

Abstract
Background
Species of Cronobacter are widespread in the environment and are occasional food-borne pathogens associated with serious neonatal diseases, including bacteraemia, meningitis, and necrotising enterocolitis. The genus is composed of seven species: C. sakazakii, C. malonaticus, C. turicensis, C. dublinensis, C. muytjensii, C. universalis, and C. condimenti. Clinical cases are associated with three species, C. malonaticus, C. turicensis and, in particular, with C. sakazakii multilocus sequence type 4. Thus, it is plausible that virulence determinants have evolved in certain lineages.
Methodology/Principal Findings
We generated high quality sequence drafts for eleven Cronobacter genomes representing the seven Cronobacter species, including an ST4 strain of C. sakazakii. Comparative analysis of these genomes together with the two publicly available genomes revealed Cronobacter has over 6,000 genes in one or more strains and over 2,000 genes shared by all Cronobacter. Considerable variation in the presence of traits such as type six secretion systems, metal resistance (tellurite, copper and silver), and adhesins were found. C. sakazakii is unique in the Cronobacter genus in encoding genes enabling the utilization of exogenous sialic acid which may have clinical significance. The C. sakazakii ST4 strain 701 contained additional genes as compared to other C. sakazakii but none of them were known specific virulence-related genes.
Conclusions/Significance
Genome comparison revealed that pair-wise DNA sequence identity varies between 89 and 97% in the seven Cronobacter species, and also suggested various degrees of divergence. Sets of universal core genes and accessory genes unique to each strain were identified. These gene sequences can be used for designing genus/species specific detection assays. Genes encoding adhesins, T6SS, and metal resistance genes as well as prophages are found in only subsets of genomes and have contributed considerably to the variation of genomic content. Differences in gene content likely contribute to differences in the clinical and environmental distribution of species and sequence types.
Introduction
The Cronobacter genus (formerly Enterobacter sakazakii) is composed of Gram-negative, motile, non-sporeforming, peritrichous rods within the Enterobacteriaceae family. This family includes the well-known enteric bacterial pathogens E. coli and Salmonella, though the Cronobacter are most closely related to the Enterobacter and Citrobacter genera. They are ubiquitous organisms present in a wide range of environments, including water, soil, and a variety of fresh and processed foods [1]. The bacterium has been isolated from factory production lines, including powdered infant formula factories, and households [2] as well as clinical sources such as cerebrospinal fluid, blood, bone marrow, sputum, urine, and faeces [3]. The organism is an opportunistic pathogen of humans that can cause infections in all age groups [4]. However, low birth weight neonates are most at risk and in this host group it has been associated with outbreaks of necrotizing enterocolitis, meningitis, and septicaemia [3], [5], [6], [7], [8], [9]. Infections with these presentations result in exceptionally high mortality rates ranging from 40 to 80 percent [10]. In recent years, several outbreaks of bacterial infection in neonatal intensive care units (NICU) have been traced to powdered formula contaminated with Cronobacter spp. [11], [12].
Cronobacter was defined as ‘yellow-pigmented Enterobacter cloacae’ until 1980 when it was designated Enterobacter sakazakii by Farmer et al [13]. Later analyses of both partial 16S rDNA and hsp60 sequences showed that E. sakazakii isolates formed at least four distinct clusters, and it was proposed that these clusters could be unique species [14]. This was confirmed using DNA-DNA hybridization and phenotyping, and subsequently Enterobacter sakazakii was re-classified as the new genus Cronobacter [15]. Initially the genus was composed of only four species, but this has been revised and it currently contains the seven species: Cronobacter sakazakii, C. malonaticus, C. turicensis, C. muytjensii, C. dublinensis, C. universalis and C. condimenti [16].
A multilocus sequence typing (MLST) scheme has been established for the entire Cronobacter genus and is available online at http://www.pubMLST.org/cronobacter [16], [17], [18]. The scheme is based on seven housekeeping genes (atpD, fusA, glnS, gltB, gyrB, infB, ppsA) with a concatenated length of 3036 nucleotides that can be used for multilocus sequence analysis (MLSA). Phylogenetic analysis based on this set of sequences estimated that the separate Cronobacter species evolved in the past 40 million years, with C. sakazakii and C. malonaticus emerging as definable species 11–23 million years ago [18].
The Cronobacter MLST scheme has been applied to more than 350 strains that were widely distributed geographically, temporally, and by source, some of which could be traced over a 50-year period. There are currently 121 defined sequence types (ST) covering all Cronobacter species, some of which are stable clones [18], [19]. Certain associations have been noted between sequence type and source. For example, C. sakazakii ST1 strains are primarily isolates from infant formula and clinical sources, whereas C. sakazakii ST8 is primarily composed of isolates from clinical sources. Of special significance is C. sakazakii ST4 which appears to have a high propensity for neonatal meningitis [18], [19]. This appears to be a very stable clone as clinical and non-clinical strains have been isolated from 7 countries for over 50 years. In addition, C. malonaticus ST7 is associated with adult infections though the source has not been identified.
To date, little is known about the mechanisms of pathogenicity in Cronobacter. Candidate virulence determinants include superoxide dismutase (SodA) for macrophage survival [20], haemolysin [21], flagella [22], a metalloprotease [23], an enterotoxin [24], and plasmid-borne virulence factors such as Cronobacter plasminogen activator (Cpa) and type six secretion systems (T6SS) [25]. The bacteria can attach to intestinal cells and survive in macrophages [20], [26]. OmpA and OmpX possibly have a role in the organism penetrating the blood-brain barrier, though the mechanism leading to the destruction of the brain cells is unknown and could, in part, be a host response [27]. However, only a few studies have described the interaction of Cronobacter spp. with human cells, and the specific receptors involved remain to be determined. It is known that the disruption of tight junctions significantly enhances association of C. sakazakii with the human intestinal cell line Caco-2 [28]. Strains from C. sakazakii and C. malonaticus show higher invasion of Caco-2 than other Cronobacter species [20]. Similarly, both C. sakazakii and C. malonaticus survive and replicate in macrophages inside phagosomes, whereas C. muytjensii dies and C. dublinensis is serum sensitive. Virulence also varies within the C. sakazakii species as evident from epidemiological studies of a NICU outbreak in France where the clinical outcome of three C. sakazakii pulsetypes differed, with only one pulsetype (now recognised as ST4 strains) causing the three deaths [8], [19]. To date, only strains from C. sakazakii, C. malonaticus and C. turicensis have been associated with reported neonatal infections, whereas C. malonaticus ST7 strains were associated with adult infections [19]. Therefore pathogenicity in humans may be an acquired trait in this genus.
Kucerova et al. [3], [29] used whole genome sequence analysis of C. sakazakii strain BAA-894 and microarray based comparative genomic hybridisation (CGH) to explore the genetic basis of virulence in Cronobacter. CGH highlighted 15 clusters of genes in C. sakazakii BAA-894 that were divergent or absent in more than half of the tested strains; six of these were of probable prophage origin. Several putative virulence factors (i.e. fimbriae and multidrug efflux systems) were identified in these variable regions.
Here we present a comparative genomic sequence analysis of eleven recently sequenced genomes from the seven Cronobacter species fully representing the genus, and including the recently recognised species C. universalis and C. condimenti [16]. These strains were chosen as representatives of the Cronobacter genus based on seven locus MLSA [17], [18]. The three C. sakazakii strains (680, 696 and 701) are well characterised clinical isolates previously reported in eight publications [3], [8], [17], [18], [19], [20], [29], [30]. In addition, strain 701 is a C. sakazakii ST4 strain that was isolated from a fatal case of neonatal meningitis and has been used for in vitro tissue culture studies of virulence [8], [26]. C. sakazakii 680 is ST8, a sequence type associated with clinical infections without being linked to infant formula consumption [17], [19]. C. malonaticus 681T is ST7, a sequence type associated with adult infections. According to MLSA, C. condimenti is distantly related to the other Cronobacter species [18]. Completed genomes of C. sakazakii BAA-894 and C. turicensis z3032 were already in the public domain [29], [32] and were used to accurately match reads to reference sequences. The incomplete draft genome of C. sakazakii E899 [32] became available during the completion of this study and was used as a comparator where appropriate. Thus, a total of 14 genomes were used in this comparative study.
The aim of this study was to determine (i) the core Cronobacter genome, (ii) specific regions related to physiological and virulence related traits, and (iii) unique regions in C. sakazakii ST4, associated with neonatal meningitis [19], [30], and in C. malonaticus ST7 associated with adult infections. Due to the severity of infant infection, a better understanding of the genomic variation between Cronobacter spp. is needed, and will be of interest to manufacturers of powdered infant formula, regulatory bodies, and those studying the evolution and diversity of bacterial pathogenicity.
Results and Discussion
Cronobacter Genomes
Using the SOLiD® 4 System and Ion Torrent PGM™ next generation sequencing platforms, we sequenced the genomes of eleven Cronobacter strains including three C. sakazakii, two C. malonaticus, one C. muytjensii, one C. turicensis, two C. dublinensis, one C. universalis, and one C. condimenti (Table 1). The sum of the contig lengths ranged from 4.4 to 4.9 Mb, in length, comparable to the genomes sizes of the previously sequenced C. sakazakii BAA-894 and C. turicensis z3032 genomes [29], [31]. The publicly available, incomplete genome of C. sakazakii E899 is only 3.96 Mb and appeared to lack plasmid sequences [32]. Genome comparison revealed that pair-wise DNA sequence identity varies between 89 and 97%. A BRIG alignment for all the 14 Cronobacter genomes using the C. sakazakii BAA-894 genome as a backbone is shown in Figure S1.
Table 1. Eleven newly assembled and three publicly available Cronobacter genomes analyzed in this study.
| Species | Strain | Source | Country | Genome size (Mbp)d | MLST Sequence Type | EBI Accession Number |
| C. sakazakii | 680 | Clinical | USA | 4.36 | 8 | CALG01000001-CALG01000201 |
| 696 | Clinical | France | 4.90 | 12 | CALF01000001-CALF01000569 | |
| 701 | Clinical | France | 4.75 | 4 | CALE01000001-CALE01000768 | |
| E899a | Clinical | USA | 3.96 | 4 | AFMO00000000 | |
| BAA-894a | Powdered formula | USA | 4.53 | 1 | NC_009778-NC_009780 | |
| C. malonaticus | 507 | Clinical | Czech Republic | 4.45 | 11 | CALD01000001-CALD01000249 |
| 681b | Clinical | USA | 4.50 | 7 | CALC01000001-CALC01000171 | |
| C. turicensis | 564 | Clinical | USA | 4.50 | 5 | CALB01000001-CALB01000114 |
| z3032a,c | Clinical | Switzerland | 4.60 | 19 | NC_013282-NC_013285 | |
| C. dublinensis | 582 | Unknown | UK | 4.68 | 36 | CALA01000001-CALA01000427 |
| 1210d | Environment | Ireland | 4.59 | 106 | CAKZ01000001-CAKZ01000221 | |
| C. muytjensii | 530 | Infant formula | Denmark | 4.53 | 49 | CAKY01000001-CAKY01000365 |
| C. universalis | NCTC 9529T | Environmental | UK | 4.45 | 54 | CAKX01000001-CAKX01000231 |
| C. condimenti | 1330e | Food | Slovakia | 4.46 | 98 | CAKW01000001-CAKW01000155 |
Strains for which genome sequence is publicly available; E899 appears to lack plasmid sequences.
C. malonaticus species type strain (LMG 23826T).
C. turicensis species type strain (LMG 23827T).
C. dublinensis species type strain (LMG 23823T).
C. condimenti species type strain (LMG 26250T).
Sizes of newly sequenced genomes were derived from the sum of the length of all contigs from de novo assembly.
Phylogenetic and Evolutionary Analysis of the Cronobacter Genus
Figure 1A represents a maximum likelihood (ML) phylogram constructed based on the 138,143 SNPs identified from a concatenated alignment of 1117 core genes (879,768 nucleotides) present in single copies in all strains. Figure 1B represents a majority rule consensus tree generated by summarizing all the individual gene trees. The topology of speciation for each Cronobacter species was similar, as predicted by the two different approaches. There were contradictions in resolving the nodes within C. sakazakii indicating that recombination events might have obscured the phylogenetic signals within individual gene trees. Within C. sakazakii there was a higher level of confidence in placing C. sakazakii 701 and C. sakazakii E899 within the same clade, indicating that these two strains might have evolved more recently from a common ancestor. MLST revealed these two strains are both ST4 (see Figure 2 for MLSA phylogenetic analysis of the fourteen sequenced). Figure 1C represents the results of Clonal Frame analysis [33] of 99 randomly chosen core loci. Clonal Frame tries to recreate the evolutionary descent of each sequence by taking into account the recombination events that may have occurred during its evolutionary history. Clonal Frame also compares the recombination and mutation rates in assessing the impact of recombination on the evolution of observed phylogeny. Consensus tree (Figure 1C) obtained from two Clonal Frame runs with 200,000 MCMC iterations had a topology similar to the ML tree obtained using core genome SNPs, with one important difference. Clonal Frame analysis predicted that C. universalis and C. turicensis directly evolved from a common ancestor, while the ML tree of core SNPs (Figure 1A) predicted that they evolved in a step-wise fashion. This correlates well with the phylogenetic analysis of the seven loci used in the Cronobacter MLST scheme [18]. Co-incidentally, among all the speciating nodes in the consensus tree of 1117 core genes, the node where C. universalis speciates has the most incongruence. Hence, based on these two observations, it can be inferred that after speciation there might have been a large scale recombination event between ancestral C. universalis and hitherto unknown source. Clonal Frame predicted that for the observed phylogeny the ratio of substitutions introduced by recombination as compared to mutation (r/m) was 0.32, while ratio of number of recombination events to mutation events (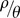 ) was 0.01.
) was 0.01.
Figure 1. Phylogentic analysis of Cronobacter spp. based on whole genome sequencing.

(A) A maximum likelihood phylogram based on 138,143 SNPs identified from a concatenated alignment of 1117 core genes (879,768 nucleotides) present in single copies in all strains. The numbers on the nodes represent the bootstrap support from 1000 replicates (in percentage). (B) A majority rule consensus tree generated by summarizing individual 1117 core gene trees. The numbers on the internal nodes indicate the fraction (in percentage) of gene trees which support the partition of the taxa into the two sets. (C) A Clonal Frame (CF) analysis based on randomly selected 99 core loci. The consensus tree was obtained from 2 CF runs with 200,000 MCMC iterations. The numbers on the nodes represent the bootstrap support in percentage.
Figure 2. Maximum Likelihood tree based on the concatenated sequences (3,036 bp) of the seven MLST loci.

The tree is drawn to scale using MEGA, with 1000 bootstrap replicates.
Comparisons between the evolutionary rates of gene families did not reveal a clear variation between the species of the Cronobacter genus. However, an enrichment analysis performed to assess the statistical over-representation of relatively fast evolving genes within certain functional categories revealed that genes related to iron acquisition, copper homeostasis, phage tail proteins, and genes related to O-antigen and fimbriae evolved faster as compared to the rest of the Cronobacter pan genome.
Core and Pan Genome Identification
A pan genome analysis of the fourteen genomes of the Cronobacter genus was used to study the diversity of the genomes, to identify genes of phenotypic and pathogenicity interest and those unique to each species. Putative prophage regions, identified using Prophinder [34], were included in the analysis. Some of the characteristics of the genomes of each of the species are discussed below. Additional detail is given in Text S1.
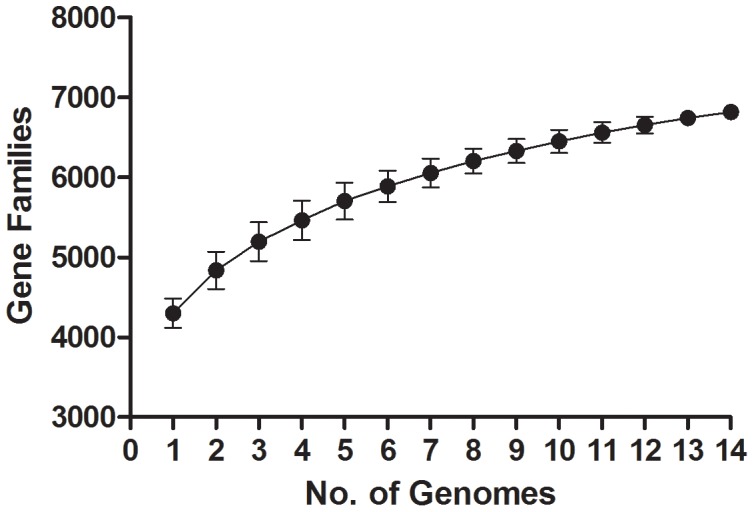
All genomes were annotated using RAST [35]. The number of annotated genes per genome varied between 3,700 and 4,200. Using the binomial mixture model approach of Snipen et al. [36], the observed pan genome was estimated to contains 6,155 genes, of which 2,271 (36.9%) genes were conserved in all fourteen Cronobacter genomes, an estimate of the core genome, and 3,884 (63.1%) were accessory genes found in at least one of the fourteen genomes studied. Because the draft genomes can have up to thousands of sequence gaps, some genes may have incomplete fragments or were unannotated. This could cause a slight under-estimation of the size of the core genome and a slight over-estimation of the size of the pan genome. A graph representing the rarefaction of the Cronobacter pan genome has been generated and is shown in Figure 3.
Figure 3. Graph representing the rarefaction of the Cronobacter pan genome, generated using the 14 Cronobacter genomes included in this study.
The published genome of C. sakazakii BAA-894 contains two plasmids pESA2 (31 kb, 51% GC content) and pESA3 (131 kb, 56% GC), encoding for 38 and 127 genes respectively. In contrast, C. turciensis z3032 contains three plasmids pCTU2 (22.5 kb, 49%GC), pCTU3 (53.8 kb, 50% GC) and pCTU1 (138 kb, 56% GC), encoding for 32, 74 and 136 genes respectively. The larger plasmids, pESA3 and pCTU1, share a common backbone and genes encoding for potential virulence traits such as iron acquisition systems (eitCBAD and iucABCD/iutA). The differences include the presence of plasminogen activator gene (cpa) and T6SS in the pESA3 plasmid, whereas in pCTU1 there is a 27 kb region with homologies to fhaB, fhaC and five adhesins (FHA locus) [25]. The cpa gene was only found in C. sakazakii strains 696 and 701, and was absent from 680. It was also absent from all other Cronobacter species with the exception of C. universalis. The BRIG alignments with pESA3 and pCTU1 as reference backbones are shown in Figures S2 and S3, respectively. It can be seen that the plasmid backbone was conserved in all the strains with varying degrees of homology. There were also clear regions of variation between the pESA3 and pCTU1 plasmids, some of which have been previously reported. However, it should be noted that matches to the plasmid do not necessarily confirm their location. To address this issue the read coverage was used to predict the location of the plasmid-borne genes. Genes homologous with those in pESA3 and pCTU1 were found in all sequenced strains of all species, though pESA3 was only partial in C. sakazakii 680 and C. condimenti 1330T, and pCTU1 had a partial match in C. sakazakii 680, 696, 701, and C. condimenti 1330T.
Previously, Kucerova et al. [29] detailed three prophages and 3 partial prophages in C. sakazakii BAA-894 contributing to the genome diversity; none of these prophages were previously reported in C. turicensis z3032 [31]. In our study of the whole genus we found a total of 27 major prophage regions (>20 ORFs) across the 13 genomes, most of which were found to be shared between the Cronobacter species (Table S1). In addition, we found a clustered regularly interspaced short palindromic repeats (CRISPR) array, which was located in the region ESA_02830-38. CRISPR arrays consist of a series of highly conserved repeats ∼20–50 bp separated by unique DNA fragments (spacers) of a similar length. It is understood that the CRISPR system provides a mechanism of resistance to infection by phage to which the bacteria have previously been exposed. Various CRISPR-associated (cas) genes and specific adjacent sequences are required to trigger these processes [37]. This specific CRISPR region was present in all Cronobacter strains except C. sakazakii 680 (ST8).
Comparison of the three C. sakazakii genomes revealed 408 ORFs, which were unique within this species. A number of regions were found to be unique to the genomes of strains 696 (ST12) and 701 (ST4). The C. sakazakii 680 (ST8) genome had unique genes encoding a region comprising iron uptake (fecRABCDE), a lac operon, and arsenate resistance on the larger plasmid. It also had large regions missing compared with the reference genome C. sakazakii BAA-894, including flagella synthesis (ESA_01224-69) and CRISPR (ESA_02831-37). The two C. malonaticus genomes revealed 92 ORFs unique to the species. Only about 15% of these ORFs were conserved between the two genomes. The C. malonaticus 681T (ST7) unique ORFs included mainly phage related proteins and two genes related to capsular polysaccharide export proteins. The C. turicensis 564 genome was analysed along with the publicly available C. turicensis z3032 genome to reveal 137 ORFs unique to the species, of which less than 10% were shared between the two genomes. Comparative analysis of C. dublinensis genomes revealed 142 ORFs unique to the species, of which less than 20% were shared between the two genomes. The C. dublinensis 582 genome also showed the presence of two additional clusters of CRISPR (938–943 and 3962–68). Only single strains were sequenced for each of the remaining Cronobacter species. The C. universalis genome had 73 unique ORFs. This included a region of 36 ORFs, which appears to be a plasmid remnant encoding for conjugation (trb genes), replication initiation, and plasmid stabilization. There were 40 ORFs found to be unique to the genome of C. muytjensii strain 530. The genome of the newly defined species C. condimenti strain 1330T contained 79 unique ORFs. These included genes related to β-xylosidase and a cluster of eight genes that are indicative of a unique fucose biosynthesis pathway.
Physiological and Phenotypic Traits in the Cronobacter Core and Pan Genome
The presence of known genes conferring physiological and phenotypic traits, categorized according to environmental stress response, cell surface composition, sugar metabolism, and metal resistance was examined.
A number of key stress response genes were investigated. A gene homologous to the universal stress protein uspA (ESA_01955) was detected in all strains. Similarly, homologues were located for stringent starvation response (ESA_03615), carbon starvation sensing protein rspA (ESA_01752), and carbon starvation protein (ESA_00801) in all Cronobacter strains. However, an additional carbon starvation homologue (ESA_00339) was found in all Cronobacter strains except C. sakazakii 680 due to the long missing region mentioned above. Cronobacter is known for its ability to survive desiccation for up to two years [38], [39]. Genes involved in desiccation resistance and osmotic stress adaption were found in all Cronobacter species. This included genes encoding the uptake of the osmoprotectants glycine, betaine, and trehalose (ESA_00587-9, ESA_01944-66), which were present in all Cronobacter. Thermal tolerance in Cronobacter has been controversial due to conflicting reports [40]–[42]. The initiation translation factor (infB) proposed by Asakura et al. [41] was present in all Cronobacter, and is one of the alleles used for MLST. In contrast, the thermoresistance gene cluster (or most components) identified by [42] was only present in C. sakazakii strains 696, 701, and in both strains of C. malonaticus. Unfortunately, this region is poorly annotated and the association with thermal resistance is not supported by experimental data in these strains. Together with biofilm formation, the ability to adapt and persist under stressed conditions enables the bacterium to survive in dry food ingredients. All Cronobacter species had genes (crtZ; ESA_00341-4) for β-carotene production, a yellow pigmentation, which we speculate helps protect the organism from light produced by oxygen radicals.
Capsular polysaccharides on the bacterial cell surface can play a crucial role in the interaction of bacteria with their environment and in their pathogenicity. The genes encoding the enzymes responsible for the production and transport of these polysaccharides are usually clustered in large operons, such as the wca operon in E. coli K-12. This gene cluster controls the biosynthesis of the exopolysaccharide colanic acid containing fucose and glucuronic acid. Cronobacter spp. can produce copious extracellular polymers, resulting in highly mucoid colony formation and can form biofilms on inert surfaces including infant feeding equipment and nasogastric feeding tubes [8], [43], [44]. Colanic acid production was encoded (ESA_01155-01175; wzABCKM) in all sequenced Cronobacter strains. The genes for capsular polysaccharide assembly and export (ESA_03350-59) were present in all Cronobacter strains except for two clinical isolates, C. sakazakii 701 (ST4) and 696 (ST12).
One highly variable genomic region (ESA_01179–89) corresponds to the O-antigen gene locus. The locus contains two genes galF (ESA_01177) and rfbB (ESA_01178) which are conserved in all the Cronobacter strains, whereas the rest of the genes from the O-antigen locus are highly divergent and were not detected previously by microarray hybridization [29]. The lipopolysaccharide (LPS) is one of the few structural features of Cronobacter which has been chemically investigated and is known to vary across the genus. In C. sakazakii and C. malonaticus the LPS are composed of various branched polymers, whereas they are unbranched in C. muytjensii [45]–[48].
Variation in the O-antigen region has been used in serotype-specific PCR-based assays [49]–[53]. The regions between UDP-glucose pyrophosporylase (galF) and 6-phosphogluconate (gnd) were amplified to generate distinguishable O-antigen groups. The genetic architecture of the O-antigen cluster in the reference genome C. sakazakii BAA-894 (ST1) corresponds to the serotype O:1 and both the ST4 strains 767 and NCTC 8155 are O:2. However, the current PCR-based serotyping assay has a significant limitation since some defined serotypes are found in more than one species [51]–[53].
It is notable that C. sakazakii 701 (ST4) also encoded an O-antigen acetylase (O-acetyl transferase; 28141.13.peg.3410) adjacent to a prophage region which, except for fragments in C. sakazakii 696 (ST12), was not present in other C. sakazakii strains. O-antigen modification enzymes are encoded by temperate bacteriophages [54]. The possibility of this in Cronobacter was proposed by Sun et al. [51] when comparing RFLP patterns from PCR amplification of the O-antigen genes of 119 C. sakazakii strains. Seroconversion is an important virulence factor since antigenic variation enhances the survival of the bacteria because the host would have to mount a specific immune response to each different serotype. Serotype-converting bacteriophages play an important role in conferring these traits, and recognising their presence in C. sakazakii is important due to the application of the serotyping scheme.
The gene cluster encoding for maltose utilisation and including α–glucosidase (ESA_02709-14) was found in all Cronobacter strains. The presence of α-glucosidase and a constitutive maltose uptake system, as well as osmotic pressure tolerance, has been used as the basis for a number of commercially available enrichment broths and chromogenic agars for the isolation of Cronobacter spp. [55]. Other sugar utilization pathways were only found in some Cronobacter species. A gene cluster for α-mannosidase utilisation (ESA_02616-18) was only found in C. sakazakii and C. turicensis, and not in the closely related C. malonaticus.
Cronobacter are ubiquitous in the environment, and their main habitat is believed to be plant material. Cronobacter are able to ferment β-glucoside sugar substrates from plants, including cellobiose, arbutin, salicin, and esculin. The genes involved in the metabolism of β-glucosides are in the cluster ESA_02544-47. ESA_02544 is probably a transcriptional antiterminator from the BglG family, which is involved in positive control of the utilization of different sugars by transcription antitermination [56]. ESA_02545 encodes a kinase which converts β-glucosides to 6-phospho-β-glucosides and ESA_02546 encodes a 6-phospho-β-glucosidase specific to arbutin-6-phosphate and salicilin-6-phosphate. ESA_02547 also encodes 6-phospho-β-glucosidase, which may have the same or similar function as ESA_02546. Malonate is also found in plants and C. malonaticus was initially a subspecies within C. sakazakii that was differentiated from C. sakazakii subsp. sakazakii by the malonate utilisation test [15]. However, the genes required for malonate metabolism - malonyl CoA acyl carrier protein transacylase, phosphoribosyl-dephospho-CoA transferase, and malonate decarboxylase (Ctu_34990-35070) - are found in all Cronobacter species except C. sakazakii and C. dublinensis strain 582. Therefore this test cannot be used solely for the purpose of identifying C. malonaticus.
Homologues of tellurite resistance genes (terACDYZ) were located in the loci ESA_01775–ESA_01804 of C. sakazakii BAA-894. As the gene cluster was absent from all other Cronobacter strains studied, the reference strain BAA-894 or the ST1 lineage probably acquired the tellurite resistance cluster recently. Homologies of these genes are also found on the IncII plasmid R478 of Serratia marcescens, pK29 of Klebsiella pneumoniae NK29, pEC-IMPQ of Enterobacter cloacae, and pAPEC-O1-R of E. coli APEC O1 [57]. In BAA-894 this cluster of genes contains an IS element (ISEhe3) just before ESA_01781 which was also identified on the pK29 plasmid as well as a fragment of an IS4 family transposase within the locus ESA_01803.
Copper is essential for bacterial growth, being required for many key enzymes, but is toxic in excess. Two copper and silver resistance gene clusters are located close to each other (ESA_04238-45 and ESA_04248–55) in the C. sakazakii BAA-894 annotated chromosome and are on the smallest plasmid (pCTU3) in C. turicensis z3032 (pCTU3_3p00600-700 and pCTU3_3p00490-590). The first region (cusRCFBA/silRECBA) was present in all C. sakazakii strains, in one C. malonaticus (681) strain, and one C. turicensis (z3032) strain. CusA is a membrane protein belonging to the resistance-nodulation-division (RND) protein superfamily. CusF is a periplasmic protein, which in E. coli interacts with the cusCBA efflux system. The second region (pcoABCDR) was present in nearly all C. sakazakii strains (except 701), in one C. malonaticus strain (681), one C. turicensis strain (z3032), and in C. universalis 581T. PcoA encodes a homologue to multicopper oxidase, and PcoB is an outer membrane protein transfering copper into the periplasm where it is oxidised by PcoA. Both these regions are encoded on the plasmids R478 and APEC-O1-R, referred to above, which carry tellurite resistance genes [57]. The copper and silver resistance genes on the reference C. sakazkaii BAA-894 genome are separate from the tellurite resistance genes. Therefore these regions do not appear to be the result of a single plasmid integration event.
Virulence Associated Genes
Putative virulence traits in Cronobacter include (i) attachment and invasion of host cells, (ii) iron acquisition, (iii) macrophage survival, and (iv) protein secretion systems. Since genes encoding for SodA, OmpA, OmpX, and a metalloprotease (zpx) were found in all genomes irrespective of source and species, they could not be connected to particular virulence-associated strains such as C. sakazakii ST4 and therefore to neonatal meningitis.
Cronobacter spp. route of infection is probably through attachment and invasion of the intestinal cells and therefore genes encoding surface appendages such as fimbriae (pili) are of importance. The genetic content of all fimbriae clusters was most similar to the type I chaperone/usher-assembled pilus system [58]. These clusters may encode complete and functional pili, as some degree of homology was found between the genes in the C. sakazakii fimbriae clusters and the remaining components necessary for type-I pilus assembly (the minor tip fibrillum fimG and fimbrial adhesin fimH genes). Type 1 fimbriae have been associated with E. coli K1 invasion of human brain cells [59] and are therefore of particular interest in Cronobacter pathogenicity studies.
A total of ten putative fimbriae gene clusters were identified and are summarised in Table 2. Curli fimbriae are believed to play an important role in the adhesion of E. coli to host cells by interacting with matrix proteins such as fibronectin, laminin, and plasminogen to initiate adherence and colonization. However, since C. sakazakii strains dominate clinical isolates, the absence of curli fimbriae genes infers this trait is not essential for Cronobacter pathogenicity. Why the region has been lost in the C. sakazakii lineage and retained in the rest of the genus is unclear.
Table 2. Cronobacter fimbriae cluster designations.
| Cluster | Loci | C. sakazakii | C. malonaticus | C. muytjensii | C. turicensis | C. dublinensis | C. universalis | C. condimenti | Comment | |||||
| BAA-894 | 680 | 696 | 701 | 507 | 681 | 530 | 564 | 582 | 1210 | NCTC 9529T | 1330 | |||
| 1 | ESA_01970-76 | + | + | − | − | − | − | − | − | − | − | − | − | Also found in C. sakazakii strain2 [Kucerova et al. 2010; 35] |
| 2 | ESA_02342-45 | + | + | + | + | + | + | + | + | + | + | + | + | Present in all Cronobacter |
| 3 | ESA_02538-42 | + | + | + | + | + | + | + | + | + | + | + | + | ESA_02541&42 not found in C. muytjensii, C. dublinensis, C. condimenti |
| 4 | ESA_02795-99 | + | + | + | + | + | + | − | − | − | − | − | − | Only found in C. sakazakii and C. malonaticus |
| 5 | ESA_03512-20 | + | + | + | + | − | − | − | − | − | − | − | − | Beta-fimbriae. Only found in C. sakazakii |
| 6 | ESA_04067-73 | + | + | + | + | + | + | + | + | + | + | + | + | Present in all Cronobacter |
| 7 | ESA_03812-15 | + | + | + | + | + | + | − | + | + | + | + | + | Absent from C. muytjensii |
| 8 | Ctu_36390-450 | − | + | + | + | + | + | + | + | + | + | + | + | π–fimbriae. Absent from C. sakazakii BAA-894 |
| 9 | ESA_03231-33 | + | + | + | + | + | + | + | + | + | + | + | + | Type IV. Gliding motility |
| 10 | Ctu-16160-230 | − | − | − | − | + | + | + | + | + | + | + | + | Curli fimbriae. Absent from C. sakazakii |
Iron is an essential growth factor and bacteria have evolved mechanisms, such as siderophores, for its acquisition. Nearly all Cronobacter strains examined possess complete operons for the production of enterobactin synthesis (entABCDEFS, ESA_00791-800), receptor, and transport (fepABCDEG; ESA_02727-31). The exception were C. sakazakii strains 701 and 696, which lacked the homologue for EntS, an enterobacterin exporter. All Cronobacter species possess a complete plasmid-borne operon for aerobactin synthesis (iucABCD) and its receptor iutA (ESA_pESA3p05547-51). Additionally, a gene cluster for hydroxamate-type siderophores (fhuABCDE, ESA_03187-90 & ESA_02242) was encoded in all Cronobacter species. As previously referred to, C. sakazakii 680 encoded a unique siderophore (fecRABCDE) which is probably plasmid-borne. According to Franco et al. [25], the only functional siderophores are those encoded on the larger plasmid. Iron uptake is often cited as a virulence mechanism, especially with respect to plausible utilisation by Cronobacter of iron in breast milk and infant formula. However, drawing further conclusions will require further work because there are a number of probable iron uptake mechanisms across the genus, including those in species not associated with neonatal infections: C. muytjensii, C. malonaticus, C. dublinensis, C. universalis, and C. condimenti.
The secretion systems ancestrally related to the bacterial conjugation machinery are referred to as the type IV secretion systems (T4SSs). The T4SSs can transfer both proteins and nucleoprotein complexes and could constitute a conjugal transfer system [60]. A T4SS, unique to C. sakazakii BAA-894 and C. turicensis z3032, was located on the smaller plasmid (pESA2 31kB and pCTU2 22.5 kB) of both strains. A second cluster of 19 genes, belonging to the Tra group of genes and annotated as IncF conjugation plasmids, was found to be present in the C. sakazakii 701 and 696 genomes and is linked to the type IV protein secretion system. This could also be a plasmid-borne region as these genes have previously been identified in the Inc group of plasmids in E. coli and Serratia.
Type VI secretion system (T6SS) is a newly described system that may be involved in competing with other bacteria in adherence, cytotoxicity, host-cell invasion, growth inside macrophages, and survival within the host. One T6SS associated gene that was located separately from the main T6SS gene cluster, was vgrG, encoding a lipoprotein (ESA_00292-4). It was present in all Cronobacter strains except for one strain of C. malonaticus (507) and one strain of C. muytjensii (530). Six putative T6SS clusters were identified in the Cronobacter genomes (Table S2), some of which were previously described by Kucerova et al. [3], [29]. These are putative T6SS clusters as it remains to be determined whether they encode functional secretion systems. Zhou et al. [61] recently reported the role of T6SS in E. coli K1 invasion of the human blood-brain barrier. Although several T6SS are found in all Cronobacter species, none were unique to C. sakazakii ST4 strain 701.
All sequenced Cronobacter genomes exhibited the presence of various hemolysin and hemolysin-related genes, scattered across the genome (Table S3). These genes were present in all the genomes with the only exceptions being C. sakazakii 701 and C. malonaticus 507, which lacked the 21 kDa hemolysin precursor gene. Most of the strains had two copies of the hemolysin gene and the hemolysin activator protein precursor gene.
One of the most interesting gene clusters, unique to C. sakazakii is ESA_03609–13 (nanKTAR) and ESA_03302 (nanC) which encode for the uptake and utilization of exogenous sialic acid, which is functional (unpublished laboratory studies). Sialic acid catabolism is limited to commensal bacteria in the intestinal tract and to pathogens [62]. In C. sakazakii ESA_03609 encodes a putative sugar isomerase (YhcH). Genes ESA_03610-13 encode the nanKTAR genes involved in the N-acetylneuraminate and N-acetylmannosamine degradation pathway. The nanK gene (ESA_03610) encodes N-acetylmannosamine kinase; nanT (ESA_03611) encodes the sialic acid transporter N-acetylneuraminate lyase; nanA (ESA_03612) encodes N-acetylneuraminate lyase, and nanR (ESA_03613) is a transcriptional regulator from the GntR family. The nanE locus encoding the enzyme N-acetylmannosamine-6-phosphate-2epimerase was located separate from this cluster at ESA_00529, and unlike the rest was found conserved across the genome of the Cronobacter genus. NanC (ESA_03302) encodes the N-acetylneuraminic acid outer membrane channel protein). It is notable that the nan cluster is adjacent to a stringent starvation gene homologue (sspA, ESA_03615) and therefore expression of the nan cluster could be responsive to environmental nutrient levels. The acquisition of genes encoding for the utilization of exogenous sialic acid may have a major role in C. sakazakii colonisation of the human intestinal tract (via mucins) and the use of sialic acid in breast milk, infant formula, and brain cells as a nutrient source [62].
Despite detailed genomic analysis, the reason for the association of C. sakazakii ST4 with neonatal meningitis remains unclear. Initially Joseph & Forsythe [19] proposed that it could be either environmental persistence, resulting in greater neonatal exposure, or the presence of specific pathogenicity traits. Other than one putative plasmid-borne T6SS, this study has not shown any conclusive unique regions in the ST4 strain C. sakazakii 701, originally isolated from the peritoneal fluid of a fatal neonatal meningitis case.
In summary, the comparison of the draft genomes representing the Cronobacter genus has revealed the core genes and accessory genes unique to each strain. It was found that C. sakazakii has acquired the ability to use exogenous sialic acid, which may be important in the colonisation of the intestinal tract. Mobile traits such as adhesins, T6SS, and metal resistance as well as prophages have contributed considerably to the variation of genomic content and probably account for much of the variation in clinical and environmental distribution of species and sequence types. The lack of clearly identifiable virulence genes unique to C. sakazakii ST4 may indicate its prevalence in neonatal meningitis cases due to environmental persistence and increasing host exposure. Further improvement of the draft genome sequences, bioinformatics searches for novel or known virulence traits present in bacteria with similar modes of infection, and in silico and in vitro ascertainment of the contribution of single nucleotide polymorphisms, genome rearrangements, and other sequence features to pathogenicity may shed light on acquired pathogenicity of ST4 strains.
Materials and Methods
Strains and Culture Conditions
Cronobacter strains were selected to represent the seven recognized species, including those from reported clinical cases as well as species type strains (Table 1). All Cronobacter strains were stored at −80°C in Nutrient Broth (Oxoid, UK) with 10% glycerol, subcultured on Trypticase Soy Agar (Oxoid ThermoFisher, UK) and checked for purity. Overnight Trypticase Soy Broth (Oxoid ThermoFisher, UK) cultures were used for DNA extraction.
Genome Sequencing and Assembly
Genomic DNA was isolated using the Qiagen DNeasy Blood and Tissue DNA Isolation Kit according to manufacturer’s instructions. Cronobacter genomes of all strains except 680, 1210T and 1330T were sequenced using the SOLiD™ 4 system (Life Technologies, Carlsbad, CA). Long mate-paired genomic DNA libraries with approximately 1.8 kb inserts were sequenced to generate 23–36 million of 2×50 bp reads for each strain, approximating 500–800 fold coverage of the genome. The colorspace reads were error-corrected and then assembled de novo into contigs and scaffolds using the Velvet assembly engine [63]. The ultimate genome assemblies contain 1,600 to 3,100 contigs with N50 of 3.7 to 5.5 kb and 260 to 1,170 scaffolds with N50 of 230 to 600 kb (Table 3).
Table 3. De novo assembly statistics for the eleven newly sequenced Cronobacter strains.
| Species | Strain | Sequencing platform | Total cont length (bp) | N50 of scaffolds (bp) | Number of scaffolds | N50 of contigs (bp) | Number of contigs | Estimated number of ORFs |
| C. sakazakii | 680 | PGM | 4,357,873 | nda | nd | 51,120 | 194 | 4,178 |
| 696 | SOLiD | 4,897,138 | 297,746 | 920 | 4,336 | 2,659 | 4,661 | |
| 701 | SOLiD | 4,752,729 | 346,235 | 1,171 | 3,538 | 3,148 | 4,509 | |
| C. malonaticus | 507 | SOLiD | 4,447,701 | 373,979 | 464 | 3,703 | 2,361 | 4,226 |
| 681 | SOLiD | 4,496,745 | 345,762 | 263 | 5,537 | 1,592 | 4,291 | |
| C. turicensis | 564 | SOLiD | 4,500,608 | 411,105 | 263 | 4,796 | 1,807 | 4,227 |
| C. dublinensis | 582 | SOLiD | 4,677,592 | 229,230 | 539 | 3,822 | 2,657 | 4,483 |
| 1210 | PGM | 4,594,228 | nd | nd | 46,941 | 210 | 4,376 | |
| C. muytjensii | 530 | SOLiD | 4,533,101 | 596,924 | 444 | 4,925 | 1,937 | 4,304 |
| C. universalis | NCTC 9529T | SOLiD | 4,450,737 | 331,248 | 389 | 4,506 | 2,085 | 4,316 |
| C. condimenti | 1330 | PGM | 4,469,5362 | nd | nd | 83,159 | 137 | 4,307 |
nd indicates that scaffolding was not performed due to lack of mate-paired libraries.
Strains 680, 1210T and 1330T were sequenced using Ion Torrent PGM system (Life Technologies). Fragment library preparation was performed with the Ion Fragment Library Kit (Life Technologies, Darmstadt, Germany). Template preparation was carried out with the Ion Xpress™ Template Kit (Life Technologies). The Ion Sequencing Kit (Life Technologies) was used with the Personal Genome Machine™ (PGM™) sequencer. A single sequencing run (65 cycles) was performed on an Ion 316™ chip for each library. Contigs were assembled from fragment reads using the MIRA 3 assembler [http://sourceforge.net/apps/mediawiki/mira-assembler/index.php].
The assembled genome scaffolds were aligned to the most closely related publicly available genomes using MUMmer [64]. The scaffolds of strains 680, 696, 701, 507, and 681T were aligned to the C. sakazakii BAA-894 complete genome (accession numbers NC009778 - NC009780). The scaffolds of strains 564, 582, 1210T, 530, 581T and 1330T were aligned to the C. turicensis z3032 complete genome (accession numbers NC013282-NC013285). Scaffolds were broken at points where non-contiguous regions of the reference genome were juxtaposed and then re-ordered so that they were syntenic with the reference genome. All scaffolds from a given strain were concatenated into a single pseudogenome, separated by the sequence, NNNNCACACACTTAATTAATTAAGTGTGTGNNNNN, which contains stop codons in all six reading frames. Scaffolds that did not match the reference genomes were concatenated in random orders at the end of the genome. The pseudogenomes were annotated using the RAST automated annotation server [35].
The genome sequences of the eleven newly sequenced strains were deposited to Genbank (see Table 1 for accession numbers).
Core and Pan Genome Identification
The genomes were compared using methods similar to those previously published [65]. Orthologous and paralogous gene families were initially constructed based on RAST annotations. An ‘all-against-all’ tblatx matrix was constructed using BLAT [66]. The blat matrix was used as an input to construct orthologous/paralogous gene families using OrthoMCL [67]. A representative nucleotide sequence from each gene family was collated with strain-specific genes not present in any gene families. This set of representative sequences was again subjected to a tblatx analysis against the whole genome nucleotide sequences to identify candidate genes that may have been missed by the annotation server RAST. A gene was considered present if it had at least 85% nucleotide sequence identity covering at least 20% of the sequence length. The phyletic table generated from the tblatx analysis was consolidated with the phyletic table generated from OrthoMCL analysis to compute a comprehensive pan genome matrix. This phyletic matrix was used as an input for the binomial mixture model software of Snipen et al. [36] to determine the Cronobacter core genome and accessory genes.
Prophage Identification
ACLAME Prophinder, available at http://aclame.ulb.ac.be/Tools/Prophinder/, was used for prophage identification [34].
Phylogenetic analysis
Core genome analysis revealed that there were 1117 annotated gene families conserved across all genomes in exactly one copy. Orthologous DNA sequences within each gene family were aligned using Muscle [68]. Individual alignments were concatenated using custom scripts, and a maximum likelihood (ML) tree was constructed using RaxML (Ver. 7.2.6) [69]. As a distinct alternative, a majority rule consensus tree of all gene family trees that were conserved across all genomes and were present in exactly one copy was constructed using Phylip (Ver 3.69) [70]. A concatenated alignment of 100 randomly chosen loci from the 1117 single copy core loci was used as an input for Clonal Frame [33]. Two independent runs, each consisting of 200,000 Markov chain – Monte Carlo (MCMC) iterations were performed, where the first 100,000 iterations were discarded as burn-in. The runs were compared for convergence using the Gelman- Rubin statistic [71], which was found to be satisfactory.
Evolutionary rate of gene families
Individual gene sequences (DNA and amino acid sequences) for all genes in gene families predicted by OrthoMCL were extracted using custom Perl scripts. Codon alignments within gene families were constructed using PAL2NAL [72]. Amino acid alignments needed as input for PAL2NAL were constructed using Muscle [68]. The dS/dN ratios for all possible pairwise comparisons of the codon alignments within a gene family were calculated using SNAP [73]. Mean dS/dN ratios were assigned for individual gene families by averaging all pairwise ratios within each family. The pan genome was ranked based on logarithmic (log10) dS/dN ratios; enrichment analysis for gene sets containing relatively fast evolving gene families was conducted using Gene Set Enrichment Analysis (GSEA) [74]. Within GSEA, controlled false discovery rates were used to adjust p values for multiple comparisons.
Multilocus Sequence Analysis (MLSA)
MLSA was undertaken by using existing sequences in the multilocus sequence typing (MLST) database (http://www.pubMLST.org/cronobacter) and for new ones obtained in this study. Multilocus sequence typing of strains was performed as previously described [17]. The sequences obtained were independently aligned with sequences of the type strains of all species of the genus Cronobacter using the ClustalW2 program [75] and MEGA (Molecular Evolutionary Genetics Analysis) software version 5 [76]. Genetic distances and clustering were determined using Kimura’s two-parameter model [77]; evolutionary trees were reconstructed by the Maximum Likelihood method [78]. Stability of the relationships was assessed by the bootstrapping method (1000 replicates).
Supporting Information
BLAST Ring Image Generator (BRIG) analysis of the Cronobacter genomes.
(DOC)
BLAST Ring Image Generator (BRIG) analysis of Cronobacter plasmid pESA3 with matching sequence content found in other Cronobacter species.
(DOC)
BLAST Ring Image Generator (BRIG) analysis of Cronobacter plasmid pCTU1 to matching content in other Cronobacter species.
(DOC)
Number of putative prophage regions in the Cronobacter genomes.
(DOC)
Presence (+) or absence (−) of type six secretion systems in Cronobacter spp.
(DOC)
Copy number variation of the hemolysin gene within the Cronobacter genus.
(DOC)
Additional regions of variation between Cronobacter genomes.
(DOC)
Funding Statement
Life Technologies funded and performed the genome sequencing. The authors thank Alain Rico for the sequencing of strains using the C. sakazakii 680, C. dublinensis 1210 and C. condimenti 1330 using the Ion Torrent PGMTM. MM and PD were supported in part by United States National Institutes of Health grants AI039557 AI052237, AI073971, AI075093, AI077645, AI083646, USDA grants 2009-03579 and 2011-67017-30127, the Binational Agricultural Research and Development Fund, and CDMRP BCRP W81XWH-08-1-0720. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Friedemann M (2007) Enterobacter sakazakii in food and beverages (other than infant formula and milk powder). Int J Food Microbiol 116: 1–10. [DOI] [PubMed] [Google Scholar]
- 2. Kandhai MC, Reij MW, Gorris LG, Guillaume-Gentil O, van Schothorst M (2004) Occurrence of Enterobacter sakazakii in food production environments and households. Lancet 363: 39–40. [DOI] [PubMed] [Google Scholar]
- 3. Kucerova E, Joseph S, Forsythe SJ (2011) The Cronobacter genus: ubiquity and diversity. Quality Assurance and Safety of Crops & Foods 3: 104–122. [Google Scholar]
- 4.FAO/WHO (2008) Enterobacter sakazakii (Cronobacter spp.) in powdered follow-up formulae. Microbiological Risk Assessment Series No. 15, Rome, 90pp. Available: http://www.who.int/foodsafety/publications/micro/mra_followup/en/.
- 5. Biering G, Karlsson S, Clark NC, Jonsdottir KE, Ludvigsson P, et al. (1989) Three cases of neonatal meningitis caused by Enterobacter sakazakii in powdered milk. J Clin Microbiol 27: 2054–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burdette JH, Santos C (2000) Enterobacter sakazakii brain abscess in the neonate: the importance of neuroradiologic imaging. Pediatr Radiol 30: 33–4. [DOI] [PubMed] [Google Scholar]
- 7. Willis J, Robinson JE (1988) Enterobacter sakazakii meningitis in neonates. Pediatr Infect Dis J 7: 196–9. [DOI] [PubMed] [Google Scholar]
- 8. Caubilla-Barron J, Hurrell E, Townsend S, Cheetham P, Loc-Carrillo C, et al. (2007) Genotypic and phenotypic analysis of Enterobacter sakazakii strains from an outbreak resulting in fatalities in a neonatal intensive care unit in France. J Clin Microbiol 45: 3979–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hariri S, Joseph S, Forsythe SJ (2012) Predominance of Cronobacter sakazakii ST4 clonal complex strains in Cronobacter neonatal meningitis infections in US 2011. Emerg Infect Dis. In Press. [DOI] [PMC free article] [PubMed]
- 10. Lai KK (2001) Enterobacter sakazakii infections among neonates, infants, children, and adults. Medicine 80: 113–122. [DOI] [PubMed] [Google Scholar]
- 11. Himelright I, Harris E, Lorch V, Anderson M (2002) Enterobacter sakazakii infections associated with the use of powdered infant formula -Tennessee, 2001. J Am Med Assoc 287: 2204–2205. [Google Scholar]
- 12. van Acker J, de Smet F, Muyldermans G, Bougatef A, Naessens A, et al. (2001) Outbreak of necrotizing enterocolitis associated with Enterobacter sakazakii in powdered milk formula. J Clin Microbiol 39: 293–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Farmer JJ, Asbury MA, Hickman FW, Brenner DJ, the Enterobacteriaceae Study Group (USA) (1980) Enterobacter sakazakii: a new species of Enterobacteriaceae isolated from clinical specimens. Int J Syst Bacteriol 30: 569–84. [Google Scholar]
- 14. Iversen C, Waddington M, On SL, Forsythe S (2004) Identification and phylogeny of Enterobacter sakazakii relative to Enterobacter and Citrobacter species. J Clin Microbiol 42: 5368–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Iversen C, Lehner A, Mullane N, Bidlas E, Cleenwerck I, et al. (2007) The taxonomy of Enterobacter sakazakii: proposal of a new genus Cronobacter gen. nov. and descriptions of Cronobacter sakazakii comb. nov. Cronobacter sakazakii subsp. sakazakii, comb. nov., Cronobacter sakazakii subsp. malonaticus subsp. nov., Cronobacter turicensis sp. nov., Cronobacter muytjensii sp.nov., Cronobacter dublinensis sp. nov. and Cronobacter genomospecies 1. BMC Evol Biol 7: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Joseph S, Cetinkaya E, Drahovska H, Levican A, Figueras M, et al. (2012) Cronobacter condimenti sp. nov., isolated from spiced meat and Cronobacter universalis sp. nov., a novel species designation for Cronobacter sp. genomospecies 1, recovered from a leg infection, water and food ingredients. Intl J Syst Evol Microl 62: 1277–83. [DOI] [PubMed] [Google Scholar]
- 17. Baldwin A, Loughlin M, Caubilla-Barron J, Kucerova E, Manning G, et al. (2009) Multilocus sequence typing of Cronobacter sakazakii and Cronobacter malonaticus reveals stable clonal structures with clinical significance which do not correlate with biotypes. BMC Microbiol 9: 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Joseph S, Sonbol H, Hariri S, Desai P, McClelland M, et al. (2012) Diversity of the Cronobacter genus as revealed by multi locus sequence typing. J. Clin. Microbiol. 50: 3031–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Joseph S, Forsythe SJ (2011) Predominance of Cronobacter sakazakii ST4 in neonatal infections. Emerg. Infect. Dis. 17: 1713–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Townsend S, Hurrell E, Forsythe S (2008) Virulence studies of Enterobacter sakazakii isolates associated with a neonatal intensive care unit outbreak. BMC Microbiol 8: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cruz A, Xicohtencatl-Cortes J, González-Pedrajo B, Bobadilla M, Eslava C, et al. (2011a) Virulence traits in Cronobacter species isolated from different sources. Can J Microbiol. 57: 735–44. [DOI] [PubMed] [Google Scholar]
- 22. Cruz AL, Rocha-Ramirez LM, Gonzalez-Pedrajo B, Ochoa SA, Eslava C, et al. (2011b) Flagella from Cronobacter sakazakii induced an inflammatory response in human monocytes. Cytokine 56: 95. [Google Scholar]
- 23. Kothary MH, McCardell BA, Frazar CD, Deer D, Tall BD (2007) Characterization of the zinc-containing metalloprotease encoded by zpx and development of a species-specific detection method for Enterobacter sakazakii . Appl Environ Microbiol 73: 4142–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pagotto FJ, Nazarowec-White M, Bidawid S, Farber JM (2003) Enterobacter sakazakii: infectivity and enterotoxin production in vitro and in vivo . J Food Prot 66: 370–5. [DOI] [PubMed] [Google Scholar]
- 25. Franco AA, Hu L, Grim CJ, Gopinath G, Sathyamoorthy V, et al. (2011) Characterization of putative virulence genes on the related RepFIB plasmids harbored by Cronobacter spp. Appl Environ Microbiol 77: 3255–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Townsend SM, Hurrell E, Gonzalez-Gomez I, Lowe J, Frye JG, et al. (2007) Enterobacter sakazakii invades brain capillary endothelial cells, persists in human macrophages influencing cytokine secretion and induces severe brain pathology in the neonatal rat. Microbiology 153: 3538–47. [DOI] [PubMed] [Google Scholar]
- 27. Kim K, Kim K-P, Choi J, Lim J-A, Lee J, et al. (2010) Outer membrane proteins A (OmpA) and × (OmpX) are essential for basolateral invasion of Cronobacter sakazakii . Appl Environ Microbiol 76: 5188–5198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim KP, Loessner MJ (2008) Enterobacter sakazakii invasion in human intestinal Caco-2 cells requires the host cell cytoskeleton and is enhanced by disruption of tight junction. Infect Immun 76: 562–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kucerova E, Clifton SW, Xia XQ, Long F, Porwollik S, et al. (2010) Genome sequence of Cronobacter sakazakii BAA-894 and comparative genomic hybridization analysis with other Cronobacter species. PLoS ONE 5: e9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hamby S, Joseph S, Forsythe SJ, Chuzhanova N (2011) In silico identification of pathogenic strains of Cronobacter from biochemical data reveals association of inositol fermentation with pathogenicity. BMC Microbiol 11: 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Stephan R, Lehner A, Tschler P, Rattei T (2011) Complete genome sequence of Cronobacter turicensis LMG 23827, a food-borne pathogen causing deaths in neonates. J Bacteriol 193: 309–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chen Y, Strain EA, Allard M, Brown EW (2011) Genome sequence of Cronobacter sakazakii E899, a strain associated with human illness. J Bacteriol 193: 581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Didelot X, Falush D (2007) Inference of bacterial microevolution using multilocus sequence data. Genetics 175: 1251–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lima-Mendez G, Van Helden J, Toussaint A, Leplae R (2008) Prophinder: a computational tool for prophage prediction in prokaryotic genomes. Bioinformatics 24: 863–5. [DOI] [PubMed] [Google Scholar]
- 35. Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, et al. (2008) The RAST Server: rapid annotations using subsystems technology. BMC Genomics 9: 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Snipen L, Almoy T, Ussery DW (2009) Microbial comparative pan-genomics using binomial mixture models. BMC Genomics 10: 385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Karginov FV, Hannon GJ (2010) The CRISPR system: small RNA-guided defence in bacteria and archaea. Molecular Cell 37: 7–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Caubilla-Barron J, Forsythe S (2007) Dry stress and survival time of Enterobacter sakazakii and other Enterobacteriaceae. J Food Protect 70: 2111–7. [DOI] [PubMed] [Google Scholar]
- 39. Osaili T, Forsythe S (2009) Desiccation resistance and persistence of Cronobacter species in infant formula. Intl J Food Microbiol 136: 214–220. [DOI] [PubMed] [Google Scholar]
- 40. Williams TL, Monday SR, Edelson-Mammel S, Buchanan R, Musser SM (2005) A top-down proteomics approach for differentiating thermal resistant strains of Enterobacter sakazakii . Proteomics 5: 4161–9. [DOI] [PubMed] [Google Scholar]
- 41. Asakura H, Morita-Ishihara T, Yamamoto S, Igimi S (2007) Genetic characterization of thermal tolerance in Enterobacter sakazakii . Microbiol Immunol 51: 671–677. [DOI] [PubMed] [Google Scholar]
- 42. Gajdosova J, Benedikovicova K, Kamodyova N, Tothova L, Kaclikova E, et al. (2011) Analysis of the DNA region mediating increased thermotolerance at 58°C in Cronobacter sp. and other enterobacterial strains. Antonie Van Leeuwenhoek. 100: 279–89. [DOI] [PubMed] [Google Scholar]
- 43. Hurrell E, Kucerova E, Loughlin M, Caubilla-Barron J, Hilton A, et al. (2009) Neonatal enteral feeding tubes as loci for colonisation by members of the Enterobacteriaceae. BMC Infect Dis 9: 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hurrell E, Kucerova E, Loughlin M, Caubilla-Barron J, Forsythe SJ (2009) Biofilm formation on enteral feeding tubes by Cronobacter sakazakii, Salmonella serovars and other Enterobacteriaceae. Intl J Food Microbiol 136: 227–231. [DOI] [PubMed] [Google Scholar]
- 45. Maclean LL, Pagotto F, Farber JM, Perry MB (2009a) Structure of the antigenic repeating pentasaccharide unit of the LPS O-polysaccharide of Cronobacter sakazakii implicated in the Tennessee outbreak. Biochem Cell Biol (Biochimie et Biologie Cellulaire) 87: 459–465. [DOI] [PubMed] [Google Scholar]
- 46. MacLean LL, Vinogradov E, Pagotto F, Farber JM, Perry MB (2009b) Characterization of the O-antigen in the lipopolysaccharide of Cronobacter (Enterobacter) malonaticus 3267. Biochem Cell Biol 87: 927–32. [DOI] [PubMed] [Google Scholar]
- 47. Czerwicka M, Forsythe SJ, Bychowska A, Dziadziuszko H, Kunikowska D, et al. (2010) Chemical structure of the O-polysaccharide isolated from Cronobacter sakazakii 767. Carbohydr Res 345: 908–913. [DOI] [PubMed] [Google Scholar]
- 48. Maclean LL, Pagotto F, Farber JM, Perry MB (2009c) The structure of the O-antigen in the endotoxin of the emerging food pathogen Cronobacter (Enterobacter) muytjensii strain 3270. Carbohydr Res 344: 667–671. [DOI] [PubMed] [Google Scholar]
- 49. Mullane N, O'Gaora P, Nally JE, Iversen C, Whyte P, et al. (2008) Molecular analysis of the Enterobacter sakazakii O-antigen gene locus. Appl Environ Microbiol 74: 3783–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Jarvis KG, Grim CJ, Franco AA, Gopinath G, Sathyamoorthy V, et al. (2011) Molecular characterization of Cronobacter lipopolysaccharide O-antigen gene clusters and development of serotype-specific PCR assays. Appl Environ Microbiol 77: 4017–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sun Y, Wang M, Liu H, Wang J, He X, et al. (2011) Development of an O-antigen serotyping scheme for Cronobacter sakazakii . Appl Environ Microbiol 77: 2209–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sun Y, Wang M, Wang Q, Cao B, He X, et al. (2012) Genetic Analysis of the Cronobacter sakazakii O4 to O7 O-antigen gene clusters and development of a PCR Assay for identification of all C. sakazakii O serotypes. Appl Environ Microbiol 78: 3966–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arbatsky NP, Sun Y, Shashkov AS, Wang M, Liu B, et al. (2012) Structure and genetics of the O-antigen of Cronobacter sakazakii G2726 (serotype O3) closely related to the O-antigen of C. muytjensii 3270. Carbohydr Res. 355: 50–55. [DOI] [PubMed] [Google Scholar]
- 54. Allison GE, Verma NK (2000) Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri . Trends Microbiol 18: 17–23. [DOI] [PubMed] [Google Scholar]
- 55. Iversen C, Druggan P, Forsythe SJ (2004) A selective differential medium for Enterobacter sakazakii . Intl J Food Microbiol 96: 133–139. [DOI] [PubMed] [Google Scholar]
- 56. Bardowski J, Ehrlich SD, Chopin A (1994) BglR protein, which belongs to the BglG family of transcriptional antiterminators, is involved in beta-glucoside utilization in Lactococcus lactis . J Bacteriol 176: 5681–5685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gilmour MW, Thompson NR, Sanders M, Parkhill J, Taylor D (2004) The complete nucleotide sequence of the resistance plasmid R478: defining the backbone components of incompatibility group H conjugative plasmids through comparative genomics. Plasmid 52: 182–202. [DOI] [PubMed] [Google Scholar]
- 58. Nucci S-P, Bäumle AJ (2007) Evolution of the chaperone/usher assembly pathway. Fibrial classification goes Greek. Microbiol Mol Biol Rev 71: 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Teng C-H, Cai M, Shin S, Zie Y, Kim K-J, et al. (2005) Escherichia coli K1 RS218 interacts with human brain microvascular endothelial cells via type 1 fimbria bacteria in the fimbriated state. Infect Immun 73: 2923–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mattick JS (2002) Type IV pili and twitching motility. Ann Rev Microbiol 56: 289–314. [DOI] [PubMed] [Google Scholar]
- 61. Zhou Y, Tao J, Yu H, Ni J, Zeng L, et al. (2012) Hcp Family proteins secreted via the Type VI secretion system coordinately regulate Escherichia coli K1 interaction with human brain microvascular endothelial cells. Infect Immun. 80: 1243–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Almagro-Moreno S, Boyd EF (2009) Insights into the evolution of sialic acid catabolism among bacteria. BMC Evol Biol 9: 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zerbino DR, Birney E (2008) Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kurtz S, Phillippy A, Delcher AL, Smoot M, Shumway M, et al. (2004) Versatile and open software for comparing large genomes. Genome Biol 5: R12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. den Bakker HC, Cummings CA, Ferreira V, Vatta P, Orsi RH, et al. (2010) Comparative genomics of the bacterial genus Listeria: Genome evolution is characterized by limited gene acquisition and limited gene loss. BMC Genomics 11: 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kent WJ (2002) BLAT-the BLAST-like alignment tool. Genome Res 12: 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Li L, Stoeckert CJ Jr, Roos DS (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res 13: 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic acids research 32: 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Stamatakis A, Hoover P, Rougemont J (2008) A rapid bootstrap algorithm for the RAxML Web servers. Systematic biology 57: 758–771. [DOI] [PubMed] [Google Scholar]
- 70. Felsenstein J (1989) PHYLIP - Phylogeny Inference Package (Version 3.2). Cladistics 5: 164–166. [Google Scholar]
- 71. Gelman A, Rubin DB (1992) Inference from iterative simulation using multiple sequences. Statistical science 7: 457–472. [Google Scholar]
- 72. Suyama M, Torrents D, Bork P (2006) PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic acids research 34: W609–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Korber B (2000) HIV Signature and Sequence Variation Analysis. In: Allen G. Rodrigo GHL, editor. Computational Analysis of HIV Molecular Sequences: Kluwer Academic Publishers. 55–72.
- 74. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, et al. (2005) Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Tamura K, Peterson D, Peterson N, Stecher G, Nei M, et al. (2011) MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol 28: 2731–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kimura M (1980) A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J Mol Evol 16: 111–120. [DOI] [PubMed] [Google Scholar]
- 78. Felsenstein J (1981) Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol 17: 368–376. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
BLAST Ring Image Generator (BRIG) analysis of the Cronobacter genomes.
(DOC)
BLAST Ring Image Generator (BRIG) analysis of Cronobacter plasmid pESA3 with matching sequence content found in other Cronobacter species.
(DOC)
BLAST Ring Image Generator (BRIG) analysis of Cronobacter plasmid pCTU1 to matching content in other Cronobacter species.
(DOC)
Number of putative prophage regions in the Cronobacter genomes.
(DOC)
Presence (+) or absence (−) of type six secretion systems in Cronobacter spp.
(DOC)
Copy number variation of the hemolysin gene within the Cronobacter genus.
(DOC)
Additional regions of variation between Cronobacter genomes.
(DOC)