

Abstract
Comparison of the expression profiles of 2,721 genes in the cerebellum, cortex and pituitary gland of three American Staffordshire terriers, one beagle and one fox hound revealed regional expression differences in the brain but failed to reveal marked differences among breeds, or even individual dogs. Approximately 85 per cent (42 of 49 orthologue comparisons) of the regional differences in the dog are similar to those that differentiate the analogous human brain regions. A smaller percentage of human differences were replicated in the dog, particularly in the cortex, which may generally be evolving more rapidly than other brain regions in mammals. This study lays the foundation for detailed analysis of the population structure of transcriptional variation as it relates to cognitive and neurological phenotypes in the domestic dog.
Keywords: canine, breed, microarray, cerebellum, cortex, pituitary
Introduction
Gene expression profiling provides a novel perspective from which to consider the degree of genetic differentiation of individuals within populations. The domestic dog, Canis familiaris, is an excellent organism for this pursuit, since phenotypic and nucleotide divergence are not highly correlated. Whereas breeds are clearly, and often discretely, differentiated morphologically and behaviourally, resolution of genetic relatedness among breeds requires a large number of anonymous microsatellite markers [1-4].
The question thus arises as to whether divergence at the gene expression level is greater within or among breeds. There is a clear expectation that some fraction of the transcriptome in particular tissues and at appropriate phases of development will correlate with phenotypic variation. Similarly, disease status ought to reflect transcriptional changes,[5,6] but any such inference must be assessed against a background knowledge of the degree of standing transcriptional variation [7].
Quantitative comparison of transcriptomes requires statistically orientated analytical methods that can partition the effects of multiple sources of variance. We and others have introduced linear analysis of variance algorithms for microarray data,[8,9] and Bayesian procedures have been employed that perform similarly [10,11]. With appropriate experimental design and moderate replication, it is straightforward to demonstrate that changes in expression smaller than twofold are significant experiment-wide. Furthermore, these approaches take into account variance contributions fromeach factor when assessing specific effects and powerfully demonstrate interaction effects. For example, in a study of the influence of sex, age and genotype on gene expression in Drosophila, we showed that a substantial fraction of the transcriptome differs more between genotypes for just one of the sexes, while age has only a very modest effect on transcription [12].
The objective of this study was to begin to assess the extent to which gene expression differs within and among breeds of dog in three parts of the brain. For many species, it has been shown that between 5 and 20 per cent of genes are differentially expressed between individuals, and the mammalian brain is no different [13,14]. The left prefrontal lobe (Brodmann area 9) of three human brains differs from the homologous region of the chimpanzee brain at over 1,000 loci, although, remarkably, one human was found to differ from the others by at least as much as all three differ from three chimpanzees [15,16]. Follow-up comparison of several regions of the human brains suggested that there is more variation among individuals than between parts of the cortex, although it is not clear whether this is due to genetic or environmental factors [17]. Similarly, an earlier study comparing normal and postseizure mouse brains highlighted strain differences among brain regions [18,19]. Recently, human Affymetrix chips were used to detect some divergence in transcript abundance between pools of brain tissue from several domestic dogs and two wild canid species [20]. Here, we conducted a complementary experiment, employing a canine brain cDNA microarray to contrast region-specific expression in five individual domestic dog brains. We discuss the nature of the genes that differentiate the cortex, cerebellum and pituitary gland of C. familiaris, and argue that differences among individuals are likely to be more prevalent than are breed-specific differences.
Methods
Microrarrays
A canine brain expressed sequence tag (EST) library consisting of approximately 4,600 unique ESTs, most of which have partial sequence and preliminary annotation, was obtained from Dr James Mickelson at the University of Minnesota [21]. Construction of our 4,224 spot cDNA microarray is described elsewhere (Thomson et al., paper submitted). Tentative annotation of many of the ESTs using BLAST matches to end sequence and GenBank accession numbers is provided online as supplementary Table 1 at http://statgen.ncsu.edu/ggibson/SupplInfo/SupplInfo9.htm, along with the raw fluorescence intensities and MIAME-compliant description of the experiment. Note that a comprehensive Affymetrix short oligonucleotide canine microarray has also just been described,[22,23] as have two small targeted cardiovascular arrays [24,25].
Table 1.
List of differentially expressed genes by brain region.
| Low in pituitary | High in pituitary | High in cerebellum | High in cortex | Low in cortex |
|---|---|---|---|---|
| Synuclein SNCA* | Ribosomal protein L6 | Na+-dependent glutamate transporter |
Doublecortin CaM kinase-like 1 |
Fat tumour suppressor homologue 2 |
| Synaptosomal-associated SNAP25 | Ribosomal protein L10* | Reticulon 4 | ||
| Neuronal glycoprotein (×3) | Ribosomal protein L11 | Fibronectin type III repeat |
||
| Ubiquinol-cytochrome c reductase core protein I |
Ribosomal protein L12 | Adenylyl cyclase mRNA |
||
| Dipeptidyl peptidase 7 (Dpp7) | Ribosomal protein L19 | Neuronal pentraxin I |
Chimerin 2 | |
| Myelin basic protein | Ribosomal protein L21 | P311 protein | ||
| N-ethylmaleimide-sensitive factor |
Ribosomal protein S11 | Sodium bicarbonate cotransporter 4* |
||
| Complexin 2* | Ribosomal protein S14 | |||
| Huntingtin-associated interacting |
Ribosomal protein S25 Iodothryonine, type II |
|||
| Glial fibrillary acidic protein (×2) |
Crystallin, mu (CRYM)* | |||
| Myelin basic protein Kinesin 2* |
Glucan (1, 4-alpha-) branching enzyme 1 |
|||
| Aldolase C, fructose bisphosphate | Thyrotropin-releasing hormone degrading ectoenzyme |
|||
| S100 calcium-binding protein, beta |
DEAD/H box polypeptide 5 |
|||
| Calcium/calmodulindependent protein kinase II |
Deiodinase type II | |||
| Dopamine-regulated neuronal phosphoprotein |
||||
| Glutamine sythetase tubulin (alpha and beta) |
||||
| GABA B receptor | ||||
| NDRG 4 | ||||
| Peanut (PNUTL2 septin) | ||||
| Fas apoptotic inhibitory molecule 2 | ||||
| Protein phosphatase 3, beta* | ||||
| Creatine kinase B subunit (×2) | ||||
| Sodium-potassium ATPase, alpha* |
*Genes that do not show the same expression bias in the Novartis Human Gene Expression Atlas (see Supplementary Information).
Dissected brains from five adult dogs that were presented to the North Carolina State University Veterinary Teaching Hospital were used as the source of mRNA from pituitary gland, cortex and cerebellum. All dogs were euthanised and subjected to necropsy at the request of the owners for medical reasons. The dogs included three American Staffordshire terriers, one beagle and one American foxhound. The brains were removed in a sterile fashion within 30 minutes of death, the meninges were dissected away and tissues were taken from the frontal cortex, lateral cerebellar hemispheres and pituitary gland. These were snap frozen in liquid nitrogen and stored at -80°C. RNA isolation was performed after addition of 1 ml per 50-100 mg tissue of TRIzol reagent (Invitrogen) following the manufacturer's instructions, with further purification using an Rneasy Mini Kit (Qiagen). The quality and purity of RNA was analysed on a 0.8 per cent agarose gel and by taking 260 nm/280 nm absorbance readings on a spectrophotometer.
The experimental design shown in Figure 1 consists of one main loop, where each tissue type was compared across different breeds, and five smaller loops in which each tissue within a single dog was compared with other tissues in that same dog. Linear RNA amplification [26] was performed using an Agilent Low RNA Input Fluorescent Linear Amplification Kit (Product number 5184-3523). A single round of amplification was performed using 500 ng of total RNA, yielding up to 50 μg of amplified complementary RNA (acRNA). Occasionally, a second amplification reaction was needed to obtain enough acRNA. When this occurred, the two reactions were pooled. First-strand cDNA was synthesised from 3 μg of acRNA using Improm II reverse transcriptase (Promega) in four separate reactions for each sample. After purification of first-strand cDNA, the four reactions were pooled and re-split to reduce variations between individual cDNA synthesis reactions. Amplified cDNA was then labelled indirectly through an aminoallyl linkage with Cy3 or Cy5 in a balanced manner (using The Institute for Genomic Research protocol SOP#M004), resulting in two Cy3 and two Cy5 reactions per sample. Hybridisations were performed for 20 hours at 42°C, followed by washing in a standard series of highstringency washes. Microarray slides were scanned using a ScanArray 4000 Microarray Analysis System Scanner (Packard Bioscience). ScanAlyze 2 http://rana.lbl.gov/EisenSoftware.htm[27] was used to generate data files from the acquired images.
Figure 1.
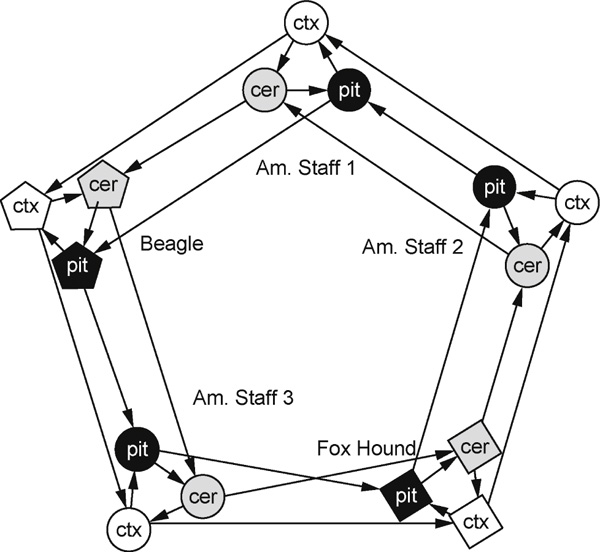
Experimental design, consisting of five loops contrasting each of the three brain tissues [cortex (ctx), cerebellum (cer) and pituitary (pit)] from a single dog, nested within three loops contrasting the same tissue across each of the five dogs. Arrowheads point to the Cy5 sample on each array, and arrow bases lead from the Cy3 sample. Each tissue in each dog is represented by four hybridisations with a balance of dye flips.
Data analysis
Raw fluorescence intensities from Scanalyze 2 were further analysed using a two-step mixed model analysis of variance procedure [8,28] in SAS Version 5.0 (SAS Institute, Cary, NC). Raw fluorescence intensities were log transformed on the base 2 scale, and the 1,503 spots with the lowest average expression across all arrays were removed from consideration. This number was selected because they lay below the inflection point of a plot of rank-ordered average raw fluorescence intensity for all of the spots on the array. Spots at or below this point (raw values 186; log2 value 7.54) are no more intense than the mean background intensity level across all array. All of the remaining 2,721 spots were then normalised with a first analysis of variance model that adjusts for overall array and dye effects. Residuals from this model are relative fluorescence intensities (log2RFI) for each gene, essentially a measure of the fold difference in expression level for each gene relative to the sample mean for the appropriate channel on each array.
These intensities were then compared on a gene by gene basis, accounting for the variance among dogs and tissues according to gene-specific mixed models of the form:
where fixed effects are represented by C for the ith individual canine (i = 1, ..., 5), T for the jth tissue (cerebellum, cortex, or pituitary) and D for the kth dye (Cy3 or Cy5). The term C × Tij fits the interaction between dog and tissue, while the random effects of the lth array (l = 1, ..., 30) are presumed to be normally distributed with mean zero and variance σ2. The mean and unexplained error are represented by μ and ε, respectively. This procedure obviates the need for a reference sample and assesses the significance of gene expression differences between samples relative to the variance in measurements of each sample type. The online Results Supplementary Table 2 shows the significance of the C, T, C × T and D terms (columns C to F), along with the amount of variance explained by each gene-specific model (column B). Subsequently, the magnitude and significance of the difference in expression of the three American Staffordshire terriers from the single foxhound or beagle individual, and of the three brain regions (cortex, cerebellum and pituitary) were computed using the DIFFS option in PROC MIXED, using SAS code that is available on request. As described in the text, the significance threshold of p < 0.0001 was adopted for gene selection, as none of the 2,721 genes in the analysis are expected to be significant at this level by chance. Clustering in Figure 2 was performed according to Ward's method on the standardised means of the four measurements from each dog, using JMP Version 5.0 software (SAS Institute, Cary, NC, USA). The genes listed in Table 1 meet the more stringent Bonferroni cutoff(p < 0.00002).
Table 2.
Number of differentially expressed genes by brain region.
| Significance level | High in pituitary |
High in cereb |
High in cortex |
Low in pituitary |
Low in cortex |
Total |
|---|---|---|---|---|---|---|
| p < 0.00002 (Bonferroni) | 30 | 13 | 20 | 95 | 3 | 142 |
| p < 0.0001 (FDR) | 73 | 49 | 22 | 135 | 11 | 290 |
| p < 0.05 (testwise) | 665 | 90 | 301 | 274 | 236 | 1,566 |
Abbreviations: Cereb, cerebellum; FDR, false discovery rate.
Figure 2.
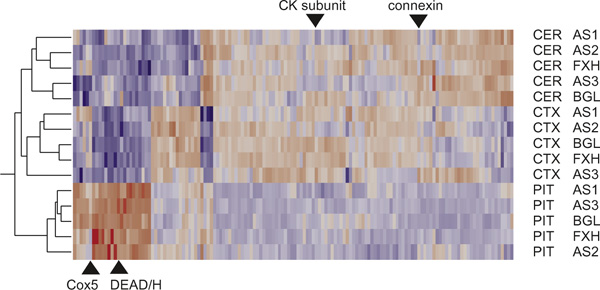
Heat map showing two-way hierarchical clustering of standardised least-square means of transcript abundance over the four hybridisations. Each row represents the indicated brain region from one dog. Each column represents one gene for which significant expression differences were observed, either among brain regions or breeds, at p < 0.0001. Red indicates relatively high expression, blue low expression. Triangles highlight genes mentioned in the Discussion showing bimodal abundance within the pituitary or cerebellum.
Comparison with the Novartis Human Gene Expression Atlas [29] was performed using the online text query feature at http://expression.gnf.org/cgi-bin/index.cgi. This resource provides the results of duplicate (cortex and cerebellum) or single (pituitary gland) human tissue hybridisations performed against the Affymetrix Human U95A platform. Since pituitary is not represented in similar mouse data, our dog results were only compared with human. Genes listed in Table 1 that were significantly differentially regulated in the dog were individually queried. Since no statistical measures are provided online, genes whose expression was twice as high (or twice as low) in the indicated tissue relative to the other two tissues, in both species, were regarded as being consistently regulated.
For the reciprocal comparison of differentially regulated human genes, we first used the online filter to identify sets of genes in the cortex, cerebellum and pituitary that are below the average, or more than twice the average, of the 46 Novartis tissues. Pairwise comparison of these lists identifies a subset of all genes that are at least twofold differentially regulated between the tissues, which numbers between 175 and 453 genes depending on the comparison. The annotations of these genes and the canine gene accessions were then scanned for exact matches. Owing to the relatively small sample of canine genes and incomplete annotation, only around 5 per cent of the human genes could be matched to canine genes. In these cases, we asked whether the difference in expression on our arrays was significantly different in the same direction (replicated), in the same direction but not significantly so (consistent), not differentially expressed (questionable) or significant in the other direction to that seen in humans (opposite effect). Human pituitary-specific genes are apparently under-represented on our canine array, so only three clones could be compared, all of which were also upregulated in the pituitary of the dog.
Results
Differential expression between brain regions
Of the 4,224 genes represented on our microarray, 2,721 were expressed above background levels in at least one tissue, with 591 genes showing nominal testwise significant differences (p < 0.05) in transcript abundance between the three brain regions. By contrast, at this 5 per cent significance level, just 139 genes differed among the five dogs, and 131 genes differed among the dogs in a region-specific manner, which is precisely the number of genes expected by chance. Consequently, the experiment provided good evidence for the differential expression of up to 15 per cent of the genes among brain regions but no strong evidence for differential expression between dogs.
The numbers of genes differentially expressed at significantly higher levels in one of the three brain tissues than in both of the other two are indicated in Table 2. At the significance cutoff of p < 0.0001, no significant differences are expected by chance, so the false discovery rate (FDR) is minimised. A total of 290 expression differences was observed, however: expression was elevated for 73 genes in the pituitary, 49 in the cerebellum and 22 in the cortex, while 135 genes were noticeably repressed in the pituitary and 11 genes showed their lowest expression in the cortex. Since no genes were lower in the cerebellum than in the cortex and pituitary there are thus five clusters of differentially expressed genes that appear in the two-way hierarchical cluster heat map in Figure 2. Table 2 further indicates that differential expression trends were also seen at more stringent (Bonferroni) or less stringent (testwise) significance cutoffs, confirming that gene expression was most divergent in the pituitary. The identities of the annotated genes in each class are listed in Table 1 and are discussed below.
Relative absence of differentiation among dogs
Two further tests for differentiation between animals and breeds failed to provide any formal evidence for global differentiation among dogs. Figure 3 presents volcano plots of significance against magnitude of expression difference for each of the three pairwise contrasts of American Staffordshire terrier (three dogs) against foxhound and beagle (one dog each). Significance on the Y-axis is plotted as the negative logarithm of the p-value, such that values exceeding the p = 0.0001 threshold are above the dotted horizontal line. Fold change in transcript abundance is shown on the log base 2 scale along the X-axis. Only a handful of genes appeared to be differentially expressed across the three brain regions between each pair of breeds.
Figure 3.
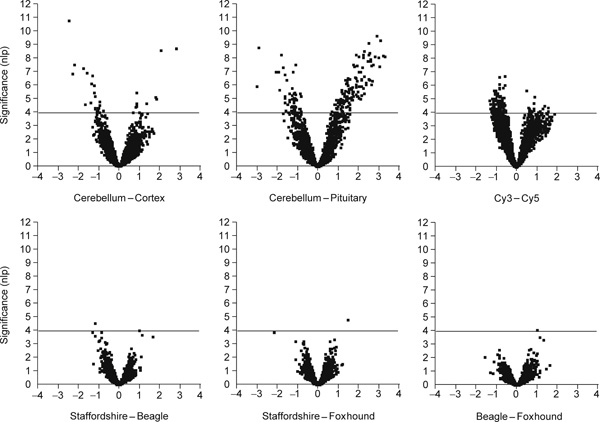
Volcano plots of significance against fold change in transcript abundance for the indicated contrasts. For example, each point in the top left panel shows the difference in log2 mean expression level for cerebellum minus cortex, so that genes more highly expressed in the cortex are to the left. Plus or minus 1 correspond to twofold differences. Higher significance is towards the top on a scale of the negative logarithm of the p-value, with the p < 0.0001 cut-off indicated by a light horizontal line on each panel. Note that a large number of genes is differentially expressed in each brain region, but that relatively few genes differentiate the breeds.
Similarly, the clustering of dogs in Figure 2 tends to indicate that any differential gene expression between dogs within brain regions also is not breed specific. Transcript abundance is remarkably uniform in the five pituitaries, while between 20 and 30 transcripts differentiate each dog from each other dog in the cerebellum. In the cortex, one of the American Staffordshire terriers is quite different from the other four dogs and there is a suggestion that the beagle and foxhound are more similar to one another than to the American Staffordshire terriers. Even though the same clustering pattern is observed when different numbers of genes are included in the analysis, it is due to just a handful of genes. Greater sampling depth and/or replication would undoubtedly elevate several percent of the genes represented in the transcriptome to the status of formally significant differential expression between individual dogs, but very few of these differences are likely to be breed specific.
Discussion
Transcriptional divergence between the pituitary and the cortex and the cerebellum generally reflects the hormonal and neuronal roles of these regions of the brain. Notable among the genes with relatively low expression in the pituitary are synaptic proteins, neuronal glycoproteins and several that encode proteins and enzymes related to neurotransmitter activity. By contrast, genes upregulated in the pituitary include a thyrotropin-releasing hormone degrading enzyme, iodothyronine and multiple ribosomal proteins, consistent with the notion that the pituitary is a site of enhanced protein synthesis. Differentiation of the cortex and cerebellum is less pronounced, but includes genes with clear neuronal functions such as a glutamate transporter, fibronectin repeat protein and pentraxin, which are upregulated in the cerebellum, and an adenylyl cyclase and calmodulindependent protein kinase which are upregulated in the cortex.
Comparison of human and canine region-specific gene expression in the brain
Comparison with online data from the Novartis Gene Expression Atlas [29] indicates that around 85 per cent (42 out of 49) of our annotated dog genes that are orthologous to unique human genes show similar differences among brain regions. This indicates that much of the functional differentiation between the cerebellum, cortex and pituitary at the gene expression level has been retained over tens of millions of years, irrespective of differences in brain size. Among the genes highlighted with asterisks in Table 1 that do not show consistent profiles across the two species, most are members of gene families, suggesting either that precise annotation of the short dog EST sequences is misleading or that subfunctionalisation among paralogous genes occurs at a reasonable frequency [30].
A reciprocal analysis, namely ascertainment of which differentially expressed genes from the Novartis Human Data Index are also differentially expressed in dogs, suggests that the cortex may be more different than the cerebellum between these species. As summarised in Table 3, over two-thirds of the 21 genes upregulated in the cerebellum relative to the pituitary that have orthologues on both sets of microarrays are also upregulated in the dog, while just one is significantly higher in the canine pituitary. By contrast, just half of the 24 orthologous genes upregulated in human cortex relative to cerebellum are also upregulated in dogs, and three genes are significantly differentially transcribed in the opposite direction. Furthermore, five of nine genes upregulated in the human cortex relative to the cerebellum are not upregulated in the dog, whereas six of seven genes more highly expressed in the human cerebellum than the cortex show the same pattern in the dog. Contrasting humans with chimpanzees similarly suggests more extensive divergence of expression in the cortex than in three other brain regions [17]. More intensive profiling, combined with molecular evolutionary analysis of sequence divergence, is a promising strategy for discovery of genes that may contribute to cognitive evolution and neuropathology [31].
Table 3.
Comparison of differential expression in human and doga
| Comparison | Totalb | Replicated | Consistent | Questionable | Opposite |
|---|---|---|---|---|---|
| Cerebellum > pituitary | 403 | 8 | 7 | 5 | 1 |
| Cortex > pituitary | 453 | 8 | 4 | 9 | 3 |
| Cerebellum > cortex | 175 | 3 | 3 | 1 | 0 |
| Cortex > cerebellum | 286 | 0 | 4 | 4 | 1 |
a Genes differentially expressed in humans showing the same direction of effect in dogs that is significant at p < 0.05 (replicated), non-significant (consistent), no change or non-significant in the opposite direction (questionable), or significant in the opposite direction.
b The total number of Affymetrix probe-sets reported as being more than two-fold higher in the first tissue than the mean of all other tissues, and less than the mean of all others in the second tissue, from the Novartis website [29]. Only around 5 per cent of these probe-sets have unambiguous orthologues in this canine cDNA array. Too few pituitary-specific human genes were detected on the canine array to report a contrast.
Expression variation in dogs and wild canids
As noted, no formally significant differences in gene expression between the dogs or breeds were detected. This is a little surprising, given that similar-sized studies in flies,[12,32,33] fish,[34] mice [19] and humans [13,15] have all found evidence for differences of approximately 10 per cent of the transcriptome between individuals. A recent comparison of pools of mRNA from three Labrador retrievers and seven German shepherds with pools from ten coyotes or five grey wolves [20] detected differential expression involving at least 114 genes between all three species in the amygdala and frontal lobe or between dogs and wild canids in the hypothalamus. Four of these genes were retested by quantitative reverse transcriptase polymerase chain reaction in samples from individual animals, and while two- to fourfold differentiation between species was confirmed, no differences between individual dogs were detected. Power computations indicate that detection of differential expression at levels less than 1.5-fold would, given the technical variance in our cDNA arrays, generally require more than the four replicates reported here. The trend detected in this study is that transcript abundance tends to be uniform among dogs and, as far as the very limited sample is concerned, across breeds of dogs. Nevertheless, it is likely that a broader survey encompassing different stages of brain development, or a larger sample of dogs with more replication, would detect genes whose expression varies among individuals either for genetic or environmental reasons.
It is well known from human genetics that behaviourally-related loci, such as monoamine oxidase and the serotonin transporter, are expressed at different levels among individuals [35,36]. These genes are not represented on our array, so it is not yet clear whether expression is polymorphic in dogs as well. Close inspection of Figure 2 reveals several dozen genes whose expression is greater in two or three of the dogs than in the others and post hoc tests suggest these as candidate genes for differential regulation across individuals. Examples indicated on Figure 2 include cytochrome c oxidase subunit COX5B (our clone identity number DG1314) and a DEAD/H box protein (DG0610) in the pituitary, and a creatine kinase subunit (DG3263) and complexin (DG3512) in the cerebellum. Most of these cases show sharing of the two transcriptional states across breeds, implying that any efforts to correlate gene expression with behavioural divergence in dogs should be conducted across a broad range of breeds to avoid the effect of population stratification on inference of genetic association.
Acknowledgements
We thank Jim Mickelson for providing the sequenced cDNA clones, the owners of the dogs that provided tissues for this study and Kevin Woollard for assistance with the craniotomies. This research was supported by NIH award R01 GM61600 to GG, and by awards from the American Kennel Club Canine Health Foundation as well as the NCSU College of Veterinary Medicine to MB.
Electronic database information
The complete list of genes on our microarray, raw fluorescence intensities, as well as the results of the Mixed Model Analysis of Variance are available online at: http://statgen.ncsu.edu/ggibson/SupplInfo/SupplInfo9.htm.
References
- Parker HG, Kim LV, Sutter NB. et al. Genetic structure of the purebred domestic dog. Science. 2004;304:1160–1164. doi: 10.1126/science.1097406. [DOI] [PubMed] [Google Scholar]
- Vilà C, Savolainen P, Maldonado JE. et al. Multiple and ancient origins of the domestic dog. Science. 1997;276:1687–1689. doi: 10.1126/science.276.5319.1687. [DOI] [PubMed] [Google Scholar]
- Savolainen P, Zhang YP, Luo J. et al. Genetic evidence for an East Asian origin of domestic dogs. Science. 2002;298:1610–1613. doi: 10.1126/science.1073906. [DOI] [PubMed] [Google Scholar]
- Irion D, Schaffer AL, Famula TR. et al. Analysis of genetic variation in 28 dog breed populations with 100 microsatellite markers. J Hered. 2003;94:81–87. doi: 10.1093/jhered/esg004. [DOI] [PubMed] [Google Scholar]
- Bunney WE, Bunney BG, Vawter MP. et al. Microarray technology: A review of new strategies to discover candidate vulnerability genes in psychiatric disorders. Am J Psychiatry. 2003;160:657–666. doi: 10.1176/appi.ajp.160.4.657. [DOI] [PubMed] [Google Scholar]
- Nevins JR, Huang ES, Dressman H. et al. Towards integrated clinico-genomic models for personalized medicine: Combining gene expression signatures and clinical factors in breast cancer outcomes prediction. Hum Mol Genet. 2003;12:R153–R157. doi: 10.1093/hmg/ddg287. [DOI] [PubMed] [Google Scholar]
- Gibson G. Population genomics: Celebrating individual expression. Heredity. 2003;90:1–2. doi: 10.1038/sj.hdy.6800195. [DOI] [PubMed] [Google Scholar]
- Wolfinger RD, Gibson G, Wolfinger ED. et al. Assessing gene significance from cDNA microarray expression data via mixed models. J Comput Biol. 2001;8:625–637. doi: 10.1089/106652701753307520. [DOI] [PubMed] [Google Scholar]
- Kerr MK, Martin M, Churchill G. Analysis of variance for gene expression microarray data. J Comput Biol. 2000;7:819–837. doi: 10.1089/10665270050514954. [DOI] [PubMed] [Google Scholar]
- Efron B, Tibshirani R. Empirical Bayes methods and false discovery rates for microarrays. Genet Epidemiol. 2002;23:70–86. doi: 10.1002/gepi.1124. [DOI] [PubMed] [Google Scholar]
- Townsend JP, Hartl DL. Bayesian analysis of gene expression levels: Statistical quantification of relative mRNA level across multiple strains or treatments. Genome Biol. 2002;3:Research0071. doi: 10.1186/gb-2002-3-12-research0071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin W, Riley R, Wolfinger RD. et al. The contributions of sex, genotype and age to transcriptional variance in Drosophila melanogaster. Nat Genet. 2001;29:389–395. doi: 10.1038/ng766. [DOI] [PubMed] [Google Scholar]
- Cheung VG, Spielman RS. The genetics of variation in gene expression. Nat Genet. 2002;32(Suppl.):522–525. doi: 10.1038/ng1036. [DOI] [PubMed] [Google Scholar]
- Vawter MP, Evans S, Choudary P. et al. Gender-specific gene expression in post-mortem human brain: Localization to sex chromosomes. Neuropsychopharmacology. 2003;29:373–384. doi: 10.1038/sj.npp.1300337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard W, Khaitorich P, Klose J. et al. Intra- and interspecific variation in primate gene expression patterns. Science. 2002;296:340–343. doi: 10.1126/science.1068996. [DOI] [PubMed] [Google Scholar]
- Hsieh WP, Chu M, Wolfinger RD, Gibson G. Mixed model reanalysis of primate data reveals species and tissue biases in gene expression. Genetics. 2003;165:747–757. doi: 10.1093/genetics/165.2.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaitovich P, Muetzel B, She X. et al. Regional patterns of gene expression in human and chimpanzee brains. Genome Res. 2004;14:1462–1473. doi: 10.1101/gr.2538704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandberg R, Yasuda R, Pankratz DG. et al. Regional and strain-specific gene expression mapping in the adult mouse brain. Proc Natl Acad Sci USA. 2000;97:11038–11043. doi: 10.1073/pnas.97.20.11038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P, Noble WS. Analysis of strain and regional variation in gene expression in mouse brain. Genome Biol. 2001;2:Research0042. doi: 10.1186/gb-2001-2-10-research0042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saetre P, Lindberg J, Leonard JA. et al. From wild wolf to domestic dog: Gene expression changes in the brain. Mol Brain Res. 2004;126:198–206. doi: 10.1016/j.molbrainres.2004.05.003. [DOI] [PubMed] [Google Scholar]
- Roberts MC, Hitte CC, Hendrickson JA. et al. Characterization and radiation hybrid mapping of expressed sequence tags from the canine brain. Mamm Genome. 2003;14:203–213. doi: 10.1007/s00335-002-2207-2. [DOI] [PubMed] [Google Scholar]
- Higgins MA, Berridge BR, Mills BJ. et al. Gene expression analysis of the acute phase response using a canine microarray. Toxicol Sci. 2003;74:470–484. doi: 10.1093/toxsci/kfg142. [DOI] [PubMed] [Google Scholar]
- Zubakov D, Hoheisel JD, Kluxen FW. et al. Late ischemic preconditioning of the myocardium alters the expression of genes involved in inflammatory response. FEBS Lett. 2003;547:51–55. doi: 10.1016/S0014-5793(03)00667-7. [DOI] [PubMed] [Google Scholar]
- Asakura M, Takashima S. et al. Canine DNA array as a potential tool for combining physiology and molecular biology. Circ J. 2003;67:788–792. doi: 10.1253/circj.67.788. [DOI] [PubMed] [Google Scholar]
- Balkovetz DF, Gerrard ER Jr, Li S. et al. Gene expression alterations during HGF-induced dedifferentiation of a renal tubular epithelial cell line (MDCK) using a novel canine DNA microarray. Am J Physiol Renal Physiol. 2004;286:F702–F710. doi: 10.1152/ajprenal.00270.2003. [DOI] [PubMed] [Google Scholar]
- Eberwine J. Amplification of mRNA populations using aRNA generated from immobilized oligo(dT)-T7 primed cDNA. Biotechniques. 1996;20:584–591. doi: 10.2144/19962004584. [DOI] [PubMed] [Google Scholar]
- Eisen MB, Spellman PT, Brown PO, Botstein D. Cluster analysis and display of genome-wide expression patterns. Proc Natl Acad Sci USA. 1998;95:14863–14868. doi: 10.1073/pnas.95.25.14863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson G, Wolfinger RD. In: Genetic Analysis of Complex Traits with SAS. Saxton AM, editor. Users Press/SAS Publishing, Cary, NC; 2004. Gene expression profiling with the SAS Microarray Solution. in press Chapter 11. [Google Scholar]
- Su AI, Cooke MP, Ching KA. et al. Large-scale analysis of the human and mouse transcriptomes. Proc Natl Acad Sci USA. 2002;99:4465–4470. doi: 10.1073/pnas.012025199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan MM, Huffaker SJ, Webster MJ. et al. Application and optimization of microarray technologies for human postmortem brain studies. Biol Psychiatry. 2004;55:329–336. doi: 10.1016/j.biopsych.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Meiklejohn CD, Parsch J, Ranz JM, Hartl DL. Rapid evolution of male-biased gene expression in Drosophila. Proc Natl Acad Sci USA. 2003;100:9894–9899. doi: 10.1073/pnas.1630690100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rifkin SA, Kim Y, White KP. Evolution of gene expression in the Drosophila melanogaster subgroup. Nat Genet. 2003;33:138–144. doi: 10.1038/ng1086. [DOI] [PubMed] [Google Scholar]
- Oleksiak MF, Churchill GA, Crawford DL. Variation in gene expression within and among natural populations. Nat Genet. 2002;32:261–266. doi: 10.1038/ng983. [DOI] [PubMed] [Google Scholar]
- Sabol SZ, Hu S, Hamer D. A functional polymorphism in the monoamine oxidase A gene promoter. Hum Genet. 1998;103:273–279. doi: 10.1007/s004390050816. [DOI] [PubMed] [Google Scholar]
- Lesch KP, Bengel D, Heils A. et al. Association of anxietyrelated traits with a polymorphism in the serotonin transporter gene regulatory region. Science. 1996;274:1527–1530. doi: 10.1126/science.274.5292.1527. [DOI] [PubMed] [Google Scholar]