

Abstract
The +1169A allele of the A/T single nucleotide polymorphism (SNP; rs2665802), located within intron 4 of the human growth hormone I (GHI) gene, has been associated with reduced levels of circulating GH and insulin-like growth factor I, a reduced risk of colorectal cancer and a predisposition to osteoporosis. Whether this intronic SNP is itself the functional polymorphism responsible for exerting a direct effect on GHI gene expression, however, or whether it is instead in linkage disequilibrium with the functional SNP, has been an open question. The evolutionary conservation of the +1169T allele (and the surrounding intronic sequence) in the bovine genome, as well as in primate genomes, is, however, suggestive of its functionality. Although a potential alternative splice site spans the location of the +1169 SNP, polymerase chain reaction-based assays failed to yield any evidence for alternative splicing associated with either allele. To determine whether the +1169 SNP, in different allelic combinations with SNPs at -278 (G/T), -57 (T/G) and +2103 (C/T), exerts a direct effect on gene expression and/or GH secretion, we performed a series of transfections of various GHI haplotype-expressing constructs into rat GC (somatotroph) cells. The results obtained provided evidence to support the contention that the +1169A allele contributes directly to the observed reduction in both GHI gene expression and GH secretion. Part of the apparent influence of the +1169A-bearing allele on GHI gene expression and GH secretion may still, however, be attributable to alleles of additional SNPs in cis to +1169A and located within either the promoter or the 3'-flanking region.
Keywords: growth hormone (GHI) gene, gene expression, protein secretion, intronic functional polymorphism, alternative splicing
Introduction
Human growth hormone (GH) plays an important role in immune function and bone turnover, in addition to its well-documented influences on stature, muscle mass, lipid and carbohydrate metabolism and postnatal growth [1]. The specificity of GH action lies in promoting the homodimerisation of its cell surface receptor (GHR), resulting in the induction of post-receptor signalling pathways [2]. Human GH synthesis is directed by the pituitary-expressed GH1 gene, which is located on chromosome 17q23 within a gene cluster that includes three paralogous placentally expressed genes (CSH1, CSH2 and GH2). The control of GH1 gene expression is regulated by the pituitary-expressed transcription factor, PIT1, which drives GH expression by binding not only to the GH1 proximal promoter, but also to a locus control region (LCR) located between 14.5 kilobases (kb) and 32 kb upstream of the GH1 gene [3].
The proximal region of the GH1 gene promoter exhibits a high level of sequence variation, with 15 single nucleotide polymorphisms (SNPs) occurring within a 450 base pair (bp) stretch of DNA [4,5]. This high level of sequence diversity is explicable in terms of a combination of gene conversion, recurrent mutations and selection [4,6,7] In the European population, these polymorphic variants manifest in at least 40 different haplotypes that display a 12-fold range of expression level in a reporter gene assay [6]. At least in this population, there is a tendency for those haplotypes associated with a markedly reduced level of reporter gene expression to be more prevalent than those haplotypes associated with an increased level, possibly as a consequence of selection [6].
In addition to the promoter polymorphisms, an A/T SNP has been reported at nucleotide +90 within intron 4 of the GH1 gene (rs2665802; chromosome 17 coordinate, 59348761 -- termed 1663 by Hasegawa et al [8]. and here, termed +1169). The +1169A allele of this SNP has been associated with reduced levels of circulating GH and insulin-like growth factor 1 (IGF-1),[8] a reduced risk of colorectal cancer [9] and a predisposition to osteoporotic bone loss [10]. It is currently unclear whether this intronic SNP performs a functional role by exerting a direct effect on GH1 gene expression or whether it is instead in linkage disequilibrium with another functional SNP. This question remained unanswered by the original authors [8]. Here, we have attempted to characterise the intronic +1169 SNP functionally, in an attempt to disentangle its potential influence upon GH structure, function and expression from that of three other potentially functional GH1 SNPs with which it is in strong linkage disequilibrium.
Materials and methods
SNP designation
All four of the SNPs studied in detail here have been reported before. Two are located in the GH1 gene promoter region (-278G/T [rs2005171] and -57G/T [rs2005172]), one is located within intron 4 (rs2665802; +1169; 90 bp from the donor splice site) and one is located in the 3' flanking region: a C/T transition at position +2103 (ss19373532). Since all four of these SNPs have been ascribed different acronyms/numbering by different authors, a guide to alternative nomenclature, as well as dbSNP numbering, is given in Table S1 (Table 6).
Table S1.
Alternative acronyms/numbering used for the five GH1 SNPs under study
| Our numberinga |
dbSNP No. |
Wagner et al [5]. |
Hasegawa et al [8]. |
Le Marchand et al [9]. |
Esteban et al [37]. |
|---|---|---|---|---|---|
| -278 | rs2005171 | -339 | P2 | - | 4886 |
| -57 | rs2005172 | -118 | P3 | - | 5107 |
| +1169 | rs2665802 | - | P1 (1663) | 1663 | P24 (6331) |
| +2103 | ss19373532 | - | - | - | - |
| +2498 | - | - | - | - | - |
aRelative to the GH1 transcriptional initiation site at +1
Polymerase chain reaction amplification and sequencing
A 3.2 kb genomic DNA fragment specific to the GH1 gene was previously amplified in a polymerase chain reaction (PCR) from 153 individuals of normal stature [11] using oligonucleotide primers GH1F (5' GGGAGCCCCAGCAATGC 3'; -615 to -599) and GH1R (5' TGTAGGAAGTCTGGGGTGC 3'; +2598 to +2616) (all numbering relative to the transcriptional initiation site at +1 [GenBank accession number J03071]). These PCR fragments were directly sequenced to identify variants within the entire coding region; introns; 5' and 3' untranslated regions; and promoter region of the GH1 gene [11].
In this study, we extended our initial sequence analysis to include 1 kb of 3' flanking region using the primers GH1SEQ4 (5' GCATCATTTTGTCTGACTAGG 3'; +1631 to +1651) and GH1SEQ5 (5' CAGACACAGCATAGGCTACC 3'; +2051 to +2070). In cases where individuals were heterozygous at two or more positions within this region, the 3.2 kb GH1 gene fragments were cloned into the cloning vector pGEM-T (Promega; Southampton, UK); four clones were sequenced unambiguously to identify the respective haplotypes.
GH1 gene constructs
The human GH1 gene was PCR amplified from the BAC clone CTC-264K15 (accession number AC127029) using primers GH1F and GH1R, cloned into pGEM-T (Promega) and sequenced using BigDye v3.1 to confirm its identity. Site-directed mutagenesis (SDM) was performed using the QuickChange kit (Agilent, Stockport, UK) according to the manufacturer's instructions, to introduce either a FLAG (5' GACTACAAAGACGATGACGACAAG 3') or a MYC (5' GAACAAAAACTCATCTCAGAAGAGGATCTG 3') tag sequence immediately 5' to the natural termination codon. The resulting vectors were termed GH-FLAG and GH-MYC, respectively. SDM was used to introduce specific sequence changes at positions -278 (T/G), -57 (T/G), +1169 (A/T) and +2103 (C/T) of the human GH1 gene (Table S2 (Table 7)). All resulting clones were sequenced to confirm that only the specified sequence changes had been introduced. The standard GH-FLAG and GH-MYC vectors used in all studies had the following haplotype, based on the four SNPs listed above: -278G, -57T, +1169T and +2103C.
Table S2.
Oligonucleotide primers used in site-directed mutagenesis reactions
| Primer | Oligonucleotide sequence (5' > 3')a |
|---|---|
| GHFLAG5 | TGGAGGGCAGCTGTGGCTTCGACTACAAAGACGATGACGACAAGTAGCTGCCCGGGTGGCATCC |
| GHMYC5 | TGGAGGGCAGCTGTGGCTTCGAACAAAAACTCATCTCAGAAGAGGATCTGTAGCTGCCCGGGTGGCATCC |
| -278T | CCACCATGGCCTGCTGCCAGAGGGCACCC |
| -57G | AGAAACAGGTGGGGGCAACAGTGGGAGAG |
| 1169A5 | CCTCTTTTTAGCAGACAGGCCCTGACCCA |
| 2103T5 | GGACATTTGAGTTGTTTGCTTGGCACTGT |
| 2498G5 | TCCAGCCTCAAAGAGCTTACAGTCTGGTA |
aOnly the sequence of the sense strand oligonucleotide is shown. Bases inserted or changed are shown in bold
Cell transfection
The rat GC pituitary cell line [12] was used in all experiments. Rat GC cells were grown in Dulbeco's Modified Eagle's Medium (DMEM) containing 15 per cent horse serum and 2.5 per cent foetal calf serum at 37°C in 5 per cent CO2. For each transfection, two GH1 gene constructs were used: one vector contained the wild-type GH-MYC sequence (normalisation control) and the second vector contained the GH-FLAG vector with or without introduced sequence changes. All transfections were performed in 24-well plates using 250 ng each plasmid construct and FuGENE HD transfection reagent (Roche; East Sussex, UK) according to the manufacturer's instructions. Twenty-four-hour post-transfection cells were washed twice with phosphate-buffered saline (PBS) and then 0.5 ml serum-free medium (0.2 per cent foetal calf serum; 2 mM glutamax in DMEM) was added to each well. Twenty-four hours later, media and cells were harvested. Each set of transfections was performed on at least three separate occasions.
DNA and RNA extraction
Medium was removed from transfected cells after 24 hours, centrifuged to remove cell debris and then stored at -80°C for subsequent analysis of secreted human GH. 200 μl RNAprotect cell reagent (Qiagen; Crawley, UK) was then added to each well. 50 μl of the resulting cell suspension was removed for extraction of genomic DNA and the remaining 150 μl was used for extraction of total RNA. All samples were centrifuged at 10,000 rpm for 5 minutes at 4°C and the supernatant removed. Samples for RNA extraction were resuspended in 200 μl TRI reagent (Sigma; Dorset, UK) and processed according to the manufacturer's instructions. Contaminating DNA was removed using the DNA-free kit (Applied Biosystems; Warrington, UK) according to the manufacturer's instructions. 100 ng of each RNA was then assessed for the presence of any residual DNA using a SYBR Green-based quantitative PCR (Q-PCR) assay (Applied Biosystems) using human GH1 gene-specific primers GHE55 (5' ATCTTCAAGCAGACCTAC AG 3'; +1366 to +1385) and GHE53 (5' CCCTCCACAGAGCGGCAC 3'; +1497 to +1514). RNA samples that exhibited no PCR amplification after 40 cycles were used for subsequent analysis.
DNA extraction was performed as described by Laird [13]. Briefly, cell pellets were resuspended in 250 μl lysis buffer (10 mM Tris-HCl pH 8.5; 5 mM ethylenediamine tetra-acetic acid (EDTA); 1 per cent sodium dodecyl sulphate (SDS); 200 mM NaCl). After resuspension, 5 mg proteinase K was added and samples were incubated overnight at 37°C. DNA was precipitated by adding an equal volume of isopropanol, washed once with 70 per cent ethanol and resuspended in 200 μl sterile water prior to quantification.
Reverse transcription
Reverse transcription was performed using 500 ng total RNA using an oligo dT(25) primer and Superscript III (Invitrogen; Paisley, UK) according to the manufacturer's instructions.
PCR amplification of GH1 cDNA
PCR amplification of cDNA derived from transfected GC cells was performed using two sets of oligonucleotide primers: GHE15 (5' CTCACCTAGCTGCAATGGC 3'; +49 to +67) and GHE53, and GHE45 (5' CGTGCAGTTCCTCAGGAGTGT 3'; +968 to +988) and GHE53. PCR was performed using the Expand™ high-fidelity system (Roche) using a hot start at 98°C for 2 minutes, followed by 95°C for 3 minutes, 35 cycles at 95°C for 30 seconds, 60°C for 30 seconds and 72°C for 1 minute 30 seconds. For the last 20 cycles, the elongation step at 72°C was increased by 5 seconds per cycle. This was followed by a further incubation at 72°C for 10 minutes.
Real-time quantitative PCR
Real-time quantitative PCR was performed on DNA samples to measure transfection efficiency and on cDNA to determine expression levels using SYBR Green PCR master mix (Applied Biosystems) and quantified on a 7500 real-time PCR machine (Applied Biosystems). Plasmid DNAs (GH-FLAG and GH-MYC) were used to generate standard curves. These vectors were diluted from 107 to 104 copies per PCR reaction. All PCRs were performed in triplicate in a final volume of 12.5 μl containing 6.25 μl 2 × SYBR Green PCR master mix, 5 ng DNA or cDNA and either primer pairs GHQ55 (5' GTGCCGCTCTGTGGAGGG 3'; +1497 to +1514) and GHFLQ3 (5'GTCGTCATCGTCTTTGTAGTC 3') or GHQ55 and GHMYQ3 (5' CAGATCCTCTTCTGAGATGAG 3') at a final concentration of 5 pmol/μl. Samples were PCR amplified as follows: 50°C for 2 minutes, 95°C for 10 minutes followed by 40 cycles of 95°C for 15 seconds, 60°C for 1 minute. Neither PCR assay showed any amplification with rat genomic DNA or cDNA.
GH enzyme-linked immunosorbent assays
A human GH enzyme-linked immunosorbent assay (ELISA) (Roche) was used to quantify cell culture supernatants from transfected GH-FLAG and GH-MYC vectors for use as standards in the assays specified below. This ELISA kit showed no cross-reactivity with rat GH.
Two separate ELISA assays were established to measure GH-FLAG and GH-MYC. 100 μl mono-clonal anti-GH antibody (Ab1954; Abcam; Cambridge, UK), diluted 1 in 1,000 in 0.05 M carbonate buffer at pH 9.6, was used to coat Nunc Maxisorp™plates overnight at 4°C. The sensitivity of measurement of GH concentration for both ELISA assays was ~80 pg/ml. The intra-assay coefficients of variation of each assay were 4.2 per cent for the GH-MYC ELISA and 4.6 per cent for the GH-FLAG ELISA. The inter-assay coefficients of variation of each assay were 10.9 per cent for the GH-MYC ELISA and 11.2 per cent for the GH-FLAG ELISA. Plates were washed four times with 200 μl PBS containing 0.05 per cent Tween 20 (PBS-T), and remaining binding sites were blocked with 5 per cent Marvel in PBS-T overnight at 4°C. Plates were washed four times with 400 μl PBS-T, and 100 μl appropriately diluted GH-containing medium, in octuplicate, were added to each well and incubated for 2 hours at room temperature. They were then washed again four times with 200 μl PBS-T and 100 μl of either DDDDK-tag (Ab1238; Abcam) or MYC-tag Worseradish peroxidase (HRP)-labelled antibodies (Ab1261; Abcam) were added to each well to detect FLAG- and MYC-tagged GH, respectively. DDDDK-tag HRP and MYC-tag HRP antibodies were used at dilutions of 1/2,000 and 1/10,000, respectively. Plates were incubated for 2 hours at room temperature. They were then washed four times with 200 μl PBS-T before 100 μl SureBlue Reserve substrate (KPL, Gaithersburg, MD, USA) was introduced and the colour allowed to develop. Finally 100 μl 3,3',5,5'-tetramethylbenzidine (TMB) stop solution (KPL) was added. Optical density at 450 nm was then measured using a 96-well plate reader. GH-FLAG and GH-MYC GH (appropriately diluted, based on GH levels determined by GH ELISA) were used to generate a standard curve. Levels of GH in each sample were then determined from the standard curve. Neither the GH-FLAG nor the GH-MYC ELISA showed any cross-reactivity to rat GH or to each other.
Statistical analysis
The absolute quantification method, with the wild-type GH-MYC or GH-FLAG vectors as standards, was used to calculate the number of GH1 molecules either transfected or expressed in each experiment. Wild-type GH-MYC was identical in all experiments and was used to normalise the transfection efficiency of the various GH-FLAG vectors, based on the number of DNA molecules transfected. The mean transfection efficiency ratio (FLAG/MYC) was then used to normalise the levels of both GH1 gene expression and secreted GH. Finally, the obtained values were divided by the value for the wild-type control. Normalised activities were interpreted as -fold changes by comparison with the wild-type. Since no significant difference was noted between plates, the expression and secretion data were combined over all plates. Differences in GH1 gene expression and GH secretion between the different haplotypes were assessed for statistical significance using Student's t-test.
Prediction of alternative splice site
The Alternative Splice Site Predictor (ASSP) software, described by Wang and Marín [14] and available at http://www.es.embnet.org/~mwang/assp.html, was used to locate the potential alternative splice site which flanks the +1169T allele in intron 4 of the GH1 gene and to predict its strength in relation to that proffered by the alternative +1169A allele.
Results
Initial evidence for the functionality of the +1169 SNP from evolutionary conservation and sequence homology data
Evidence supporting the potential functionality of the +1169 SNP initially came from two different sources: evolutionary conservation and sequence homology with a known transcription factor binding site. First, with respect to evolutionary conservation, nucleotide +1169T is invariant in the four human GH paralogues (GH2, CSH1, CSH2 and CSHP1), as well as in the GH orthologues of five primate species and Bos taurus (cow) (Figure 1). In addition, the intron 4 sequence immediately flanking the +1169 SNP in the human genome (CTCTTTTTAGCAGTCAGGCCCTGACCCA) is also highly conserved in these six mammals (Figure 1). Such conservation over what corresponds to a ~50 Myr period of evolutionary time is strongly suggestive of functionality. Second, using TRANSFAC [15] to search for known transcription factor binding sites, a decanucleotide sequence (AGACAGGCCC) corresponding to the A allele of the +1169 SNP, and spanning the site of the polymorphism, was found to be highly homologous (one mismatch) to the consensus sequence of a p53-responsive element (RRRCWWGYYY; Figure 1) [16]. The T allele of the +1169 SNP, however, exhibits lower similarity to this consensus.
Figure 1.
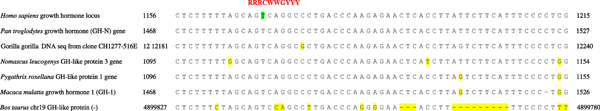
Intron 4 sequence from the orthologous GH genes of human, five other primates and Bos taurus (cow). The polymorphic nucleotide at +1169 in the human GH1 gene is marked in green. Mismatched positions are highlighted in yellow. GenBank accession nos: Homo sapiens J03071; Pan troglodytes, chimpanzee (AF374232.1); Gorilla gorilla, gorilla (FP245407.5); Nomascus leucogenys, white-cheeked gibbon (AY621637.1); Pygathrix roxellana, Sichuan-snub-nosed monkey (AY621647.1); Macaca mulatta, rhesus macaque (DQ002799.1); Bos taurus, cow (Bos taurus release Btau_4.0). The location of the consensus sequence of the p53-responsive element (RRRCWWGYYY) is shown in red [16].
Linkage disequilibrium between the +1169 SNP and two GH1 promoter SNPs
The +1169T allele previously has been found to be associated with higher circulating GH and IGF-1 levels than the +1169A allele in a cohort of prepubertal short children with or without GH insufficiency [8]. Since these authors also showed that there was significant linkage disequilibrium between the +1169 SNP and two promoter SNPs at positions -278 (rs2005171; SNP66) and -57 (rs2005172; SNP96), however, it was unclear from the outset which SNP (or SNPs) was actually responsible for the observed variation in GH level. In order to determine whether the +1169T intronic SNP was itself functional, or whether it was instead in linkage disequilibrium with another SNP that was actually responsible for exerting an effect upon GH structure, function or expression, we set out to identify additional potentially functional SNPs (in the European population) from within the GH1 gene region that are in linkage disequilibrium with the +1169T allele with a view to characterising them individually.
The sequence analysis of the entire human GH1 gene in 153 individuals of European descent has been reported previously [6,11]. The frequencies of the SNPs at positions -278 (T = 0.402; G = 0.598), -57 (G = 0.637; T = 0.363) and +1169 (T = 0.585; A = 0.415) in the European population [6,11] were very similar to those seen by Hasegawa et al [8]. in the Japanese population, although -- unlike the latter authors -- we did not find complete linkage disequilibrium between the -278T and -57G SNPs. In agreement with Hasegawa et al.,[8] however, we found that the -278G allele was linked in cis with +1169T in 168/179 (f = 0.94) alleles, whereas the -57T allele was linked in cis with +1169T in 104/179 (f = 0.58) alleles. By contrast, the +1169A allele was linked in cis with -57G (120/127; f = 0.94) and -278T in 112/127 alleles (f = 0.88). Further, -278T/-57G was found to be linked in cis with +1169A in 111/127 alleles (f = 0.874), whereas +1169T was linked in cis with either -278G/-57T (98/179; f = 0.55) or -278G/-57G (70/179; f = 0.39) [Table 1]. A matrix showing the linkage disequilibrium measures, D' and r2, relating to the biallelic SNP frequencies of the -278, -57, +1169, +2103 and +2498 polymorphisms identified in our controls of north European origin is shown in Table S3 (Table 8).
Table 1.
Relationship between the different -278/-57 GH1 gene haplotypes and the alleles of the GH1 intron 4 polymorphism at position +1169 in 153 individuals of European descent [6]
| -278 | -57 | +1169T | +1169A |
|---|---|---|---|
| G | T | 98 | 6 |
| T | G | 5 | 111 |
| G | G | 70 | 9 |
| T | T | 6 | 1 |
Table S3.
Matrix showing the linkage disequilibrium measures, D' and r2, based on the biallelic SNP frequencies of the -278, -57, +1169, +2103 and +2498 polymorphisms identified in 153 Caucasian controls
|
GH1 SNP alleles |
-278 | -57 | +1169 | +2103 | +2498 |
|---|---|---|---|---|---|
| -278 | - | D' = 0.843 | D' = 0.847 | D' = 0.847 | D' = 0.847 |
| r2 = 0.272 | r2 = 0.679 | r2 = 0.679 | r2 = 0.310 | ||
| -57 | - | D' = 0.848 | D' = 0.848 | D' = 0.483 | |
| r2 = 0.288 | r2 = 0.288 | r2 = 0.186 | |||
| +1169 | - | D' = 1.000 | D' = 1.000 | ||
| r2 = 1.000 | r2 = 0.513 | ||||
| +2103 | - | D' = 1.000 | |||
| r2 = 0.513 | |||||
| +2498 | - |
Sequence analysis of the GH1 gene 3' flanking region
In an attempt to identify further SNPs in linkage disequilibrium with +1169, the initial sequence analysis of the GH1 gene was extended to include an additional 1 kb of 3' flanking region. This region includes a putative silencer element [17]. Two further SNPs were identified: a C/T transition at position +2103 (ss19373532) and a novel G/A transition at position +2498. No other sequence changes were found within this 1 kb region.
These two SNPs were found 101 bp (+2103) and 496 bp (+2498), respectively, 3' to the putative silencer element [17]. Using TRANSFAC [15] to search for known transcription factor binding sites, a heptanucleotide sequence (TGTTTGC) corresponding to the T allele of the +2103 SNP and spanning the site of the polymorphism (highlighted in bold) was found to be homologous with the consensus sequence of HNF3β (GCAAACA) [18]. The C allele of the +2103 SNP exhibits lower similarity with this consensus. Neither the A nor the G alleles of the +2498 SNP displayed any homology to a known transcription factor binding site. The +2103C allele (f = 0.585) was found to be in total linkage disequilibrium with the +1169T allele of the intron 4 SNP, whereas the +2498A allele (f = 0.415) showed no evidence of linkage disequilibrium with either +1169 or +2103. The +2498 SNP was therefore not considered further in this analysis.
GH1 gene expression and GH secretion in GC cells
To determine whether different combinations of the SNPs at -278, -57, +1169 and +2103 exerted an effect on gene expression and/or GH secretion, we performed a series of transfections of various GH1 constructs into rat GC (somatotroph) cells. All transfections were performed with two vectors: a standard vector, containing the MYC tag sequence and a haplotype defined by alleles -278G, -57T, +1169T and +2103C; and test vectors containing the FLAG tag and the four different combinations of the GH1 gene promoter SNPs (-278G/T and -57G/T), and two different combinations of the intron 4 (+1169T/A) and 3' flanking region (+2103C/T) SNPs, resulting in a total of eight different combinations of alleles at four sites. Twenty-four hours after transfection, the medium was replaced with serum-free medium and the experiment was continued for an additional 24 hours. After this time the medium was collected for GH measurement and cells were collected for extraction of both DNA and total RNA. Using a serum-free medium ensured that only basal levels of GH1 gene expression and GH secretion were measured. DNA extracted from transfected GC cells was used for Q-PCR with primer sets GHQ55/GHFLQ3 and GHQ55/GHMYQ3 to determine transfection efficiency relative to the standard vector. Total RNA was extracted from transfected GC cells and then reverse transcribed with an oligo dT(25) primer. The resulting cDNA was then used for quantitative PCR using GHQ55/GHFLQ3 and GHQ55/GHMYQ3 primer sets. Gene expression levels were measured relative to that of the control FLAG vector (-278G/-57T/+1169T/+2103C). The results obtained are shown in Table 2. Comparison of the different allele combinations of promoter SNPs with the +1169T/+2103C haplotype indicated there to be no statistically significant difference in GH1 gene expression between -278G/-57T and -278G/-57G (p = 0.739), whereas there was a 6 per cent difference in expression between the -278G/-57T and -278T/-57G haplotypes (p = 0.047) and a 6 per cent difference in GH1 gene expression between -278G/-57T and -278T/-57T (p < 0.001). By contrast, none of the promoter SNP allele combinations bearing the +1169A/+2103T haplotype showed any significant difference in GH1 gene expression that was attributable to a specific allele at these locations. Comparison of the +1169T/+2103C and +1169A/+2103T haplotypes bearing the same -278/-57 promoter genotype revealed that neither -278T/-57G (p = 0.61) nor -278G/-57G (p = 0.19) exhibited any significant difference in GH1 gene expression, whereas both the -278G/-57T (p = 0.033) and -278T/-57T (p < 0.001) displayed significant decreases in expression of 4 per cent and 12 per cent, respectively.
Table 2.
Comparison of GH1 gene expression between eight different allele combinations involving four different SNPs from the GH1 gene region
| -278 | -57 | +1169 | +2103 | Expressiona | SEM |
p value versus 1 |
p value versus 2 |
p value (TC versus AT) |
|
|---|---|---|---|---|---|---|---|---|---|
| 1 | G | T | T | C | 1.0000 | 0.014 | - | 0.033 | 0.033 |
| 2 | G | T | A | T | 0.9597 | 0.012 | 0.033 | - | |
| 3 | T | G | T | C | 0.9387 | 0.026 | 0.047 | 0.466 | 0.611 |
| 4 | T | G | A | T | 0.9540 | 0.015 | 0.029 | 0.759 | |
| 5 | G | G | T | C | 1.0088 | 0.022 | 0.739 | 0.061 | 0.194 |
| 6 | G | G | A | T | 0.9574 | 0.022 | 0.114 | 0.928 | |
| 7 | T | T | T | C | 1.0644 | 0.008 | <0.001 | <0.001 | <0.001 |
| 8 | T | T | A | T | 0.9371 | 0.012 | 0.002 | 0.184 |
aGH1 expression levels relative to 1. P values exhibiting statistically significant difference (< 0.05) are shown in bold
Although this GH1 gene expression analysis only represents the situation pertaining at the end of the experiment (rather than during the experiment), it is clear that the different combinations of the -278 and -57 SNPs do indeed influence GH1 gene expression, but only in combination with the +1169T/+2103C allele, not the +1169A/+2103T allele (Table 2). The -57T allele may therefore be of functional significance, independently of the -278G/T SNP, but in a way that is critically dependent on the identity of the +1169/+2103 allele combination in cis (Table 2; rows 1 vs. 2 [p = 0.033] and 7 vs. 8 [p < 0.001]). No significant difference was observed when comparing the -57G allele and different +1169/+2103 allele combinations (Table 2; rows 3 vs. 4 [p = 0.611] and 5 vs. 6 [p < 0.194]). Luciferase reporter gene assays employing GH1 gene promoter constructs containing identical combinations of -278/-57 SNPs as used in this study and under similar conditions, failed to demonstrate any significant difference in luciferase gene expression between the four -278/-57 haplotypes [19]. Although the assay of Giordano et al [19]. employed a human breast cancer cell line (MCF7), their results are nevertheless consistent with the observed differences in GH1 gene expression being due to the different +1169/+2103 allele combinations.
GH secretion was then measured in the cell culture supernatant from transfected cells in serum-free medium over a 24-hour period. It has been shown that GH is stable in cell culture supernatants from GH3 cells over 72 hours [20]. On this basis, it is likely that the GH measured in our experiments represents the accumulated total GH secreted over the entire 24-hour period, unlike the GH1 gene expression analyses, which reflect the steady-state levels of expression measured at the end of the experiment. Normalised secretion levels are shown in Table 3. Comparison of the different combinations of promoter SNP alleles (-278T/G and -57G/T) with the +1169T/+2103C haplotype indicated that there were significant statistical differences (p < 0.001) in all cases. GH secretion levels were found to be increased when compared with the -278G/-57T standard, by 13 per cent, 12 per cent and 9 per cent for the -278T/-57G, -278G/-57G and -278T/-57T promoter allele combinations, respectively. A similar picture was seen with the different allele combinations of the promoter SNPs with the +1169A/+2103T haplotype; the levels of secreted GH were increased by 14 per cent, 18 per cent and 28 per cent for -278T/-57G, -278G/-57G and -278T/-57T, respectively, compared with -278G/-57T. Comparison of the +1169T/+2103C and +1169A/+2103T haplotypes bearing the same -278/-57 promoter genotype indicated that in all cases GH secretion was significantly reduced in the +1169A/+2103T haplotype. The reductions in GH secretion for the +1169A/+2103T haplotype compared with +1169T/+2103C were 20 per cent (-278G/-57T), 19 per cent (-278T/-57G), 15 per cent (-278G/-57G) and 6 per cent (-278T/-57T), respectively; however, no significant difference was observed between haplotypes -278G/-57T/+1169T/+2103C and -278T/-57T/+1169A/+2103T (p = 0.192). These results reinforce the conclusions drawn from the GH1 gene expression experiments, in that different combinations of the four SNPs differentially affect the amount of GH secreted from the somatotroph cells. Unlike in the expression analysis, however, the -57T SNP had no independent effect on GH secretion. Although it is clear that the two SNPs in the GH1 promoter region exert an effect on both GH1 gene expression and GH secretion, it is also evident that specific +1169/+2103 allele combinations play a significant role in determining the level of GH secretion. This is in agreement with the data of Hasegawa et al.,[8] who reported a significant difference in peak GH levels following standard GH provocation tests between +1169T (18.2 ± 9.8 ng/ml) and +1169A (10.3 ± 5.8 ng/ml) in a cohort of prepubertal short children with either mild or no GH insufficiency. Taken together, these data argue that either the +1169 or the +2103 SNP plays a substantial role in determining the level of secreted GH.
Table 3.
Comparison of GH secretion levels between four different SNPs located in different parts of the GH1 gene
| -278 | -57 | +1169 | +2103 | Secretiona | SEM |
p value versus 1 |
p value versus 2 |
p value (TC versus AT) |
|
|---|---|---|---|---|---|---|---|---|---|
| 1 | G | T | T | C | 1.0000 | 0.015 | - | <0.001 | <0.001 |
| 2 | G | T | A | T | 0.8023 | 0.010 | <0.001 | - | |
| 3 | T | G | T | C | 1.1341 | 0.014 | <0.001 | <0.001 | <0.001 |
| 4 | T | G | A | T | 0.9155 | 0.013 | <0.001 | <0.001 | |
| 5 | G | G | T | C | 1.1181 | 0.010 | <0.001 | <0.001 | <0.001 |
| 6 | G | G | A | T | 0.9466 | 0.013 | 0.008 | <0.001 | |
| 7 | T | T | T | C | 1.0887 | 0.013 | <0.001 | <0.001 | 0.001 |
| 8 | T | T | A | T | 1.0267 | 0.014 | 0.192 | <0.001 |
aGH secretion levels relative to 1. P values exhibiting statistically significant difference (< 0.05) are shown in bold
To determine if the +2103C/T SNP plays a role in either GH1 expression or GH secretion, we compared different allele combinations of the +1169/+2103 SNPs in association with the -278G/-57T promoter genotype (Tables 4 and 5). The +1169A/+2103C haplotype does not occur naturally but was created by site-directed mutagenesis by converting the haplotype -278G/-57T/+1169A/+2103T to -278G/-57T/+1169A/+2103C. As was to be expected from our previous results, GH1 gene expression was found to be reduced by 5 per cent (p = 0.002) for the +1169A/+2103T and 7 per cent (p < 0.001) for the +1169A/+2103C haplotypes when compared with +1169T/+2103C (Table 4). No significant difference in gene expression was found between the +1169A/+2103T and +1169A/+2103C haplotypes (p = 0.309), suggesting that the +2103 SNP has no influence on GH1 gene expression. Hence, we may conclude that the +1169 SNP is important in its own right in determining GH1 gene expression.
Table 4.
Comparison of GH1 expression levels between the C and T alleles of the +2103 SNP in the GH1 gene 3' flanking region
| -278 | -57 | +1169 | +2103 | Expressiona | SEM |
p value versus 1 |
p value (AT versus AC) |
|
|---|---|---|---|---|---|---|---|---|
| 1 | G | T | T | C | 1.0000 | 0.0113 | - | |
| 2 | G | T | A | T | 0.9494 | 0.0060 | 0.002 | 0.309 |
| 3 | G | T | A | C | 0.9310 | 0.0166 | <0.001 |
aGH1 expression levels relative to 1. P values exhibiting statistically significant differences (< 0.05) are shown in bold
Table 5.
Comparison of GH secretion levels between the C and T alleles of the +2103 SNP in the GH1 gene 3' flanking region
| -278 | -57 | +1169 | +2103 | Secretiona | SEM |
p value versus 1 |
p value (AT versus AC) |
|
|---|---|---|---|---|---|---|---|---|
| 1 | G | T | T | C | 1.0000 | 0.0180 | - | |
| 2 | G | T | A | T | 0.7825 | 0.0119 | <0.001 | 0.479 |
| 3 | G | T | A | C | 0.7994 | 0.0203 | <0.001 |
aGH secretion levels relative to 1. P values exhibiting statistically significant differences (< 0.05) are shown in bold
Similar results were obtained when GH secretion was analysed (Table 5); no significant difference was noted between the +1169A/+2103T and +1169A/+2103C haplotypes (p = 0.479), whereas there was a 22 per cent (p < 0.001) reduction in GH secretion for the +1169A/+2103T haplotype and a 20 per cent (p < 0.001) reduction for the +1169A/+ 2103C haplotype when compared with the standard +1169T/+2103C haplotype. These results indicate that the +1169T/A SNP within intron 4 of the GH1 gene is likely to be directly responsible for the observed reduction in both GH1 gene expression and GH secretion.
It is, however, still unclear precisely how the +1169 SNP exerts its effect on GH1 gene expression and GH secretion. The GH1 gene expression assay utilised total cellular RNA, which was then reverse transcribed with an oligo dT primer. This assay therefore measures the level of the fully processed (transcribed, spliced and polyadenylated) mRNA species, and hence it is not possible at the current time to say at which stage(s) in the gene expression pathway the +1169 polymorphism exerts its effect. We did, though, determine the extent to which the level of GH secretion was predictable from the GH1 gene expression level by comparing mean GH secretion values with mean GH1 gene expression values using a Pearson correlation. A positive correlation was noted between GH secretion and GH1 gene expression, although this trend did not reach statistical significance (Pearson correlation = 0.485; p = 0.13). The absence of a correlation may be due to GH secretion reflecting the total amount of GH produced over a 24-hour period, whereas GH1 gene expression only measures the amount of mRNA transcribed at one point in time and hence will not take into account any changes (if any) in gene expression during the 24-hour time course of the experiment. The positive correlation observed does, however, suggest that the +1169 SNP has little or no effect on GH translation.
The +1169T allele may contribute to an alternative splice site
Using the Alternative Splice Site Predictor software described by Wang and Marín,[14] we located a potential alternative splice site spanning the location of the +1169 SNP. This alternative (donor) splice site predicts an exon-intron junction just 2 bp upstream of the polymorphic +1169 nucleotide [CTTTTTAGCAgtcaggccct -- nucleotide +1169 shown in bold] with a probability of 0.907 (cf. probability of it being a constitutive splice site = 0.068). Hence, the +1169T allele is predicted to contribute to the obligate gt dinucleotide at this cryptic splice site, while, unsurprisingly, the program predicts that this site would not be recognised as an alternative splice site in the +1169A allele.
To determine whether this potential alternative splice site was used, we designed two gel-based PCR assays. One assay amplified the entire GH1 cDNA with PCR primers located in the 5' UTR/exon 1 and exon 5; the second assay amplified a smaller GH1 cDNA using primers located in exons 4 and 5. Total RNA was isolated from rat GC cells transfected with the GH1 gene construct bearing either the +1169T or +1169A allele and were reverse transcribed using oligo dT and then subjected to PCR amplication. No differences between the PCR products were observed with either the +1169T or the +1169A alleles. This suggests that either: (i) no alternative splicing occurs in the specific cell type employed in this assay; (ii) alternative splicing occurs in only a small proportion of transcripts which are not visible in a gel-based assay; or (iii) any alternatively spliced product is unstable and hence is rapidly degraded.
Discussion
GH, encoded by the GH1 gene, plays an important role in human development, growth and metabolism [1]. The biological effects of GH are mediated through binding and dimerisation of a cell surface-expressed GHR, initiating a signalling cascade which results in an increased plasma level of IGF-1. We and others have previously documented an unparalleled level of GH1 gene variation in people within the range of normal stature [4-6,8,11,19,21]. Such variation may be very important in a number of different clinical contexts. Thus, an increased risk of colorectal cancer may be mediated by GH and IGF-1 levels [9,22-24]. Cardiovascular disease, hypertension and large artery stiffening are multi-factorial conditions with a polygenic basis that are all known to be influenced by GH [25-27]. An association may also exist between plasma GH level and survival in critically ill people, since non-surviving children with meningococcal sepsis have been reported to exhibit significantly higher GH levels than those who survived,[28] while critically ill adults treated with GH were found to experience increased mortality [29].
With this clinical backdrop, the potential importance of functional polymorphisms in the GH1 gene is apparent. The +1169A allele of the A/T SNP, located within intron 4 of the human GH1 gene, has been associated with reduced levels of circulating GH and IGF-1, a reduced risk of colorectal cancer and a predisposition to osteoporosis. Whether this intronic SNP is itself a functional polymorphism that exerts a direct effect on GH1 gene expression, however, or whether it is instead in linkage disequilibrium with another potentially functional SNP (e.g. -278G/T, -57G/T, +2103C) has been an open question. The -278 SNP is located within a putative nuclear factor 1 (NF1) binding site (-286 to -274)[30] but has not so far been functionally characterised. The -57 SNP is located within a functional vitamin D response element (-60 to -46);[31,32] the -57T allele has been found to be associated with an increased risk of isolated GH deficiency,[19] while the -57G allele is associated with the complete loss of the vitamin D-induced inhibitory response [19]. These findings suggest that specific alleles of either the -278 and/or the -57 SNPs could have been responsible for the observed effect on GH secretion that was originally attributed to +1169.
We attempted here to untangle the potential influences of the intronic SNP at +1169, and three additional SNPs identified as being in linkage disequilibrium with +1169, upon GH1 gene expression and GH secretion. Evolutionary conservation of the +1169T allele (and its surrounding sequence) provided tentative evidence for its functionality. In order to determine whether +1169, in combination with the SNPs at -278, -57 and +2103C, exerts a direct effect on gene expression and/or GH secretion, however, we performed a series of transfections of various GH1 constructs into rat GC (somatotroph) cells. The results obtained have provided evidence to support the contention that the +1169A allele contributes directly to the observed reduction in both GH1 gene expression and GH secretion. The above notwithstanding, however, part of the influence of the +1169A-bearing allele on GH1 gene expression and GH secretion may still be attributable to alleles of additional SNPs in cis to +1169A and located within either the promoter or the 3' flanking region. It remains to be seen whether the +1169T SNP might exert an effect on the GH1 mRNA splicing phenotype; owing to the presence of exon- and intron-splice enhancers within this gene, confirmation or otherwise of this possibility would require the use of whole gene constructs in in vitro assays.
Intronic SNPs residing at some distance from splice sites may often go undetected unless they induce either alternative or aberrant splicing that is readily distinguishable qualitatively or quantitatively from 'normal' splicing [33,34]. Indeed, introns probably represent substantially larger mutational targets than has hitherto been appreciated, on account of their harbouring a multiplicity of functional elements including intron splice enhancers and silencers, as well as the cis-acting RNA elements that regulate alternative splicing. Some deep intronic polymorphic variants are known to have the potential to confer susceptibility to disease (eg Choi et al [35].) and these may be much more common than hitherto realised. In this context, it is pertinent to note that diseaseassociated intronic SNPs have recently been found to impact upon long-range regulatory mechanisms involving highly conserved non-coding elements [36]. Although the precise mechanisms through which it exerts its effect on GH1 gene expression and GH secretion remain unclear, the +1169 A/T SNP studied here appears to represent a new example of an intron-located functional polymorphism with potential clinical significance.
References
- Kaplan SL. In: Handbook of Physiology, Section 7, The Endocrine System Hormonal Control of Growth. Kostyo JL, HM, Goodman HM, editor. V. Oxford University Press, New York, NY; 1999. 'Hormonal regulation of growth and metabolic effects of growth hormone'; pp. 129–143. [Google Scholar]
- Waters MJ, Hoang HN, Fairlie DP, Pelekanos RA. et al. 'New insights into growth hormone action'. J Mol Endocrinol. 2006;36:1–7. doi: 10.1677/jme.1.01933. [DOI] [PubMed] [Google Scholar]
- Procter AM, Phillips JA, Cooper DN. 'The molecular genetics of growth hormone deficiency'. Hum Genet. 1998;103:255–272. doi: 10.1007/s004390050815. [DOI] [PubMed] [Google Scholar]
- Giordano M, Marchetti C, Chiorboli E, Bona G. et al. 'Evidence for gene conversion in the generation of extensive polymorphism in the promoter of the growth hormone gene'. Hum Genet. 1997;100:249–255. doi: 10.1007/s004390050500. [DOI] [PubMed] [Google Scholar]
- Wagner JK, Eblé A, Cogan JD, Prince MA. et al. 'Allelic variations in the human growth hormone-1 gene promoter of growth hormone-deficient patients and normal controls'. Eur J Endocrinol. 1997;137:474–481. doi: 10.1530/eje.0.1370474. [DOI] [PubMed] [Google Scholar]
- Horan M, Millar DS, Hedderich J, Lewis G. et al. 'Human growth hormone 1 (GH1) gene expression: Complex haplotype-dependent influence of polymorphic variation in the proximal promoter and locus control region'. Hum Mutat. 2003;21:408–423. doi: 10.1002/humu.10167. [DOI] [PubMed] [Google Scholar]
- Wolf A, Millar DS, Caliebe A, Horan M. et al. 'A gene conversion hotspot in the human growth hormone (GH1) gene promoter'. Hum Mutat. 2009;30:239–247. doi: 10.1002/humu.20850. [DOI] [PubMed] [Google Scholar]
- Hasegawa Y, Fujii K, Yamada M, Igarashi Y. et al. 'Identification of novel human GH-1 gene polymorphisms that are associated with growth hormone secretion and height'. J Clin Endocrinol Metab. 2000;85:1290–1295. doi: 10.1210/jc.85.3.1290. [DOI] [PubMed] [Google Scholar]
- Le Marchand L, Donlon T, Seifried A, Kaaks R. et al. 'Association of a common polymorphism in the human GH1 gene with colorectal neoplasia'. J Natl Cancer Inst. 2002;94:454–460. doi: 10.1093/jnci/94.6.454. [DOI] [PubMed] [Google Scholar]
- Dennison EM, Syddall HE, Rodriguez S, Voropanov A. et al. 'Polymorphism in the growth hormone gene, weight in infancy, and adult bone mass'. J Clin Endocrinol Metab. 2004;89:4898–4903. doi: 10.1210/jc.2004-0151. [DOI] [PubMed] [Google Scholar]
- Millar DS, Lewis MD, Horan M, Newsway V. et al. 'Novel mutations of the growth hormone 1 (GH1) gene disclosed by modulations of the clinical selection criteria for individuals with short stature'. Hum Mut. 2003;21:424–440. doi: 10.1002/humu.10168. [DOI] [PubMed] [Google Scholar]
- Tashjian AH, Yasumura Y, Levine L, Sato GH. et al. 'Establishment of clonal strains of rat pituitary tumor cells that secrete growth hormone'. Endocrinology. 1968;82:342–352. doi: 10.1210/endo-82-2-342. [DOI] [PubMed] [Google Scholar]
- Laird PW, Zijderveld A, Linders K, Rudnicki MA. et al. 'Simplified mammalian DNA isolation procedure'. Nucleic Acids Res. 1991;19:4293. doi: 10.1093/nar/19.15.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Marín A. 'Characterization and prediction of alternative splice sites'. Gene. 2006;366:219–227. doi: 10.1016/j.gene.2005.07.015. [DOI] [PubMed] [Google Scholar]
- Matys V, Kel-Margoulis OV, Fricke E, Liebich I, 'TRANSFAC and its module TRANSCompel: Transcriptional gene regulation in eukaryotes'. Nucleic Acids Res. 2006. pp. D108–110. [DOI] [PMC free article] [PubMed]
- Bourdon JC, Deguin-Chambon V, Lelong JC, Dessen P. et al. 'Further characterisation of the p53 responsive element -- Identification of new candidate genes for trans-activation by p53'. Oncogene. 1997;14:85–94. doi: 10.1038/sj.onc.1200804. [DOI] [PubMed] [Google Scholar]
- Trujillo MA, Sakagashira M, Eberhardt NL. 'The human growth hormone gene contains a silencer embedded within an Alu repeat in the 3'-flanking region'. Mol Endocrinol. 2006;20:2559–2575. doi: 10.1210/me.2006-0147. [DOI] [PubMed] [Google Scholar]
- Cardinaux J-R, Chapel S, Wahli W. 'Complex organization of CTF/NF-I, C/EBP, and HNF3 binding sites within the promoter of the liver-specific vitellogenin gene'. J Biol Chem. 1994;269:32947–32956. [PubMed] [Google Scholar]
- Giordano M, Godi M, Mellone S, Petri A. et al. 'A functional common polymorphism in the vitamin D-responsive element of the GH1 promoter contributes to isolated growth hormone deficiency'. J Clin Endocrinol Metab. 2008;93:1005–1012. doi: 10.1210/jc.2007-1918. [DOI] [PubMed] [Google Scholar]
- Bancroft FC, Levine L, Tashjian AH. 'Control of growth hormone production by a clonal strain of rat pituitary cells'. J Cell Biol. 1969;43:432–441. doi: 10.1083/jcb.43.3.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giordano M, Godi M, Giacopelli F, Lessi M. et al. 'A variation in a Pit-1 site in the growth hormone gene (GH1) promoter induces a differential transcriptional activity'. Mol Cell Endocrinol. 2006;249:51–57. doi: 10.1016/j.mce.2006.01.006. [DOI] [PubMed] [Google Scholar]
- Baris D, Gridley G, Ron E, Weiderpass E. et al. 'Acromegaly and cancer risk: A cohort study in Sweden and Denmark'. Cancer Causes Control. 2002;13:395–400. doi: 10.1023/A:1015713732717. [DOI] [PubMed] [Google Scholar]
- Jenkins PJ, Frajese V, Jones AM, Camacho-Hubner C. et al. 'Insulin-like growth factor I and the development of colorectal neoplasia in acromegaly'. J Clin Endocrinol Metab. 2000;85:3218–3221. doi: 10.1210/jc.85.9.3218. [DOI] [PubMed] [Google Scholar]
- Cohen P, Clemmons DR, Rosenfeld RG. 'Does the GH-IGF axis play a role in cancer pathogenesis?'. Growth Horm IGF Res. 2000;10:297–305. doi: 10.1054/ghir.2000.0171. [DOI] [PubMed] [Google Scholar]
- Smith JC, Evans LM, Wilkinson I, Goodfellow J. et al. 'Effects of GH replacement on endothelial function and large-artery stiffness in GH-deficient adults: A randomized double-blind, placebo-controlled study'. Clin Endocrinol. 2002;56:493–501. doi: 10.1046/j.1365-2265.2002.01514.x. [DOI] [PubMed] [Google Scholar]
- Napoli R, Guardasole V, Angelini V, D'Amico F. et al. 'Acute effects of growth hormone on vascular function in human subjects'. J Clin Endocrinol Metab. 2003;88:2817–2820. doi: 10.1210/jc.2003-030144. [DOI] [PubMed] [Google Scholar]
- Horan M, Newsway V, Lewis MD, Easter TE. et al. 'Genetic variation at the growth hormone (GH1) and growth hormone receptor (GHR) loci as a risk factor for hypertension and stroke'. Hum Genet. 2006;119:527–540. doi: 10.1007/s00439-006-0166-5. [DOI] [PubMed] [Google Scholar]
- De Groof F, Joosten KFM, Janssen JAMJL, De Kleijn ED. et al. 'Acute stress response in children with meningococcal sepsis: Important differences in the growth hormone/insulin-like growth factor I axis between nonsurvivors and survivors'. J Clin Endocrinol Metab. 2002;87:3118–3124. doi: 10.1210/jc.87.7.3118. [DOI] [PubMed] [Google Scholar]
- Takala J, Ruokonen E, Webster NR, Nielsen MS. et al. 'Increased mortality associated with growth hormone treatment in critically ill adults'. N Engl J Med. 1999;341:785–792. doi: 10.1056/NEJM199909093411102. [DOI] [PubMed] [Google Scholar]
- Courtois SJ, Lafontaine DA, Lemaigre FP, Durviaux SM. et al. 'Nuclear factor-1 and activator protein-2 bind in a mutually exclusive way to overlapping promoter sequences and trans-activate the human growth hormone gene'. Nucleic Acids Res. 1990;18:57–64. doi: 10.1093/nar/18.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso M, Segura C, Dieguez C, Perez-Fernandez R. 'High-affinity binding sites to the vitamin D receptor DNA binding domain in the human growth hormone promoter'. Biochem Biophys Res Commun. 1998;247:882–887. doi: 10.1006/bbrc.1998.8884. [DOI] [PubMed] [Google Scholar]
- Seoane S, Alonso M, Segura C, Perez-Fernandez R. 'Localization of a negative vitamin D response sequence in the human growth hormone gene'. Biochem Biophys Res Commun. 2002;292:250–255. doi: 10.1006/bbrc.2002.6641. [DOI] [PubMed] [Google Scholar]
- Hull J, Campino S, Rowlands K, Chan MS. et al. 'Identification of common genetic variation that modulates alternative splicing'. PLoS Genet. 2007;3:e99. doi: 10.1371/journal.pgen.0030099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwan T, Benovoy D, Dias C, Gurd S. et al. 'Genome-wide analysis of transcript isoform variation in humans'. Nat Genet. 2008;40:225–231. doi: 10.1038/ng.2007.57. [DOI] [PubMed] [Google Scholar]
- Choi JW, Park CS, Hwang M, Nam HY. et al. 'A common intronic variant of CXCR3 is functionally associated with gene expression levels and the polymorphic immune cell responses to stimuli'. J Allergy Clin Immunol. 2008;122:1119–1126. doi: 10.1016/j.jaci.2008.09.026. e7. [DOI] [PubMed] [Google Scholar]
- Ragvin A, Moro E, Fredman D, Navratilova P. et al. 'Long-range gene regulation links genomic type 2 diabetes and obesity risk regions to HHEX, SOX4, and IRX3'. Proc Natl Acad Sci USA. 2010;107:775–780. doi: 10.1073/pnas.0911591107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteban C, Audi L, Carrascosa A, Fernandez-Cancio M. et al. 'Human growth hormone (GH1) gene polymorphism map in a normal-statured adult population'. Clin Endocrinol (Oxf) 2007;66:258–268. doi: 10.1111/j.1365-2265.2006.02718.x. [DOI] [PMC free article] [PubMed] [Google Scholar]