

Abstract
Cerebral amyloid angiopathy (CAA) is an age-associated disease characterized by amyloid deposition in cerebral and meningeal vessel walls. CAA is detected in the majority of the individuals with dementia and also in a large number of non-demented elderly individuals. In addition, CAA is strongly associated with Alzheimer’s disease (AD) pathology. Mechanical consequences including intra-cerebral or subarachnoid hemorrhage remains CAA most feared complication, but only a small fraction of CAA results in severe bleeding. On the hand the non-mechanical consequences in cerebrovascular regulation are prevalent and may be even more deleterious. Studies of animal models have provided strong evidence linking the vasoactive Aβ 1–40, the main species found in CAA, to disturbances in endothelial-dependent factors, disrupting cerebrovascular regulation Here, we aimed to review experimental findings regarding the non-mechanical consequences of CAA for cerebrovascular regulation and discuss the implications of these results to clinical practice.
Keywords: amyloid, angiopathy, dementia, Alzheimer’s disease, animal models, endothelium, cerebrovascular dysfunction, blood brain barrier
Introduction
Approximately 20% of the left ventricular cardiac output is directed to the brain, even though it only represents about 2% of the total body mass. This clearly illustrates that brain function depends on the continuous and highly-regulated delivery of blood to meet energy requirements and to clear deleterious metabolic by-products. Even short periods of cerebrovascular impairment can lead to neuronal damage by compromising the steady supply of essential metabolites such as oxygen, amino acids and glucose (Bell and Zlokovic, 2009; Kalaria, 2009). This is demonstrated by the dramatic and irreversible neurological deficits caused by even a quickly reversed thromboembolism. The brain vascular system is regulated independently of the systemic circulation (van Beek et al., 2008). This auto-regulatory ability delays cerebral ischemia during low blood pressure states, and diminishes the risk of blood–brain barrier (BBB) disruption and brain edema during systemic hypertensive episodes. Ischemic and hemorrhagic lesions resulting from large brain vessel disease and their consequences are easily detected, even by low resolution imaging or simple gross inspection during autopsy (Yoshiura et al., 1999). On the other hand, identification of small vessel (arterioles, capillaries and venules) changes often requires special histological processing and comprehensive, meticulous microscopic investigation. Small vessels comprise less than 10% of the total brain volume, but are of paramount importance in regulating cerebral blood flow, oxygen supply, and metabolite exchange.
Cerebral amyloid angiopathy (CAA) is an age-associated disease characterized by amyloid deposition in vessel walls of meningeal arteries and capillaries, as well as intra-cortical vessels. Experimental evidence suggests that the non-mechanical consequences of CAA are even more deleterious than the classically acknowledged mechanical consequences of this disease, which include lethal cerebral hemorrhage. Here, we aimed to review experimental findings regarding the non-mechanical consequences of CAA for cerebrovascular regulation and discuss the implications of these results to clinical practice.
Epidemiology and neuropathological characteristics of CAA
CAA neuropathology was described over a century ago, and the condition received its current nomenclature several decades later (Biffi and Greenberg, 2011; Fischer, 1910; Oppenheim, 1909). However, it only gained importance and became the subject of systematic investigation when its association with lethal cerebral hemorrhage was identified (Jellinger, 1977; Okazaki et al., 1979). Current evidence indicates that CAA is much more prevalent than hitherto appreciated, detected in one third of non-demented elderly individuals and two thirds of elderly individuals with dementia in population-based clinicopathological studies (Charidimou et al., 2012). CAA is strongly associated with Alzheimer’s disease (AD) pathology, although it is not clear what impact CAA has on cognitive decline in these cases (Jellinger, 2002; Thal et al., 2003).
CAA is characterized by deposition of amyloid formed mainly from beta-amyloid peptides (Aβ) in the walls of leptomeningeal and parenchymal vessels’ (Jellinger, 2002). Aβ is a product of amyloid precursor protein proteolytic cleavage by β-secretase and γ-secretase. The resulting peptides predominantly have 40 (Aβ 1-40) or 42 (Aβ 1-42) amino acid residues. Aβ may polymerize into the insoluble fibrils found in AD plaques, mainly composed of Aβ 1-42, and in the deposits found in vessel walls in CAA, mainly composed of Aβ 1-40. In CAA, immunohistochemical and electron microscopic investigations show Aβ fibrils first deposited in the abluminal aspect of the basal lamina around smooth muscle cells, followed by a gradual spreading towards the internal elastic lamina of arteries and the endothelium of arterioles. The smooth muscle cells degenerate as the disease progresses (Wisniewski et al., 1994). In capillaries, which are devoid of smooth muscle, Aβ fibrils are found within the basal lamina and adjacent neuropil. Endothelial cells are apparently well preserved, even in severe CAA (Pezzini et al., 2009).
Some of the open questions related to CAA concern the origin or source of the amyloid deposited in the vessel walls (Smith and Greenberg, 2009) and the reason for its predilection for occipital, parietal and frontal cortices, whereas the deep central gray nuclei, white matter and brainstem are rarely affected (Pezzini et al., 2009; Vinters, 1987).
CAA complications
CAA complications can occur depending on the extent and location of disease pathology. Intra-cerebral or subarachnoid hemorrhage remains CAA most feared complication, caused by Aβ accumulation in the leptomeningeal and intracranial arteries, with smooth muscle loss and adventitial involvement, leading to aneurysm formation, fibrinoid necrosis, and rupture. However, the proportionally low number of CAA-associated hemorrhages suggests that only a small fraction of CAA cases result in severe bleeding (Jellinger et al., 2007). This does not mean, however, that CAA is benign. For instance, in capillaries, amyloid accumulation leads to wall brittleness and lumen obstruction, causes of small ischemic lesions and microbleeds (Thal et al., 2009), which are emerging as potential contributors to cognitive decline (De Reuck et al., 2011; Nakata-Kudo et al., 2006).
In addition, non-mechanical changes in cerebrovascular regulation are emerging as important CAA complications. Several independent studies of animal models have provided strong evidence linking the vasoactive Aβ 1–40 to disturbances in endothelial-dependent factors, disrupting cerebrovascular regulation (Bamberger et al., 2003; Niwa et al., 2000) (Farkas and Luiten, 2001; Frackowiak et al., 1994).
Aβ and cerebrovascular dysregulation
Decreased cerebral blood flow negatively affects brain metabolism and synthesis of proteins required for memory and learning (Bell and Zlokovic, 2009). Cerebral flow dysregulation is an early feature of AD. The mechanisms causing such dysregulation are poorly understood, but some evidence suggests Aβ as an important culprit. The first clues suggesting that Aβ altered cerebral autoregulation came from neuroimaging studies in the early stages of AD and in asymptomatic, but genetically predisposed individuals (Iadecola, 2003; Paris et al., 2003). On the experimental side, several investigations using transgenic animals or human cell lines established that both soluble and insoluble forms of Aβ, in particular the CAA-associated Aβ 1-40, were strongly vasoactive.
Attempts to understand the mechanisms mediating Aβ-associated vascular dysregulation using isolated cerebral arteries suggested that brain parenchyma participation was not required, indicating a direct effect of Aβ on the vessels (Crawford et al., 1998; Niwa et al., 2001; Thomas et al., 1996). To investigate Aβ effects on cerebrovascular regulation further, transgenic mice Tg2576 overexpressing a mutated form of human APP, which show age-dependent elevations in brain Aβ and behavioral deficits (Hsiao et al., 1996), have been extensively studied. This APP mutation leads to the development of CAA in older mice, who also show severe vasodilatory dysfunction compared to young Tg2576 free of CAA (Christie et al., 2001). These mice start showing progressive cerebrovascular dysfunction when the levels of soluble Aβ rise, in a dose-dependent fashion which is independent of the presence of Aβ deposits (Niwa et al., 2000; Zhang et al., 1997). Further observations established endothelium-dependent pathways as important targets of Aβ-mediated vascular toxicity (Price et al., 2004). A series of elegant experiments comparing APP and wild-type mice showed that intrathecal injection of Aβ 1–40 altered the response of the cerebral vessels to several endothelium-dependent vasodilators, while no effect was seen on the response to endothelium-independent vasodilators (Iadecola et al., 1999). Indeed, application of exogenous Aβ caused endothelium-dependent vasoconstriction in normal blood vessels ex vivo (Thomas et al., 1996; Townsend et al., 2002). Since cerebral endothelial cells remain ultrastructurally intact in APP mice and in human CAA, these authors suggested that changes in a common pathway utilized by endothelium-dependent vasodilators, rather than endothelium structural problems, are the cause of this dysfunction. Impaired endothelium-dependent vasodilatation is found in many cardiovascular conditions, such as atherosclerosis, hypertension, heart failure and diabetes mellitus (Cai and Harrison, 2000). In the presence of Aβ 1-40, dysfunction of endothelium-dependent vasodilatation is often mediated by pathways involving production of reactive oxygen species (ROS) mediated by nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Iadecola, 2003), a multi-unit enzyme that requires p67phox, p47phox, p40phox and GTPase Rac for activation (Ray and Shah, 2005). One of its main functions is to catalyze the production of superoxide from oxygen (Babior, 2004), by coupling the latter to electrons transferred from intracellular NADPH. NADPH oxidase activation occurs through multiple mechanisms, such as angiotensin II (Li and Shah, 2003), growth factors (Ushio-Fukai et al., 2002), cytokines (Frey et al., 2002) and reduced bioavailability of nitric oxide (Park et al., 2011). CD36 is an endothelial membrane glycoprotein acting as a scavenger receptor. Aβ-induced ROS production through a signaling pathway culminating in Rac activation is mediated by a molecular complex that includes CD36 (Coraci et al., 2002). In experimental studies using CD36-null mice or APP mice lacking CD36, Aβ 1–40 did not cause endothelial dysfunction, regardless of the brain Aβ levels (Park et al., 2011). In addition, the use of CD36 blocking antibodies reversed endothelial dysfunction in APP mice (Park et al., 2011). These results suggest that CD36 inactivation is vasoprotective against Aβ toxicity, and its suppression could potentially ameliorate Aβ-mediated cerebrovascular dysfunction in humans.
Besides impaired vasodilatation, other Aβ-mediated endothelial effects have been proposed. Permeability of the BBB is controlled by endothelial cells, which are kept together by tight junction (TJ) proteins. BBB defects are associated with reduced cognition in AD (Zlokovic, 2008). Experimental studies show a detrimental effect of Aβ on some TJ proteins, with increased BBB permeability. In rat endothelial cells treated with Aβ 1–42, TJ transmembrane proteins claudin-5 and ZO-2 are mislocalized to the cytoplasm (Marco and Skaper, 2006). Human brain endothelial cell lines exposed to Aβ 1–40 showed increased permeability associated with a decrease of occludin levels and no changes in claudin-5 and ZO-1 levels (Tai et al., 2010). On the other hand, another study found Aβ-induced changes in claudin levels (Hartz et al., 2012). Therefore, although TJ proteins are probably involved in Aβ-induced cerebrovascular dysfunction, the mechanisms are not yet clear.
CAA-affected capillaries demonstrate loss of TJ proteins associated with a pericapillary increase in activated microglia expressing NADPH oxidase-2 (Carrano et al., 2011).
Several agents have been shown to ameliorate Aβ-mediated vascular dysfunction in vitro. Aβ 1-42 dose-dependent generation of ROS was halted by the administration of antioxidants, NOX-2 inhibitors, and by blocking receptors for advanced glycation end-products (RAGE) in human brain endothelial cell lines (Carrano et al., 2011). TJ protein changes were ameliorated by inhibition of c-Jun N-terminal kinases (JNK) and p38 mitogen-activated protein kinase (p38 MAPK) pathways, suggesting that these could represent potential therapeutic targets (Tai et al., 2010; Vukic et al., 2009).
Aβ-mediated cerebrovascular dysfunction in humans
The findings of the experimental studies reported above cannot yet be confirmed in humans, using currently available methods. However, several indirect observations indicate that increased Aβ levels are also detrimental to human cerebrovascular regulation, and population-based studies found an association between CAA and cognitive decline (2001; Pfeifer et al., 2002).
Imaging studies have shown that resting cerebral blood flow is reduced in selected neocortical regions in AD (Jagust et al., 1997; Johnson et al., 1998; Prohovnik, 1998). This effect could be partially mediated by vascular dysfunction. Doppler ultrasound studies found that CAA was associated with decreased vascular reactivity in response to visual stimulation (Smith et al., 2008). Another study using functional MRI identified robust differences in both amplitude and timing of the response to visual stimulation in advanced CAA (Dumas et al., 2012). On the other hand, some CAA-associated effects found in mouse models, such as changes in evoked response to CO2 measured in the middle cerebral artery (Shin et al., 2007), could not be reproduced consistently in humans (Bar et al., 2007; Jagust et al., 1997; Yamaguchi et al., 1980). It is yet to be determined if these findings reflect true differences between CAA pathogenesis in humans and rodents, or if they are due to the occipital lobe predilection in humans, which makes the middle cerebral artery a poor target for such investigations. Finally, a lack of power in the statistical analyses has to be considered as a limitation of the human studies.
NADPH oxidase was implicated in Aβ-mediated vascular dysfunction in experimental models (Iadecola, 2003) and this enzyme is up-regulated in AD (Shimohama et al., 2000).
CAA was also associated with ischemic lesions, especially in small vessels (Haglund et al., 2006; Olichney et al., 2000; Suter et al., 2002). This observation is corroborated by animal models, where increased Aβ levels increase brain vulnerability to ischemic damage via disturbances in endothelium-dependent vascular reactivity (Zhang et al., 1997). In turn, ischemic brain lesions induced in an APPswe:PS1dE9 mouse model (Jankowsky et al., 2001) were associated with increased Aβ deposition (Garcia-Alloza et al., 2011), suggesting a bi-directional relationship between Aβ and ischemia. Such a relationship would perpetuate the detrimental effects of both phenomena.
Further studies are needed to show if Aβ-induced vascular dysfunction is as deleterious in humans as it is in experimental models, and if the pathways involved are similar. If similarities are detected, substances proven to ameliorate this damage in experimental models are already in use for other human conditions such as Vitamin E (Venugopal et al., 2004) or SS31 peptide (Cho et al., 2007) for inhibiting CD36, and apocynin, a NADPH oxidase inhibitor (Lull et al., 2011). These could therefore be prioritized as potential therapeutic agents targeting vascular dysfunction in AD clinical trials.
Conclusions
In this paper, we aimed to highlight soluble and insoluble Aβ-mediated cerebrovascular dysfunction, mediated via endothelial pathways. Although direct observations were made using animal models, the results are robust enough to suggest that the same effects may occur in humans. If human brain endothelial cells are prone to the same harmful effects of Aβ 1–40 as are seen in experimental models, dysfunctional cerebrovascular regulation and BBB deficits would be much more important than mechanical CAA complications and would explain part of the cognitive decline observed in individuals with CAA.
Figure 1.
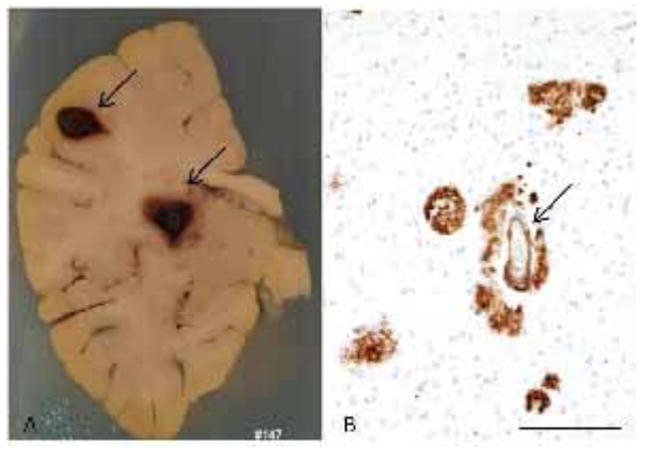
(A) Formalin fixed brain slice of a 60 years old male with two focus of cerebral hemorrhage (arrows). (B) Histological examination showed Aβ deposition in form of plaques and in vessels (arrow)(immonohistochemistry for Aβ – scale bar:100 μm). In this case, the bleeding was probably a CAA complication
Highlights.
Cerebral amyloid angiopathy (CAA) is an age-associated disease characterized by amyloid deposition in cerebral and meningeal vessel walls
CAA causes mechanical and non-mechanical complications
The non-mechanical deleterious consequences in cerebrovascular regulation are prevalent
Studies of animal models have linked the vasoactive Aβ 1–40, the main species found in CAA, to disturbances in endothelial-dependent factors, disrupting cerebrovascular regulation
If human brain endothelial cells are prone to the same harmful effects of Aβ 1–40 as are seen in experimental models, dysfunctional cerebrovascular regulation and BBB deficits would be much more important than mechanical CAA complications and would explain part of the cognitive decline observed in individuals with CAA
Acknowledgments
Lea Tenenholz Grinberg receives research grants from the National Institute of Health (2R01AG040311-01A1 and 2 P50 AG023501-06), John Douglas French Alzheimer Foundation and Albert Einstein Research Institute – São Paulo. We want to thank for his critical reading of the manuscript and Dr. Janet Johnston (www.output.ie) for language editing.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Pathological correlates of late-onset dementia in a multicentre, community-based population in England and Wales. Neuropathology Group of the Medical Research Council Cognitive Function and Ageing Study (MRC CFAS) Lancet. 2001;357:169–175. doi: 10.1016/s0140-6736(00)03589-3. [DOI] [PubMed] [Google Scholar]
- Babior BM. NADPH oxidase. Curr Opin Immunol. 2004;16:42–47. doi: 10.1016/j.coi.2003.12.001. [DOI] [PubMed] [Google Scholar]
- Bamberger ME, Harris ME, McDonald DR, Husemann J, Landreth GE. A cell surface receptor complex for fibrillar beta-amyloid mediates microglial activation. J Neurosci. 2003;23:2665–2674. doi: 10.1523/JNEUROSCI.23-07-02665.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar KJ, Boettger MK, Seidler N, Mentzel HJ, Terborg C, Sauer H. Influence of galantamine on vasomotor reactivity in Alzheimer’s disease and vascular dementia due to cerebral microangiopathy. Stroke. 2007;38:3186–3192. doi: 10.1161/STROKEAHA.107.492033. [DOI] [PubMed] [Google Scholar]
- Bell RD, Zlokovic BV. Neurovascular mechanisms and blood-brain barrier disorder in Alzheimer’s disease. Acta Neuropathol. 2009;118:103–113. doi: 10.1007/s00401-009-0522-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biffi A, Greenberg SM. Cerebral amyloid angiopathy: a systematic review. J Clin Neurol. 2011;7:1–9. doi: 10.3988/jcn.2011.7.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai H, Harrison DG. Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res. 2000;87:840–844. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- Carrano A, Hoozemans JJ, van der Vies SM, Rozemuller AJ, van Horssen J, de Vries HE. Amyloid Beta induces oxidative stress-mediated blood-brain barrier changes in capillary amyloid angiopathy. Antioxid Redox Signal. 2011;15:1167–1178. doi: 10.1089/ars.2011.3895. [DOI] [PubMed] [Google Scholar]
- Charidimou A, Gang Q, Werring DJ. Sporadic cerebral amyloid angiopathy revisited: recent insights into pathophysiology and clinical spectrum. J Neurol Neurosurg Psychiatry. 2012;83:124–137. doi: 10.1136/jnnp-2011-301308. [DOI] [PubMed] [Google Scholar]
- Cho S, Szeto HH, Kim E, Kim H, Tolhurst AT, Pinto JT. A novel cell-permeable antioxidant peptide, SS31, attenuates ischemic brain injury by down-regulating CD36. J Biol Chem. 2007;282:4634–4642. doi: 10.1074/jbc.M609388200. [DOI] [PubMed] [Google Scholar]
- Christie R, Yamada M, Moskowitz M, Hyman B. Structural and functional disruption of vascular smooth muscle cells in a transgenic mouse model of amyloid angiopathy. Am J Pathol. 2001;158:1065–1071. doi: 10.1016/S0002-9440(10)64053-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coraci IS, Husemann J, Berman JW, Hulette C, Dufour JH, Campanella GK, Luster AD, Silverstein SC, El-Khoury JB. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford F, Soto C, Suo Z, Fang C, Parker T, Sawar A, Frangione B, Mullan M. Alzheimer’s beta-amyloid vasoactivity: identification of a novel beta-amyloid conformational intermediate. FEBS Lett. 1998;436:445–448. doi: 10.1016/s0014-5793(98)01170-3. [DOI] [PubMed] [Google Scholar]
- De Reuck J, Deramecourt V, Cordonnier C, Leys D, Maurage CA, Pasquier F. The impact of cerebral amyloid angiopathy on the occurrence of cerebrovascular lesions in demented patients with Alzheimer features: a neuropathological study. Eur J Neurol. 2011;18:913–918. doi: 10.1111/j.1468-1331.2010.03329.x. [DOI] [PubMed] [Google Scholar]
- Dumas A, Dierksen GA, Gurol ME, Halpin A, Martinez-Ramirez S, Schwab K, Rosand J, Viswanathan A, Salat DH, Polimeni JR, Greenberg SM. Functional magnetic resonance imaging detection of vascular reactivity in cerebral amyloid angiopathy. Ann Neurol. 2012;72:76–81. doi: 10.1002/ana.23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farkas E, Luiten PG. Cerebral microvascular pathology in aging and Alzheimer’s disease. Prog Neurobiol. 2001;64:575–611. doi: 10.1016/s0301-0082(00)00068-x. [DOI] [PubMed] [Google Scholar]
- Fischer OZ. Die presbyophreneDemenz, derenanatomische Grundlage und klinische Abgrenzung. Gesamte Neurol Psychiatr. 1910;3:371–471. [Google Scholar]
- Frackowiak J, Zoltowska A, Wisniewski HM. Non-fibrillar beta-amyloid protein is associated with smooth muscle cells of vessel walls in Alzheimer disease. J Neuropathol Exp Neurol. 1994;53:637–645. doi: 10.1097/00005072-199411000-00011. [DOI] [PubMed] [Google Scholar]
- Frey RS, Rahman A, Kefer JC, Minshall RD, Malik AB. PKCzeta regulates TNF-alpha-induced activation of NADPH oxidase in endothelial cells. Circ Res. 2002;90:1012–1019. doi: 10.1161/01.res.0000017631.28815.8e. [DOI] [PubMed] [Google Scholar]
- Garcia-Alloza M, Gregory J, Kuchibhotla KV, Fine S, Wei Y, Ayata C, Frosch MP, Greenberg SM, Bacskai BJ. Cerebrovascular lesions induce transient beta-amyloid deposition. Brain. 2011;134:3697–3707. doi: 10.1093/brain/awr300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haglund M, Passant U, Sjobeck M, Ghebremedhin E, Englund E. Cerebral amyloid angiopathy and cortical microinfarcts as putative substrates of vascular dementia. Int J Geriatr Psychiatry. 2006;21:681–687. doi: 10.1002/gps.1550. [DOI] [PubMed] [Google Scholar]
- Hartz AM, Bauer B, Soldner EL, Wolf A, Boy S, Backhaus R, Mihaljevic I, Bogdahn U, Klunemann HH, Schuierer G, Schlachetzki F. Amyloid-beta contributes to blood-brain barrier leakage in transgenic human amyloid precursor protein mice and in humans with cerebral amyloid angiopathy. Stroke. 2012;43:514–523. doi: 10.1161/STROKEAHA.111.627562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Iadecola C. Cerebrovascular effects of amyloid-beta peptides: mechanisms and implications for Alzheimer’s dementia. CellMolNeurobiol. 2003;23:681–689. doi: 10.1023/A:1025092617651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Zhang F, Niwa K, Eckman C, Turner SK, Fischer E, Younkin S, Borchelt DR, Hsiao KK, Carlson GA. SOD1 rescues cerebral endothelial dysfunction in mice overexpressing amyloid precursor protein. Nat Neurosci. 1999;2:157–161. doi: 10.1038/5715. [DOI] [PubMed] [Google Scholar]
- Jagust WJ, Eberling JL, Reed BR, Mathis CA, Budinger TF. Clinical studies of cerebral blood flow in Alzheimer’s disease. Ann N Y Acad Sci. 1997;826:254–262. doi: 10.1111/j.1749-6632.1997.tb48477.x. [DOI] [PubMed] [Google Scholar]
- Jankowsky JL, Slunt HH, Ratovitski T, Jenkins NA, Copeland NG, Borchelt DR. Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol Eng. 2001;17:157–165. doi: 10.1016/s1389-0344(01)00067-3. [DOI] [PubMed] [Google Scholar]
- Jellinger K. Cerebrovascular amyloidosis with cerebral hemorrhage. J Neurol. 1977;214:195–206. doi: 10.1007/BF00316150. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Alzheimer disease and cerebrovascular pathology: an update. JNeural Transm. 2002;109:813–836. doi: 10.1007/s007020200068. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Lauda F, Attems J. Sporadic cerebral amyloid angiopathy is not a frequent cause of spontaneous brain hemorrhage. Eur J Neurol. 2007;14:923–928. doi: 10.1111/j.1468-1331.2007.01880.x. [DOI] [PubMed] [Google Scholar]
- Johnson KA, Jones K, Holman BL, Becker JA, Spiers PA, Satlin A, Albert MS. Preclinical prediction of Alzheimer’s disease using SPECT. Neurology. 1998;50:1563–1571. doi: 10.1212/wnl.50.6.1563. [DOI] [PubMed] [Google Scholar]
- Kalaria RN. Linking cerebrovascular defense mechanisms in brain ageing and Alzheimer’s disease. Neurobiol Aging. 2009;30:1512–1514. doi: 10.1016/j.neurobiolaging.2007.10.020. [DOI] [PubMed] [Google Scholar]
- Li JM, Shah AM. Mechanism of endothelial cell NADPH oxidase activation by angiotensin II. Role of the p47phox subunit. J Biol Chem. 2003;278:12094–12100. doi: 10.1074/jbc.M209793200. [DOI] [PubMed] [Google Scholar]
- Lull ME, Levesque S, Surace MJ, Block ML. Chronic apocynin treatment attenuates beta amyloid plaque size and microglial number in hAPP(751)(SL) mice. PLoS One. 2011;6:e20153. doi: 10.1371/journal.pone.0020153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marco S, Skaper SD. Amyloid beta-peptide1-42 alters tight junction protein distribution and expression in brain microvessel endothelial cells. Neurosci Lett. 2006;401:219–224. doi: 10.1016/j.neulet.2006.03.047. [DOI] [PubMed] [Google Scholar]
- Nakata-Kudo Y, Mizuno T, Yamada K, Shiga K, Yoshikawa K, Mori S, Nishimura T, Nakajima K, Nakagawa M. Microbleeds in Alzheimer disease are more related to cerebral amyloid angiopathy than cerebrovascular disease. Dement Geriatr Cogn Disord. 2006;22:8–14. doi: 10.1159/000092958. [DOI] [PubMed] [Google Scholar]
- Niwa K, Porter VA, Kazama K, Cornfield D, Carlson GA, Iadecola C. A beta-peptides enhance vasoconstriction in cerebral circulation. Am J Physiol Heart Circ Physiol. 2001;281:H2417–2424. doi: 10.1152/ajpheart.2001.281.6.H2417. [DOI] [PubMed] [Google Scholar]
- Niwa K, Younkin L, Ebeling C, Turner SK, Westaway D, Younkin S, Ashe KH, Carlson GA, Iadecola C. Abeta 1-40-related reduction in functional hyperemia in mouse neocortex during somatosensory activation. Proc Natl Acad Sci U S A. 2000;97:9735–9740. doi: 10.1073/pnas.97.17.9735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okazaki H, Reagan TJ, Campbell RJ. Clinicopathologic studies of primary cerebral amyloid angiopathy. Mayo Clin Proc. 1979;54:22–31. [PubMed] [Google Scholar]
- Olichney JM, Hansen LA, Hofstetter CR, Lee JH, Katzman R, Thal LJ. Association between severe cerebral amyloid angiopathy and cerebrovascular lesions in Alzheimer disease is not a spurious one attributable to apolipoprotein E4. Arch Neurol. 2000;57:869–874. doi: 10.1001/archneur.57.6.869. [DOI] [PubMed] [Google Scholar]
- Oppenheim G. Über “drusigeNekrosen” in der Grosshirnrinde. Neurol Centralbl. 1909;28:410–413. [Google Scholar]
- Paris D, Humphrey J, Quadros A, Patel N, Crescentini R, Crawford F, Mullan M. Vasoactive effects of A beta in isolated human cerebrovessels and in a transgenic mouse model of Alzheimer’s disease: role of inflammation. Neurol Res. 2003;25:642–651. doi: 10.1179/016164103101201940. [DOI] [PubMed] [Google Scholar]
- Park L, Wang G, Zhou P, Zhou J, Pitstick R, Previti ML, Younkin L, Younkin SG, Van Nostrand WE, Cho S, Anrather J, Carlson GA, Iadecola C. Scavenger receptor CD36 is essential for the cerebrovascular oxidative stress and neurovascular dysfunction induced by amyloid-beta. Proc Natl Acad Sci U S A. 2011;108:5063–5068. doi: 10.1073/pnas.1015413108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pezzini A, Del Zotto E, Volonghi I, Giossi A, Costa P, Padovani A. Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Curr Med Chem. 2009;16:2498–2513. doi: 10.2174/092986709788682047. [DOI] [PubMed] [Google Scholar]
- Pfeifer LA, White LR, Ross GW, Petrovitch H, Launer LJ. Cerebral amyloid angiopathy and cognitive function: the HAAS autopsy study. Neurology. 2002;58:1629–1634. doi: 10.1212/wnl.58.11.1629. [DOI] [PubMed] [Google Scholar]
- Price JM, Hellermann A, Hellermann G, Sutton ET. Aging enhances vascular dysfunction induced by the Alzheimer’s peptide beta-amyloid. Neurol Res. 2004;26:305–311. doi: 10.1179/016164104225014003. [DOI] [PubMed] [Google Scholar]
- Prohovnik I. Iodine-123-iomazenil SPECT in Alzheimer’s disease. J Nucl Med. 1998;39:927. [PubMed] [Google Scholar]
- Ray R, Shah AM. NADPH oxidase and endothelial cell function. Clin Sci (Lond) 2005;109:217–226. doi: 10.1042/CS20050067. [DOI] [PubMed] [Google Scholar]
- Shimohama S, Tanino H, Kawakami N, Okamura N, Kodama H, Yamaguchi T, Hayakawa T, Nunomura A, Chiba S, Perry G, Smith MA, Fujimoto S. Activation of NADPH oxidase in Alzheimer’s disease brains. Biochem Biophys Res Commun. 2000;273:5–9. doi: 10.1006/bbrc.2000.2897. [DOI] [PubMed] [Google Scholar]
- Shin HK, Jones PB, Garcia-Alloza M, Borrelli L, Greenberg SM, Bacskai BJ, Frosch MP, Hyman BT, Moskowitz MA, Ayata C. Age-dependent cerebrovascular dysfunction in a transgenic mouse model of cerebral amyloid angiopathy. Brain. 2007;130:2310–2319. doi: 10.1093/brain/awm156. [DOI] [PubMed] [Google Scholar]
- Smith EE, Greenberg SM. Beta-amyloid, blood vessels, and brain function. Stroke. 2009;40:2601–2606. doi: 10.1161/STROKEAHA.108.536839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EE, Vijayappa M, Lima F, Delgado P, Wendell L, Rosand J, Greenberg SM. Impaired visual evoked flow velocity response in cerebral amyloid angiopathy. Neurology. 2008;71:1424–1430. doi: 10.1212/01.wnl.0000327887.64299.a4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suter OC, Sunthorn T, Kraftsik R, Straubel J, Darekar P, Khalili K, Miklossy J. Cerebral hypoperfusion generates cortical watershed microinfarcts in Alzheimer disease. Stroke. 2002;33:1986–1992. doi: 10.1161/01.str.0000024523.82311.77. [DOI] [PubMed] [Google Scholar]
- Tai LM, Holloway KA, Male DK, Loughlin AJ, Romero IA. Amyloid-beta-induced occludin down-regulation and increased permeability in human brain endothelial cells is mediated by MAPK activation. J Cell Mol Med. 2010;14:1101–1112. doi: 10.1111/j.1582-4934.2009.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thal DR, Capetillo-Zarate E, Larionov S, Staufenbiel M, Zurbruegg S, Beckmann N. Capillary cerebral amyloid angiopathy is associated with vessel occlusion and cerebral blood flow disturbances. Neurobiol Aging. 2009;30:1936–1948. doi: 10.1016/j.neurobiolaging.2008.01.017. [DOI] [PubMed] [Google Scholar]
- Thal DR, Ghebremedhin E, Orantes M, Wiestler OD. Vascular pathology in Alzheimer disease: correlation of cerebral amyloid angiopathy and arteriosclerosis/lipohyalinosis with cognitive decline. J Neuropathol Exp Neurol. 2003;62:1287–1301. doi: 10.1093/jnen/62.12.1287. [DOI] [PubMed] [Google Scholar]
- Thomas T, Thomas G, McLendon C, Sutton T, Mullan M. Beta-amyloid-mediated vasoactivity and vascular endothelial damage. Nature. 1996;380:168–171. doi: 10.1038/380168a0. [DOI] [PubMed] [Google Scholar]
- Townsend KP, Obregon D, Quadros A, Patel N, Volmar C, Paris D, Mullan M. Proinflammatory and vasoactive effects of Abeta in the cerebrovasculature. Ann N Y Acad Sci. 2002;977:65–76. doi: 10.1111/j.1749-6632.2002.tb04799.x. [DOI] [PubMed] [Google Scholar]
- Ushio-Fukai M, Tang Y, Fukai T, Dikalov SI, Ma Y, Fujimoto M, Quinn MT, Pagano PJ, Johnson C, Alexander RW. Novel role of gp91(phox)-containing NAD(P)H oxidase in vascular endothelial growth factor-induced signaling and angiogenesis. Circ Res. 2002;91:1160–1167. doi: 10.1161/01.res.0000046227.65158.f8. [DOI] [PubMed] [Google Scholar]
- van Beek AH, Claassen JA, Rikkert MG, Jansen RW. Cerebral autoregulation: an overview of current concepts and methodology with special focus on the elderly. J Cereb Blood Flow Metab. 2008;28:1071–1085. doi: 10.1038/jcbfm.2008.13. [DOI] [PubMed] [Google Scholar]
- Venugopal SK, Devaraj S, Jialal I. RRR-alpha-tocopherol decreases the expression of the major scavenger receptor, CD36, in human macrophages via inhibition of tyrosine kinase (Tyk2) Atherosclerosis. 2004;175:213–220. doi: 10.1016/j.atherosclerosis.2004.03.012. [DOI] [PubMed] [Google Scholar]
- Vinters HV. Cerebral amyloid angiopathy. A critical review. Stroke. 1987;18:311–324. doi: 10.1161/01.str.18.2.311. [DOI] [PubMed] [Google Scholar]
- Vukic V, Callaghan D, Walker D, Lue LF, Liu QY, Couraud PO, Romero IA, Weksler B, Stanimirovic DB, Zhang W. Expression of inflammatory genes induced by beta-amyloid peptides in human brain endothelial cells and in Alzheimer’s brain is mediated by the JNK-AP1 signaling pathway. Neurobiol Dis. 2009;34:95–106. doi: 10.1016/j.nbd.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski HM, Frackowiak J, Zoltowska A, Kim KS. Vascular beta-amyloid in Alzheimer’s disease angiopathy is produced by proliferating and degenerating smooth muscle cells. Amyloid. 1994;1:8–16. [Google Scholar]
- Yamaguchi F, Meyer JS, Yamamoto M, Sakai F, Shaw T. Noninvasive regional cerebral blood flow measurements in dementia. Arch Neurol. 1980;37:410–418. doi: 10.1001/archneur.1980.00500560040003. [DOI] [PubMed] [Google Scholar]
- Yoshiura T, Wu O, Sorensen AG. Advanced MR techniques: diffusion MR imaging, perfusion MR imaging, and spectroscopy. Neuroimaging Clin N Am. 1999;9:439–453. [PubMed] [Google Scholar]
- Zhang F, Eckman C, Younkin S, Hsiao KK, Iadecola C. Increased susceptibility to ischemic brain damage in transgenic mice overexpressing the amyloid precursor protein. J Neurosci. 1997;17:7655–7661. doi: 10.1523/JNEUROSCI.17-20-07655.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlokovic BV. The blood-brain barrier in health and chronic neurodegenerative disorders. Neuron. 2008;57:178–201. doi: 10.1016/j.neuron.2008.01.003. [DOI] [PubMed] [Google Scholar]