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Saraiva M., Smith P., Fallon P.G., Alcami A. Inhibition of type 1 cytokine-mediated inflammation by a soluble CD30 homologue encoded by ectromelia (mousepox) virus. J. Exp. Med. 2002;196:829–839. doi: 10.1084/jem.20020319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alejo A., Saraiva M., Ruiz-Arguello M.B., Viejo-Borbolla A., de Marco M.F., Salguero F.J., Alcami A. A method for the generation of ectromelia virus (ECTV) recombinants: in vivo analysis of ECTV vCD30 deletion mutants. PloS One. 2009;4:e5175. doi: 10.1371/journal.pone.0005175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pickup D.J., Ink B.S., Hu W., Ray C.A., Joklik W.K. Hemorrhage in lesions caused by cowpox virus is induced by a viral protein that is related to plasma protein inhibitors of serine proteases. Proc. Natl. Acad. Sci. U S A. 1986;83:7698–7702. doi: 10.1073/pnas.83.20.7698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Theofilopoulos A.N., Baccala R., Beutler B., Kono D.H. Type I interferons (alpha/beta) in immunity and autoimmunity. Annu. Rev. Immunol. 2005;23:307–336. doi: 10.1146/annurev.immunol.23.021704.115843. [DOI] [PubMed] [Google Scholar]
- 48.Perdiguero B., Esteban M. The interferon system and vaccinia virus evasion mechanisms. J. Interferon Cytokine Res. 2009;29:581–598. doi: 10.1089/jir.2009.0073. [DOI] [PubMed] [Google Scholar]
- 49.Smith G.L., Chan Y.S. Two vaccinia virus proteins structurally related to the interleukin-1 receptor and the immunoglobulin superfamily. J. Gen. Virol. 1991;72(Pt 3):511–518. doi: 10.1099/0022-1317-72-3-511. [DOI] [PubMed] [Google Scholar]
- 50.Waibler Z., Anzaghe M., Frenz T., Schwantes A., Pohlmann C., Ludwig H., Palomo-Otero M., Alcami A., Sutter G., Kalinke U. Vaccinia virus-mediated inhibition of type I interferon responses is a multifactorial process involving the soluble type I interferon receptor B18 and intracellular components. J. Virology. 2009;83:1563–1571. doi: 10.1128/JVI.01617-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alcami A., Smith G.L. Vaccinia, cowpox, and camelpox viruses encode soluble gamma interferon receptors with novel broad species specificity. J. Virology. 1995;69:4633–4639. doi: 10.1128/jvi.69.8.4633-4639.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Symons J.A., Tscharke D.C., Price N., Smith G.L. A study of the vaccinia virus interferon-gamma receptor and its contribution to virus virulence. J. Gen. Virol. 2002;83:1953–1964. doi: 10.1099/0022-1317-83-8-1953. [DOI] [PubMed] [Google Scholar]
- 53.Smith V.P., Bryant N.A., Alcami A. Ectromelia, vaccinia and cowpox viruses encode secreted interleukin-18-binding proteins. J. Gen. Virol. 2000;81:1223–1230. doi: 10.1099/0022-1317-81-5-1223. [DOI] [PubMed] [Google Scholar]
- 54.Liptakova H., Kontsekova E., Alcami A., Smith G.L., Kontsek P. Analysis of an interaction between the soluble vaccinia virus-coded type I interferon (IFN)-receptor and human IFN-alpha1 and IFN-alpha2. Virology. 1997;232:86–90. doi: 10.1006/viro.1997.8527. [DOI] [PubMed] [Google Scholar]
- 55.Jonjic S., Babic M., Polic B., Krmpotic A. Immune evasion of natural killer cells by viruses. Curr. Opin. Immunol. 2008;20:30–38. doi: 10.1016/j.coi.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ogasawara K., Lanier L.L. NKG2D in NK and T cell-mediated immunity. J. Clin. Immunol. 2005;25:534–540. doi: 10.1007/s10875-005-8786-4. [DOI] [PubMed] [Google Scholar]
- 57.Fang M., Lanier L.L., Sigal L.J. A role for NKG2D in NK cell-mediated resistance to poxvirus disease. PLoS Pathog. 2008;4:e30. doi: 10.1371/journal.ppat.0040030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Campbell J.A., Trossman D.S., Yokoyama W.M., Carayannopoulos L.N. Zoonotic orthopoxviruses encode a high-affinity antagonist of NKG2D. J. Exp. Med. 2007;204:1311–1317. doi: 10.1084/jem.20062026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Campbell J.A., Davis R.S., Lilly L.M., Fremont D.H., French A.R., Carayannopoulos L.N. Cutting edge: FcR-like 5 on innate B cells is targeted by a poxvirus MHC class I-like immunoevasin. J. Immunol. 2010;185:28–32. doi: 10.4049/jimmunol.1000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Miller I., Hatzivassiliou G., Cattoretti G., Mendelsohn C., Dalla-Favera R. IRTAs: a new family of immunoglobulinlike receptors differentially expressed in B cells. Blood. 2002;99:2662–2669. doi: 10.1182/blood.v99.8.2662. [DOI] [PubMed] [Google Scholar]
- 61.Polson A.G., Zheng B., Elkins K., Chang W., Du C., Dowd P., Yen L., Tan C., Hongo J.A., Koeppen H., Ebens A. Expression pattern of the human FcRH/IRTA receptors in normal tissue and in B-chronic lymphocytic leukemia. Int. Immunol. 2006;18:1363–1373. doi: 10.1093/intimm/dxl069. [DOI] [PubMed] [Google Scholar]
- 62.Haga C.L., Ehrhardt G.R., Boohaker R.J., Davis R.S., Cooper M.D. Fc receptor-like 5 inhibits B cell activation via SHP-1 tyrosine phosphatase recruitment. Proc. Natl. Acad. Sci. U S A. 2007;104:9770–9775. doi: 10.1073/pnas.0703354104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Peaper D.R., Cresswell P. Regulation of MHC class I assembly and peptide binding. Annu. Rev. Cell Dev. Biol. 2008;24:343–368. doi: 10.1146/annurev.cellbio.24.110707.175347. [DOI] [PubMed] [Google Scholar]
- 64.Hansen T.H., Bouvier M. MHC class I antigen presentation: learning from viral evasion strategies. Nat. Rev. Immunol. 2009;9:503–513. doi: 10.1038/nri2575. [DOI] [PubMed] [Google Scholar]
- 65.Dasgupta A., Hammarlund E., Slifka M.K., Fruh K. Cowpox virus evades CTL recognition and inhibits the intracellular transport of MHC class I molecules. J. Immunol. 2007;178:1654–1661. doi: 10.4049/jimmunol.178.3.1654. [DOI] [PubMed] [Google Scholar]
- 66.Byun M., Verweij M.C., Pickup D.J., Wiertz E.J., Hansen T.H., Yokoyama W.M. Two mechanistically distinct immune evasion proteins of cowpox virus combine to avoid antiviral CD8 T cells. Cell Host Microbe. 2009;6:422–432. doi: 10.1016/j.chom.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Byun M., Wang X., Pak M., Hansen T.H., Yokoyama W.M. Cowpox virus exploits the endoplasmic reticulum retention pathway to inhibit MHC class I transport to the cell surface. Cell Host Microbe. 2007;2:306–315. doi: 10.1016/j.chom.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 68.Alzhanova D., Edwards D.M., Hammarlund E., Scholz I.G., Horst D., Wagner M.J., Upton C., Wiertz E.J., Slifka M.K., Fruh K. Cowpox virus inhibits the transporter associated with antigen processing to evade T cell recognition. Cell Host Microbe. 2009;6:433–445. doi: 10.1016/j.chom.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.da Fonseca F.G., Silva R.L., Marques J.T., Ferreira P.C., Kroon E.G. The genome of cowpox virus contains a gene related to those encoding the epidermal growth factor, transforming growth factor alpha and vaccinia growth factor. Virus Genes. 1999;18:151–160. doi: 10.1023/a:1008072720217. [DOI] [PubMed] [Google Scholar]