

This review discusses how crizotinib has changed the paradigm of future drug development for targeted therapies by targeting a molecular-defined subtype of non-small cell lung cancer and affected the practice of personalized medicine in oncology.
Keywords: Crizotinib, ALK inhibitor, MET inhibitor, ROS1 inhibitor, Chromosomal rearrangements, Receptor tyrosine kinase fusion positive malignancies, Non-small cell lung cancer (NSCLC)
Abstract
Crizotinib, an ALK/MET/ROS1 inhibitor, was approved by the U.S. Food and Drug Administration for the treatment of anaplastic lymphoma kinase (ALK)-rearranged non-small cell lung cancer (NSCLC) in August 2011, merely 4 years after the first publication of ALK-rearranged NSCLC. The crizotinib approval was accompanied by the simultaneous approval of an ALK companion diagnostic fluorescent in situ hybridization assay for the detection of ALK-rearranged NSCLC. Crizotinib continued to be developed as an ALK and MET inhibitor in other tumor types driven by alteration in ALK and MET. Crizotinib has recently been shown to be an effective ROS1 inhibitor in ROS1-rearranged NSCLC, with potential future clinical applications in ROS1-rearranged tumors. Here we summarize the heterogeneity within the ALK- and ROS1-rearranged molecular subtypes of NSCLC. We review the past and future clinical development of crizotinib for ALK-rearranged NSCLC and the diagnostic assays to detect ALK-rearranged NSCLC. We highlight how the success of crizotinib has changed the paradigm of future drug development for targeted therapies by targeting a molecular-defined subtype of NSCLC despite its rarity and affected the practice of personalized medicine in oncology, emphasizing close collaboration between clinical oncologists, pathologists, and translational scientists.
Introduction
The era of molecularly targeted therapy in lung cancer began in 2004 with the discovery that activating epidermal growth factor receptor (EGFR) mutations in non-small cell lung cancer (NSCLC) correlated with clinical response to EGFR tyrosine kinase inhibitors (TKIs) [1–3], although EGFR TKIs were developed and approved for clinical use prior to the knowledge of these activating EGFR mutations [4, 5]. Subsequently, five randomized trials in patients with NSCLC with activating EGFR mutations have demonstrated statistically significant superior response rates and progression-free survival for EGFR TKIs compared with standard doublet chemotherapy [6–10]. Activating EGFR mutations provide a partial molecular explanation for the observation that NSCLC in never-smokers is considered a distinct disease with a high proportion of Asian female adenocarcinoma patients who have a better survival outcome than that of smokers [11, 12]. This observation also provided the impetus for the identification of other driver mutations in NSCLC. In the Iressa Pan-Asia Study (IPASS) [13, 14] and First-SIGNAL [15] trials, despite the enrichment of Asian female never-smokers with adenocarcinoma and the employment of sophisticated sequencing techniques, only approximately 60% and 44% of patients in each study, respectively, were found to carry activating EGFR mutations, indicating that other potential driver mutations remain to be discovered [16].
Discovery of Anaplastic Lymphoma Kinase Rearrangement in the Pathogenesis of NSCLC
ALK rearrangements were identified in NSCLC in 2007 by two independent groups. Soda et al. developed retroviral-based cDNA expression libraries to screen for novel oncogenes [17, 18]. They transfected a cDNA library derived from a lung adenocarcinoma from a 62-year-old Japanese male smoker who was prescreened to be negative for KRAS and EGFR mutations [17]. They identified an echinoderm microtubule associated protein-like 4 (EML4)-anaplastic lymphoma kinase (EML4-ALK) fusion transcript that possessed transforming activity in 3T3 cells [17]. 3T3 cells transfected with EML4-ALK and implanted in nude mice resulted in rapid tumor growth [17]; an ALK inhibitor inhibited the growth of BA/F3 cells transfected with EML4-ALK. Finally, a preliminary survey of a panel of 33 NSCLC tumors revealed that EML4-ALK rearrangement occurs independently of EGFR and KRAS mutations [17]. To further demonstrate the role of EML4-ALK in the pathogenesis of NSCLC, Soda et al. generated transgenic mice engineered to specifically express EML4-ALK in lung alveolar cells, which resulted in hundreds of lung adenocarcinoma nodules. Treatment of these transgenic mice with an ALK inhibitor resulted in reduced tumor burden compared with untreated mice. Intravenous injection of EML4-ALK/3T3 cells resulted in massive infiltration of EML4-ALK/3T3 cells in the lungs of nude mice and rapid death of the majority of the mice within 1 month [18]. Treatment with the same ALK inhibitor resulted in the absence of EML4-ALK/3T3 cells in the lung and prolonged survival [18]. In summary, Soda et al. convincingly demonstrated that EML4-ALK is a unique driver mutation in NSCLC and that inhibition of EML4-ALK activity in vivo led to the reduction of lung cancer burden.
Contemporaneously, Rikova et al. characterized the phosphotyrosine profile in 191 NSCLC cell lines and tumor samples using a phosphoproteomic approach to identify “driver kinases” in NSCLC [19]. They identified a high level of ALK phosphorylation in several NSCLC tumor samples and H2228 NSCLC cells. Rapid amplification of the 5′ complementary DNA ends (5′ RACE) of the RNA transcripts isolated from these samples with highly phosphorylated ALK revealed the EML4-ALK fusion transcript [19]. No mutations were found in the ALK kinase domain [19]. Thus, two groups using two different approaches independently identified ALK translocation, the first of its kind in a common solid malignancy.
The Normal Physiological Role of ALK and Signal Transduction Pathways Activated by EML4-ALK
The full-length ALK cDNA contains 29 exons. The full-length ALK protein contains 1,620 amino acids, with a predicted molecular weight of 177 kilodaltons (kDa) [20]. The 254-amino acid kinase domain comprises amino acid residues 1123–1376 preceded by a short transmembrane region of amino acids 1031–1058 [20]. ALK is temporally and spatially expressed during development of the murine neonatal central nervous system with its expression highest in the neonatal brain, and is not expressed in any non-neural tissues during any stage of development of the mouse [20, 21]. ALK was so named when it was discovered to be translocated in anaplastic large cell lymphoma (ALCL) [22]. Subsequently, ALK rearrangement with various fusion partners has been discovered in diffuse large B-cell lymphoma and inflammatory myofibroblastic tumor (IMT) prior to the discovery of ALK rearrangement in NSCLC. All three of the major signaling pathways (PI3K-AKT, RAS-ERK, JAK-STAT3) have been identified as being engaged by the various ALK fusion proteins [23, 24].
Much less is known about the normal function of the native ALK protein. Based on its expression pattern [21, 22], ALK is believed to be involved in early neurogenesis. In Drosophila, the ligand for ALK is jelly belly, but no human homolog of jelly belly has been identified [25]. Instead, there are two known ALK ligands: pleiotrophin [26] and midkine [27]. Both are polypeptide nerve growth factors that can bind to other receptors besides ALK. Binding of pleiotrophin to ALK led to both MAPK- [28] and phosphatidylinositol 3-kinase (PI3K) mediated cell growth and MAPK-mediated antiapoptotic signals [28, 29]. The MAPK signaling pathway, especially MEK, is important for the promotion of cell growth and neurite outgrowth of the SK-N-SH neuroblastoma cell line [30].
ALK is also negatively regulated by receptor protein tyrosine phosphatase (RPTP) β/ζ, which dephosphorylates ALK. RPTP β/ζ is also a receptor for pleiotrophin and is inactivated by pleiotrophin upon binding to RPTP β/ζ, allowing the continual autophosphorylation of ALK [30]. Thus, pleiotrophin can activate ALK both directly and indirectly. Expression of midkine during central nervous development correlates with the expression of ALK [31]. Midkine activates ALK, resulting in increased N-myc and trkB expression and the proliferation of immature sympathetic neurons. Midkine also (through a different pathway) increases the expression of a transcription factor, Hand2, that results in the upregulation of ALK expression, resulting in a positive autoregulatory pathway [31].
ALK has recently been recognized as a “dependence receptor,” where in its inactive form (without ligand binding) it is proapoptotic, whereas ALK is antiapoptotic in its active form (with ligand binding or aberrantly translocated in malignancies) [32, 33]. ALK can enhance apoptosis following caspase-mediated cleavage at position D1160 in the ALK juxtamembrane region, disrupting the ALK kinase domain and exposing a proapoptotic region encompassing amino acids 1090–1125 in the intracellular juxtamembrane domain [33]. Activation/phosphorylation prevents caspase-mediated cleavage, resulting in an autoregulatory cycle. Overexpression of ALK led to apoptosis of primary cortical neural cultures and neuroblasts [33]. ALK knockout mice are viable and exhibited no phenotypic abnormalities. They do exhibit an age-dependent increase in basal hippocampal neurogenesis consistent with its normal role in proapoptotic enhancement and the restriction of ALK expression largely to the central nervous system [34]. Consistent with the role of ALK in neurogenesis, activating mutations in ALK lead to both familial [35] and sporadic neuroblastoma [36–38], and provide the rationale for an ALK inhibitor in the treatment of neuroblastoma [37].
One of the major differences between the signaling pathways engaged by native ALK and EML4-ALK is the difference in the subcellular location. Native ALK is transmembrane and generally not activated, whereas EML4-ALK is cytoplasmic in location and in a constitutively activated form. EML4-ALK had been shown to engage all three major signaling pathways involved in receptor tyrosine kinases (RTKs): MAPK/MEK/ERK, PI3K/AKT, and RAS/STAT3 [39–41], but it remains relatively unclear which pathway(s) are critical to pathogenesis of NSCLC by EML4-ALK. Takezawa et al. recently demonstrated that both the MAP kinase (MEK/ERK) and JAK/STAT3 pathways but not the PI3K/AKT pathway are engaged by EML4-ALK to induce apoptosis. Cell lines with stably transfected EML4-ALK variants 1 and 3 had markedly increased phosphorylation of MEK, ERK, and STAT3 but not AKT [42].
Inhibition of EML4-ALK with TAE684, a small-molecule ALK inhibitor, led to decreased phosphorylation of ERK and increased levels of BIM, a pro-apoptotic protein. BIM is a member of the BCL-2 family of proteins and is degraded via phosphorylation by ERK. It has been recently shown that the level of BIM corresponds to the ability of tyrosine kinase inhibitors to induce apoptosis in various cell lines harboring driver mutations with the higher levels BIM associated with higher levels of apoptosis. Simultaneously and by an independent mechanism, TAE684 led to decreased phosphorylation of STAT3 and reduced levels of survivin [42]. Survivin directly and indirectly inhibits caspases, leading to apoptosis. Thus, EML4-ALK in NSCLC results in activation of ERK and STAT3, which leads to decreased BIM and increased survivin, allowing for the synergistic effects of antiapoptosis and promotion of tumor growth (Fig. 1).
Figure 1.

Signaling pathways transduced by EML4–ALK.
Abbreviations: ALK, anaplastic lymphoma kinase; EGFR, epidermal growth factor receptor.
Clinicopathologic Characteristics of in Patients with ALK-Rearranged NSCLC
Many retrospective tissue bank studies have demonstrated the incidence of ALK-rearranged NSCLC to be approximately 3%–5% in NSCLC, with no apparent difference in incidence by ethnicity. Patients with ALK-rearranged NSCLC tend to be young (approximately 50 years of age at diagnosis) and never-smokers (∼70%–75%) or light smokers. The vast majority present with adenocarcinoma and there is no sex preference for males or females. Compared to patients with EGFR mutations, ALK-positive patients had a younger median age of diagnosis (61 vs. 57 years old, respectively) and a higher proportion of never-smokers or light smokers (<100 cigarettes per lifetime; 48% vs. 67%) [43]. The vast majority of ALK-rearranged NSCLCs possess wild-type EGFR and KRAS genes [44, 45], although coexisting ALK rearrangements and other driver mutations have been described (Table 1) [46–56].
Table 1.
Patients with simultaneous ALK and EGFR rearrangements reported in the literature

Abbreviations: FISH, fluorescent in situ hybridization; IHC, immunohistochemistry; NA, not applicable; NR, not reported; NSCLC, non-small cell lung cancer; PD, progressive disease; PR, partial response; pt, patient; RT-PCR, reverse-transcriptase polymerase chain reaction; TKI, tyrosine kinase inhibitor.
Several large prospective molecular profiling epidemiology studies (e.g., the Lung Cancer Mutation Consortium in the USA [47], the Institut National Du Cancer in France, and LUNGSCAPE in Europe [57]) will provide data on the incidence and clinical characteristics of ALK rearrangement as groups begin to report their findings. Most recently, the Lung Cancer Mutation Consortium reported that, among 901 patients using ALK breakapart assay, ALK positivity was 8.3% in adenocarinoma in a clinically enriched population (younger, with more women and never smokers) [58]. In addition, Li et al. reported a rate of 3.2% of ALK-positivity after large-scale screening 4,500 patients with NSCLC formalin-fixed paraffin-embedded (FFPE) tissue using multiplex reverse transcriptase-polymerase chain reaction (RT-PCR) assays that included nine known EML4-ALK fusion transcripts and ALK RNA levels [59].
Many histologic features have been ascribed to ALK-rearranged NSCLC, including acinar [60, 61], papillary [60, 62–63], micropapillary [63], bronchioalveolar [60, 62–63], and the presence of signet ring cells [64–66]. Yoshida et al. performed an extensive histology review of 54 ALK-rearranged NSCLC resections and identified two major and different histologic patterns: a solid growth pattern with focal signet ring cell component and a mucinous cribriform pattern associated with significant extracellular mucus materials [67]. Several studies have shown that signet ring cell differentiation is a common specific feature of ALK-positive NSCLC within up to two-thirds of tumors exhibiting this morphology [64–67]. Interestingly, “mucinous cribiform” component has recently been shown to be much more common in receptor tyrosine kinase fusion-positive NSCLC and may be used as another feature to draw attention to the potential presence of ALK rearrangement [68]. However, none of the documented histologic features were sufficiently sensitive or specific to predict for ALK rearrangement [69]. Likewise, although lymphovascular invasion and necrosis, TTF-1 expression, and p63 expression (generally considered a “squamous cell” marker but found to be expressed in ALK-positive adenocarcinoma) were all found significantly more often in ALK-positive than in ALK-negative samples, such features should not be used to replace current diagnostic tests for ALK rearrangement [67].
There are now at least 27 different ALK fusion variants in NSCLC reported in the literature: 21 EML4-ALK isoforms (including one with fusion to exon 19 of ALK) [17, 39, 63, 66, 70–75, 75a, 75b], 3 KIF5B-ALK isoforms [68, 75, 75a, 76], 1 KCL1-ALK isoform [77], 1 TFG-ALK isoform [19] and 1 ALK-PTPN3 isoform [17, 19, 39, 63, 66, 68, 70–75, 75a, 76–78] (Fig. 2A, Table 2). The distribution of EML4-ALK variants has been previously reported [89] and we have performed an updated analysis based on a total of 371 published EML4-ALK variants. Variant 1 (49.6%) made up the most common type of the EML4-ALK fusion transcripts, followed by variant 3a/b (25.6%) and variant 2 (10%) [17, 39, 41, 49, 61, 63, 66, 71–75, 75a, 79, 80, 90–94] (Fig. 2B).
Figure 2.

List of ALK fusion variants in NSCLC and proportion of various of EML4-ALK fusion variants. (A): Schematic of various forms of ALK fusions in ALK-rearranged NSCLC [17, 19, 39, 63, 66, 68, 70–78, 75a]. Adapted from Sasaki T, Rodig SJ, Chirieac LR et al. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer 2010;46:1773–1780, with permission. (B): Pie chart of distribution of EML4–ALK various fusions [17, 39, 41, 49, 61, 63, 66, 71–75, 75a, 79, 80, 90–94].
Table 2.
List of ALK fusion variants in NSCLC reported in the literature

Only the reference that first reported the variant is cited here.
aThese variants have not been officially named.
bVariant 8a contains an internal stop codon within the 30 bp insertion. Variant 8a was discovered in the same sample as variant 8b.
cOnly reference that first reported the variant is cited here.
Despite these various EML4-ALK fusion transcripts in NSCLC, Li et al. did not find any significant difference in the median age of diagnosis and gender composition among variants V1–5a in 152 patients with ALK-rearranged NSCLC [59]. However, there is in vitro evidence that various ALK fusions have different sensivitivites to ALK inhibitors and may have different intrinsic protein stability within the cell [78a], although EML4-ALK variants 1 and 3a had similar clinical outcome treated with first-line platinum-based chemotherapy [78b]. EML4-ALK fusion transcripts have been found in solid tumors other than NSCLC [79, 80]. Using a tumor repository, Lin et al. reported EML4-ALK in 2.4% of breast cancer (5/209) and colorectal cancer (2/83) [80]. Recently, EML4-ALK has also been detected in non-clear cell renal cell carcinoma using an immunohistochemical screening method [81]. The frequency of KIF5B-ALK in adenocarcinoma of the lung estimated from the three reports was 4 of 1,331 patients or approximately 0.3% [68, 75, 76]. It is important to note that the three fusion partners to ALK—EML4, KIF5B, and KLC1—all interact with the microtubule apparatus [82, 86]. Therefore, agents that interrupt the assembly of microtubules such as taxanes or vinca alkaloids may be useful in the treatment of ALK-rearranged NSCLC alone and/or in combination with an ALK inhibitor.
Finally, an ALK-PTPN3 variant has been observed from the insertion of genomic DNA encompassing exons 10 and 11 of ALK into the intronic region between exons 2 and 3 of protein tyrosine phosphatase, nonreceptor type 3 gene (PTPN3) [78]. Of note, ALK-PTPN3 can theoretically generate a split-apart fluorescent in situ hybridization (FISH) signal but will not respond to crizotinib as the ALK kinase domain is absent.
The clinical presentation and course of patients with ALK-rearranged NSCLC have not been extensively described. One study reported that patients with ALK-rearranged NSCLC tended to present with a higher frequency of pericardial and pleural-based disease, an increased propensity for liver but not brain metastases, and an increased number of sites of metastasis compared with EGFR+ and ALK−/EGFR− patients [46]. Similarly of the 901 patients with stage IIIB/IV adenocarcinoma profiled to date for ALK rearrangement by the Lung Cancer Mutation Consortium, ALK-positive disease was significantly associated with liver metastasis compared with ALK-negative disease (23% vs. 10%; p = .004), but no difference was detected in bone, brain, or adrenal gland metastases [58].
Patients with ALK-rearranged NSCLC may also present with uncommon sites of metastasis such as retinal metastasis [46]. In a retrospective study, Shaw et al. have demonstrated among contemporaneously diagnosed patients with ALK-rearranged NSCLC that those treated with crizotinib had a significant survival advantage over those who did not receive crizotinib (Table 3) [95]. In fact, crizotinib-treated patients with ALK-rearranged NSCLC achieved excellent survival outcomes comparable with EGFR mutation-positive patients who received EGFR TKIs (Table 4) [95]. Taken together, these studies indicate that it is important to diagnose patients with ALK-rearranged NSCLC early in the treatment course to initiate effective antitumor treatment.
Table 3.
Comparison of overall survival of patients with ALK-rearranged NSCLC treated with crizotinib as second-line or third-line treatment versus crizotinib-naïve patients with ALK-rearranged NSCLC [95]

Abbreviations: CI, confidence interval; NR, not reached.
Table 4.
Comparison of overall survival of crizotinib-treated patients with ALK-rearranged NSCLC and tyrosine kinase inhibitor-treated patients with EGFR-mutated NSCLC [95]

Abbreviations: CI, confidence interval; NR, not reached; TKI, tyrosine kinase inhibitor.
Recently, several retrospective studies have shown that pemetrexed-based chemotherapy may confer high tumor response either as first-line combination with platinum [96], as single agent in second-line or beyond treatment [97, 98] or longer progression-free survival when first-line platinum/pemetrexed combination is used in patients with ALK-rearranged NSCLC [98a]. However, these analyses may have confounded by some of the clinical demographics of the ALK-positive population such as younger age and/or never-smoking status [98a]. On the other hand, a larger retrospective analysis of treatment histories from patients enrolled in the PROFILE 1005 study, which is a global phase II single arm study of crizotinib in patients with ALK-rearranged NSCLC, showed that among the 711 patients who had received pemetrexed as combination therapy or as a single agent, overall response rate (ORR) and time-to-progression outcomes were similar to unselected patients with NSCLC who were enrolled in phase III randomized trials comparing the use of first-line platinum/pemetrexed or second-line pemetrexed and treated with pemetrexed [99]. It will be important to complete the enrollment and await the results of the randomized trials comparing crizotinib to platinum pemetrexed-based chemotherapy in the first-line treatment setting (PROFILE 1014) or to standard second-line chemotherapy (pemetrexed or docetaxel; PROFILE 1007), which was recently reported to meet its primary endpoint [100].
The prognostic significance of ALK rearrangement in NSCLC has not been settled. In two separate reports [65, 95], Shaw et al. did not demonstrate any significant differences in overall survival (OS) for patients with NSCLC by EML4-ALK status in the era before crizotinib. Similarly, Zhang et al. reported no survival difference according to ALK status after adjusting for disease stage, histology, and EGFR/KRAS mutant status [101]. Lee et al. showed that patients with ALK-rearranged NSCLC had the shortest overall survival compared to wild-type patients, but the difference was not significant [102]. On the other hand, Kim et al. were able to demonstrate that patients with ALK-rearranged NSCLC had a significant worse OS outcomes after factoring in age, sex, histology, stage, and performance status [103]. Similarly, Yang et al. found that EML4-ALK status is a poor prognostic factor for relapse-free survival after factoring in stage, sex, age, and treatment [104].
Conversely, Wu et al. found that patients with EML4-ALK NSCLC identified from pleural effusion cytology had a significantly improved survival outcome compared with patients without EML4-ALK NSCLC [94]. Furthermore, Takeuchi et al. found that receptor kinase fusion-positive NSCLC is an independent favorable prognostic factor after taking into consideration age, sex, stage, and smoking status [68]. All of these studies are limited by the small number of ALK-positive patients, different comparison group of patients, the heterogeneous treatment patients received, and differences in the balance of prognostic factors (e.g., smoking status, surgical treatment, age) compared with ALK-rearranged patients.
Original Design and Synthesis of Crizotinib
Crizotinib Development and Preclinical Activity
Crizotinib is a competitive ATP inhibitor against both wild-type MET and against MET ATP-binding site mutants (V1092I, H1094R), P-loop mutant (M1250T), and juxtamembrane domains mutants (R988C, T1010I), but is not active against activation loop mutants (Y1230C, Y1235D) [105]. The cellular effects from MET inhibition by crizotinib are multifold: induction of apoptosis, decrease in proliferation, and decrease in angiogenesis [106]. Crizotinib inhibited the growth of the MET-dependent GTL-16 gastric carcinoma cell line, and inhibited cell migration and invasion of the HGF-stimulated NCI-H441 lung cancer cell line and the growth of multiple human carcinoma xenograft models including gastric carcinoma (GTL-16), renal cell (Caki-1), glioblastoma (U87MG), prostate (PC-3), and NSCLC (NCI-H441) [106].
After its synthesis as a MET inhibitor [107–109] (Figures 3A and 3B [107–109]), crizotinib was evaluated against a panel of >120 kinases in biochemical assays and 12 cell-based phosphorylation assays revealed that it also inhibited phosphorylation of NPM-ALK in both Karpas 299 and SU-DHL-1 ALCL cells with a mean IC50 of 24 nmol/L [110]. Since both Karpas 299 and SU-DHL-1 cell lines express detectable MET levels, the lack of inhibitory activity of two specific MET inhibitors (PHA-665752 and PF-4217903) indicated that the inhibitory effect of crizotinib on these two cell lines was not due to its anti-MET activity. Crizotinib also resulted in dose-dependent growth inhibition of the Karpas299 xenograft in SCID-Beige mice. This inhibition was again accomplished by inhibition of NPM-ALK phosphorylation and the phosphorylation of NPM-ALK's downstream signaling mediators: PLC-γ and STAT3 over a wide range of crizotinib concentrations, and AKT and ERK kinases at a relatively high concentration of crizotinib [110].
Figure 3.

Synthesis of crizotinib. (A): Design and synthesis of crizotinib. Figure 3A provided by Jean Cui. Please refer to reference 107 for detailed synthesis of crizotinib. (B): Crystallography of crizotinib in unphosphorylated MET and ALK [108, 109]. PDB ID 2wgj for PF-02341066/c-MET complex; PDB ID 2xp2 for PF-02341066/ALK complex. Figure 3B provided Jean Cui.
Abbreviation: MW, molecular weight.
Clinical Data for Crizotinib
First In-Human Phase I Trial of Crizotinib
The first in-human crizotinib trial (PROFILE 1001, NCT00585195) was written as an open-label, multicenter trial that was activated in 2006 with an initial standard dose-escalation pharmacokinetic (PK) portion followed by a clinical efficacy portion that aimed to enroll a small number of molecularly enriched patients to assess the antitumor activity of crizotinib. The initial PK portion used the standard dose-escalation finding schema to determine the maximal tolerated dose (MTD) and the recommended phase II dose (RP2D). There were several substudies to investigate the effect of food on crizotinib PK, a midazolam substudy to assess the effect of CYP3A isozyme inhibition by crizotinib, and a study to assess the feasibility of using fluoro-L-thymidine positron emission tomography to monitor response to crizotinib as an alternative to fluorodeoxyglucose positron emission tomography [111].
Pharmacokinectics of Crizotinib
Crizotinib was first dosed at 50 mg orally once daily and eventually escalated to 300 mg orally b.i.d., at which dose two patients experienced grade 3 fatigue [112]. The dose of crizotinib was reduced to 250 mg orally b.i.d. and was found to be tolerable; this dose was determined to be the MTD and RP2D dose [112].
Eleven patients were enrolled onto the food effect substudy. The absorption of a single dose of 250 mg of crizotinib after a high-fat, high-calorie meal was 14% lower than the concentration of a single dose of 250 mg of crizotinib taken on an empty stomach [112, 113]. Thus crizotinib could be taken with or without food. The peak plasma concentration was reached 4–6 hours after a single dose of crizotinib [113]. There was linear PK from 100 mg of crizotinib once daily to 300 mg of crizotinib b.i.d. [114]. Steady-state concentration of crizotinib was reached within 15 days of repeated administration of crizotinib at 250 mg orally b.i.d., with a half-life of approximately 43–51 hours [114]. The mean steady-state trough plasma level for 250 mg crizotinib b.i.d. (the recommended phase II dose) is 274 ng/mL or 57 nM of free drug, which exceeded the target efficacy levels predicted for the inhibition of MET (∼13 nM) and ALK (∼26 nM) based on preclinical mouse models [114].
Metabolism and Drug Interactions
Thirteen patients were enrolled in the midazolam substudy. The PK of a single 2 mg midazolam dose was evaluated before and 28 days after 250 mg of crizotinib orally b.i.d.. Midazolam is metabolized by CYP3A isozymes and there was a 3.6-fold increase in the midazolam level (90% confidence interval [CI]: 2.7–4.9) after 28 days of crizotinib administration, indicating that crizotinib is a moderate CYP3A inhibitor [114]. Coadministration of strong CYP3A inhibitors or inducers should be avoided.
Ethnicity
The phase I study revealed higher exposure to crizotinib in Asian patients at steady state after crizotinib 250 mg b.i.d. than in non-Asian patients (Fig. 4) [115]. Exposure to crizotinib remained higher in Asian patients even after correction for ideal body weight, body surface area, or body mass index [115]. Recently, exposure-response analyses from both PROFILE 1001 and 1005 showed that Asian patients had higher ORR and longer progression-free survival (PFS) times, associated with higher crizotinib exposure than non-Asian patients [116].
Figure 4.

Steady-state crizotinib levels in Asians and non-Asians receiving repeated doses of crizotinib 250 mg twice daily [115].
Clinical Efficacy of Crizotinib in Patients with ALK-Rearranged NSCLC
Following the discovery of the EML4-ALK translocation in NSCLC in 2007 [17, 19] and in the context of crizotinib's ALK inhibitor activity [110], a commercially available break-apart FISH assay for detecting ALK rearrangement in ALCL was modified to detect ALK rearrangement in NSCLC. The first patient with EML4-ALK NSCLC was treated with crizotinib on the phase I dose-escalating cohort (300 mg b.i.d.) at Massachusetts General Hospital on December 26, 2007 with rapid clinical benefit, but was taken off trial due to liver enzyme elevations despite dose reduction. Six months later, the second patient with EML4-ALK NSCLC was treated with crizotinib, with clinical benefit and stable disease. Based on the clinical benefit from crizotinib for these two patients, there was a concerted shift among the phase I clinical sites to screen for NSCLC patients with ALK-rearrangement.
The clinical efficacy data for crizotinib in ALK-rearranged NSCLC patients have been remarkably consistent at multiple data cutoff dates, with an ORR of approximately 60% over the 4 years since the first report from 19 patients in 2009 (Table 5) [90, 112, 117–118]. The updated estimated PFS is stable at approximately 9.7 months [118]. The clinicopathologic characteristics of patients with ALK-rearranged NSCLC enrolled onto the study remained remarkably constant with a median age of 51, although patients as young as 21 and as old as 86 were enrolled again underlying the heterogeneous population of patients with NSCLC harboring ALK rearrangement. Only ∼70% of these patients were never- or light-smokers. The vast majority of patients with ALK-rearranged NSCLC presented with adenocarcinoma (Table 5).
Table 5.
Summary of clinicopathologic characteristics of patients with ALK-rearranged NSCLC and clinical activity of crizotinib in patients enrolled in the first in-human trial (PROFILE 1001) over a period of 4 years

aMean.
Abbreviations: CI, confidence interval; DCR, disease control rate; ECOG PS, Eastern Cooperative Oncology Group performance status; NR, not reported; PFS, progression-free survival; ORR, overall response rate; SD, stable disease.
An ongoing single-arm, global phase II study of crizotinib was launched in 2009 (PROFILE 1005, NCT00932451) based on the initial data from the phase I study, with central diagnosis of ALK rearrangement using the Vysis FISH test (Abbott Molecular, Abbott Park, IL; at the time also under evaluation by the U.S. Food and Drug Administration [FDA]) for the first 250 patients enrolled. (The protocol was subsequently amended to allow NSCLC patients with any ALK-positive test performed outside the central laboratory to be enrolled on a case-by-case basis.) This trial also provides an avenue for patients with ALK-rearranged NSCLC randomized to the control chemotherapy arm of the second-line phase III PROFILE 1007 (NCT00932893) trial to access crizotinib if they experience progression.
Data from PROFILE 1005 showed that patients had very similar clinicopathologic characteristics (Table 6) to those enrolled in PROFILE 1001, and the first 261 patients enrolled (whose ALK positivity was centrally confirmed by FISH) achieved a response rate of 60% (95% CI: 54%–66%) with a median PFS of 8.1 months (95% CI: 6.8–9.7) and median duration of response of 46 weeks [119]. In addition, best overall response appeared to be independent of the percentage of cells positive for ALK rearrangement by FISH in the diagnosis sample [120]. ORR was independent of age, sex, and number of prior metastatic treatment regimens. Responses were achieved rapidly, with approximately half of the observed responses occurring at the time of first follow-up scan: within 8 weeks (PROFILE 1001) and 6 weeks (PROFILE 1005) [118, 119]).
Table 6.
Clinicopathologic characteristics of patients with ALK-rearranged NSCLC enrolled in the PROFILE 1005 trial [119]

Data are n (%) unless noted.
aThree patients were not eligible due to prior adjuvant treatment only.
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; FISH, fluorescense in situ hybridization.
Adverse Events with Crizotinib
As of January 2012, more than 1,000 patients had been treated in PROFILE 1001 and PROFILE 1005, and the safety profile for crizotinib has been consistent between these studies [118, 119]. Crizotinib was generally well tolerated with the majority of adverse events being grade 1 or 2, and low rates of withdrawal due to adverse events. The most commonly reported treatment-emergent, all-causality adverse events were nausea, diarrhea, vomiting, constipation, and visual effects [113]. Other events reported at frequencies ≥20% in one or both studies included peripheral edema, dizziness, fatigue, and decreased appetite [113]. Visual effects with crizotinib are particularly noteworthy as such events are not commonly associated with cancer therapies; however, no patient required dosing interruption, dose reduction, or permanent discontinuation of crizotinib treatment because of visual effects. Patient-reported symptom data reported from patients enrolled on PROFILE 1005 showed that visual events were short-lived (the majority of events lasted <1 minute), occurred less frequently as treatment continues, and were not bothersome, with no effect on patient's activities of daily living [119]. Further detail will be elucidated as these data mature and more patients are included. Based on data from ophthalmologic examinations performed at baseline and post-treatment in clinical trials, no clinically meaningful changes in visual acuity, biomicroscopy, or ophthalmoscopy in patients experiencing visual disturbances occurred [121].
Hepatic and Pulmonary Adverse Events with Crizotinib
Transaminase elevations have been noted with crizotinib, generally occurring within the first 2 months of treatment, with a median onset of 40 days for any grade ALT elevation. Among 1,054 patients treated with crizotinib in both PROFILE 1001 and PROFILE 1005, ALT elevations of greater than 3, 5, and 10 times the upper limit of normal occurred in 15%, 7.4%, and 3% of patients, respectively. The incidences of alanine aminotransferase (ALT), aspartate aminotransferase, and alkaline phosphatase (AP) elevation were comparable for grade 1 and grade 2 events, but grade 3 and grade 4 events were more frequent for ALT elevations. Total bilirubin elevation was less common, suggesting that hepatic function is less affected in most cases of liver injury.
Temporary discontinuation or dose reduction occurred in 5.3% of all patients, and permanent discontinuation rate due to hepatic adverse events was 1.3%. Transaminase elevations were usually reversible and patients were able to resume the treatment at the same or at a lower dose. Fatal drug-induced hepatotoxicity occurred in <1% of patients in these studies [122]. Concurrent elevations in ALT greater than 3 times the upper limit of normal and total bilirubin greater than 2 times the upper limit of normal, with normal AP, occurred in less than 1% of patients in clinical trials. Liver function test monitoring is recommended, including ALT and total bilirubin once a month and as clinically indicated, with more frequent repeat testing for increased liver transaminases, AP, or total bilirubin in patients who develop transaminase elevations [113].
Crizotinib has been associated with severe, life-threatening, or fatal treatment-related pneumonitis/interstitial lung disease (ILD). Four cases among 255 patients (1.6%) in PROFILE 1005 and PROFILE 1001 have been reported [113]. Patients should therefore be monitored for pulmonary symptoms indicative of pneumonitis/ILD in addition to NSCLC progression. Other causes of pulmonary symptoms, such as other pulmonary disease, infection, or radiation effects, should be excluded [113].
Endocrine Adverse Events
Recently, the University of Colorado reported that 13 men with NSCLC experienced a rapid decrease in total testosterone levels after initiating crizotinib potentially via a hypothalamic or pituitary effect, which may account for some of the fatigue experience by some patients [123]. Testosterone promptly returned to the normal level after crizotinib discontinuation. However, hypogonadism is multifactorial in its etiology in advanced lung cancer. Further investigation in an appropriate control setting will provide a more comprehensive description and understanding of this phenomenon.
Specific Clinical Situations (Treatment Beyond Progression/Brain Metastasis)
Both the PROFILE 1001 and 1005 protocols allow crizotinib to be continued if the investigators deem the patient will continue to derive “continual clinical benefit.” Of 138 patients who continued on crizotinib for >2 weeks postprogression on PROFILE 1001 and PROFILE 1005, a total of 53 patients (46%) had progression in the brain as a new lesion or nontarget lesion [125]. Among the 42 patients who received more than 6 months treatment postprogression, half had brain metastases and some patients had received 40 weeks of postprogression treatment, with treatment still ongoing [125]. The level of crizotinib achieved in cerebrospinal fluid (CSF) has been reported in one patient who received crizotinib 250 mg orally b.i.d. [124]. The CSF level was only 0.26% of the plasma level (0.616 ng/mL; 0.0014 μmol/L), a level expected to be insufficient to inhibit ALK. Thus, progression in the brain alone likely reflect a “santuary site” for crizotinib rather than true resistance to crizotinib given many patients continued to be on crizotinib after progression in the brain.
Additionally, PROFILE 1001 and PROFILE 1005 protocols empirically specify that crizotinib be withheld the day before, during, and the day after any radiation treatment, allowing patients who received definitive radiation to the brain to continue crizotinib without undue interruption. This stratey has been successfully employed by the University of Colorado to provide additional 6 months of PFS for selected patients with ALK-rearranged NSCLC who presented with oligoprogressive disease [126].
FDA Approval of Crizotinib and the Break-Apart FISH Assay
The FDA agreed to accept clinical efficacy and safety data of crizotinib from the PROFILE 1001 and PROFILE 1005 trials as the basis for the new drug application (NDA) filing in April 2010. Crizotinib was granted orphan drug designation for ALK-positive NSCLC in September 2010. Fast-track designation for crizotinib was granted in December 2010 and review of the NDA on a rolling basis began in January 2011.
The break-apart FISH assay kit manufactured by Abbott Molecular was required to satisfy two criteria: an analytical component and clinical utility. The exclusive use of the Abbott kit in PROFILE 1005 satisfied the clinical utility requirement based on response rates in that study [115]. For the analytical component, Abbott Molecular and the Department of Pathology at Massachusetts General Hospital (which served as the central laboratory for PROFILE 1001) simultaneously and independently re-evaluated the patients enrolled in PROFILE 1001 using the Abbott Vysis probes with excellent concordance (>90%). The Abbott Molecular break-apart FISH testing regulatory package was submitted in May 2011, completing the regulatory submission package to the FDA.
Crizotinib was approved in the USA on August 26, 2011, primarily based on response rates of 50% from the first 136 patients with ALK-rearranged NSCLC enrolled on PROFILE 1005 [127] and secondarily on a response rate of 61% from the first 119 patients with ALK-rearranged NSCLC enrolled on PROFILE 1001 [117]. The approval language states: “Xalkori is a kinase inhibitor indicated for the treatment of patients with locally advanced or metastatic non-small cell lung cancer (NSCLC) that is anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test. This indication is based on response rate. There are no data available demonstrating improvement in patient reported outcomes or survival with Xalkori” [113], with no restriction as to which line of treatment crizotinib could be used to treat patients with ALK-positive NSCLC. Crizotinib is now also approved in many countries, including Argentina, Canada, Israel, Japan, Korea, Macau, Mexico, and Switzerland and imminently by the European Union.
What Is the Best Companion Diagnostic Test for Detecting ALK Rearrangements?
Break-apart FISH Assay as the “Gold Standard”
Molecularly targeted therapy in oncology is critically dependent on a validated test to detect the molecular alteration in question, especially when molecular alterations constitute a small subgroup of patients. Ideally, the test should be both highly sensitive and highly specific, relatively inexpensive, and feasible in most diagnostic laboratories to facilitate worldwide adoption. FISH is routinely used in oncology to detect chromosomal translocations which play an important role in soft tissue and hematologic malignancies, including the diagnosis of ALK-positive ALCL. Until recently, chromosomal translocations were considered uncommon in epithelial tumors [128]. The same ALK break-apart FISH assay used for ALCL was the basis for the ALK diagnostic test eventually approved by the FDA to detect ALK-rearranged NSCLC in conjunction with the approval of crizotinib in the USA [113].
The performance of the FISH assay has been rigorously evaluated and shown to be effective in analyzing FFPE NSCLC specimens. Technically, ALK FISH can be more challenging than most break-apart assays, because ALK and its most common fusion partner EML4 are situated very near to each other on chromosome 2 (approximately 12 megabases apart). EML4-ALK rearrangement is generated most commonly from an intrachromosomal inversion, which results in “split” signals that may not be very far apart in positive cases and thus occasionally difficult to interpret. FISH positivity is defined by the separation between the 5′ and 3′ signals of greater than two signal diameters (Fig. 5A, 5B) [129] and at least 50 cells to be counted to ensure 100% sensitivity and specificity [130].
Figure 5.

ALK breakapart FISH assay. (A): Greater than 2 signal diameter separation as a criterion for anaplastic lymphoma kinase (ALK) break-apart fluorescense in situ hybridization (FISH) positivity. (B): 5′3′ break-apart ALK FISH. Figure provided by John Iafrate. (C): Isolated 3′ break-apart ALK FISH. (D): Using the 15% criterion for ALK break-apart, FISH resulted in a clear separation of two groups of NSCLC patients. Adapted and modified from figure 2 of reference [130], with permission.
A second common pattern in positive cases is the presence of isolated ALK 3′ probes (red signals only; Fig. 5C), likely reflecting the presence of an ALK rearrangement with loss of the nonfunctional derivative chromosome. A careful analysis of known ALK-negative cases revealed a cutoff of 15% of tumors cells to have split signals, above which a case will be called positive (15% is 2 standard deviations above the mean cell count in negative cases). Some cells may be falsely positive because the tumor cells counted may have artificial split signals due to sectioning artifact or poor nucleus morphology. The mean proportion of cells with split signals in ALK-positive NSCLC (n = 13) was 53.8% (range: 22.25%–86.62%) and 5.98% (range: 3.51–9.45) in ALK-negative NSCLC (n = 56; p < .0001; Table 7, Fig. 5D) [130–132]. With the inclusion of more ALK-positive NSCLC samples (n = 90), the mean percentage positive cells remained similar at 56% (range: 18%–100%) [133].
Table 7.
Mean percentage of FISH-positive cells in ALK-positive and ALK-negative non-small cell lung cancer

aPositive FISH signal was defined as >1 signal diameter apart.
bStandard deviation.
Abbreviations: FISH, fluorescense in situ hybridization; NR, not reported.
The reason the percentage of positive cells in ALK-positive samples is not always 100% is due to the inherent technical limitations of the assay to detect a rearrangement on a cell-by-cell basis and not because of tumor heterogeneity. Furthermore, 5′ and 3′ split signals are more common than isolated 3′ signals in FISH-positive samples (Table 8) [131, 133–134]. Interestingly, NSCLC with isolated 3′ signals has on average more percentage cells positive than NSCLC with split signals, likely due to the difficulty in calling a split signal with the inversion pattern (Table 9) [133]. Importantly, there was no correlation between the percentage of cells positive by FISH and response to crizotinib from analyses of both PROFILE 1001 [133] and PROFILE 1005 [120].
Table 8.
Distribution of patterns of break-apart FISH-positivity in patients with ALK-rearranged non-small cell lung cancer

Abbreviation: FISH, fluorescense in situ hybridization.
Table 9.
Correlation of FISH pattern positivity and percentage of ALK-positive cells (n = 90) [133]

Abbreviation: FISH, fluorescense in situ hybridization.
There are advantages and disadvantages to the break-apart FISH assay. As mentioned, the break-apart FISH assay is currently the only ALK assay that has been clinically validated. This assay can be performed on FFPE tissue, which is how the vast majority of lung cancer tissue is processed. Furthermore, it is not necessary to know the specific fusion partner to perform break-apart FISH, an important factor considering that more fusion partners may be discovered in NSCLC and other solid tumors, such as VCL-ALK in sickle cell trait-associated renal medullary carcinoma [135, 136] and C2orf44-ALK in colorectal cancer [84]. A standardized break-apart FISH protocol will therefore allow detection of ALK rearrangements beyond NSCLC.
The major challenge with the ALK FISH assay is that interpretation of the ALK break-apart FISH assay requires experience, patience, and technical expertise. For example, there is potential to misinterpret ALK amplification (copy number gain) as positive for ALK rearrangement due to the existence of multiple single red signals; however, this is not an indication for crizotinib use based on current evidence. The close involvement or supervision by an anatomic pathologist familiar with NSCLC morphology and FISH techniques will allow optimal FISH assessment.
The College of American Pathologists (CAP) has recommended that break-apart FISH analysis be performed by two independent observers and confirmed by a cytogeneticist or pathologist with training in FISH [137]. The Internal System of Human Cytogenetic Nomenclature (ISCN) may also be recommended for use with all ALK break-apart reports (examples of ISCN reports are given in Table 10) [137]. The break-apart FISH assay is generally more expensive than some other assays, and so its primary role in the detection of ALK rearrangement will need to be carefully evaluated for both performance and cost-effectiveness against other techniques that are currently being introduced.
Table 10.
Examples of Internal System of Human Cytogenetic Nomenclature (ISCN) for ALK FISH results

Abbreviations: ISCN, International System of Human Cytogenetic Nomenclature; nuc ish, interphase in situ hybridization; 5′ALK con 3′ALK, fusion of ALK probes; 5′ALK sep 3′ALK, separation of ALK probes.
ALK FISH has been validated in pathologic specimens but not on cytologic specimens as part of the PROFILE 1005 trial, although the package insert of the Abbott ALK FISH Vysis test kit does not make this fine distinction. If the FISH test is to be performed on cell blocks from tumor cells spun down and collected from pleural, pericardial, or ascitic fluids and not from tumor cells, it is recommended to use cell blocks made of spun down tumor cells or to spread the specimens isolated from fine needle biopsy on glass slides to prevent cells from overlapping one another.
Dual-Color Break-apart Chromogenic In Situ Hybridization
Chromogenic in-situ hybridization (CISH) has been developed in recent years to simplify FISH as a detection assay for HER2 overexpression in breast cancer; it has shown high concordance with FISH in clinical studies [138]. Instead of using fluorescent probes as in FISH, CISH uses probes that can be detected by routine cytochemical techniques. The advantage of CISH is that it has been automated, can be assessed under light microscope (thus enabling simultaneous cytomorphologic examination), and the signals are stable in room temperature, meaning slides can be stored as permanent records.
Using the adapted standard criteria of FISH positivity, Kim et al. demonstrated 94% sensitivity (17/18) and a 100% specificity (425/425) for dual-color break-apart CISH compared with break-apart FISH [139]. Separately, Yoshida et al. also compared CISH with FISH using RT-PCR as the criterion standard. In 15 ALK-positive NSCLC and 30 ALK-negative NSCLC samples identified by RT-PCR, CISH and FISH were able to detect the same 14 ALK-positive samples and all of the 30 ALK-negative samples were FISH- and CISH-negative [132]. However, one of the criteria for FISH/CISH positivity in the study was separation of the probes by greater than only one signal diameter rather than the standard criterion of greater than two signal diameter separations. Based on these two studies, dual-color break-apart CISH has the potential to be an alternative to FISH as the diagnostic choice for ALK-rearrangement in NSCLC, but the criteria for positivity will likely have to be different from FISH and will need to be validated to reach a consensus.
Reverse-Transcriptase Polymerase Chain Reaction
ALK rearrangement in NSCLC may also be detected by RT-PCR. RT-PCR is easy to perform, and the majority of current ALK fusion variants were detected by RT-PCR in fresh frozen tumor tissue [17, 19, 39, 63, 66, 71, 73–75]. However, RT-PCR requires ALK fusion variants to be known so that primers to all variants are included in the reaction. With an ever-expanding list of ALK fusion variants (Fig. 2A), primers for RT-PCR must be constantly updated to keep pace with published literature. Further, in daily clinical practice, most of the tumor tissue available for molecular profiling is from FFPE tissue, where the integrity of RNAs is likely to be greatly compromised compared with fresh/frozen tissue. Although FISH and immunohistochemistry (IHC) can be performed on a single FFPE slide, RT-PCR requires multiple slides in order to extract sufficient RNA for a successful reaction.
Li et al. demonstrated that RT-PCR can be successfully performed to detect the common EML4-ALK fusion variants from 4,750 archived FFPE NSCLC samples at a commercial diagnostic laboratory, but the false-negative rate is unknown [59]. Therefore, head-to-head comparison with FISH and/or IHC is still needed before RT-PCR can become a useful mass screening test. Thus, although RT-PCR remains a critical laboratory technique to investigate the various ALK fusion variants, to identify new fusion variants and to ultimately multiplex other markers, it remains to be optimized as a definite method to detect ALK rearrangement in NSCLC.
Immunohistochemistry
Although ALK FISH is currently considered the criterion standard for diagnosing ALK-positive NSCLC, ALK IHC holds promise as a rapid and affordable method that could be preferred for routine screening and diagnosis by pathology laboratories worldwide. Similar to ALK FISH, IHC requires one unstained slide cut from an FFPE block as long as there are at least a few clusters of viable tumor cells. IHC can be performed successfully on a variety of different tumor specimens, including FNA cell blocks. The major challenge for ALK IHC is the low level of ALK fusion protein expression in ALK-rearranged NSCLC (at least fivefold lower than ALK fusion protein expression in ALCL; Fig. 6A–D) [140]. It is most likely due to the weaker transcription activity of the EML4 promoter compared with the transcriptional activity of the nucleophosmin (NPM) promoter in the generation of NPM-ALK protein in ALCL. There are three ALK antibodies (ALK1, 5A4, and D5F3) that have been studied in depth in NSCLC (Table 11) [131, 134].
Figure 6.

Comparison of immunohistochemistry (IHC) using anaplastic lymphoma kinase (ALK) 1 and 5A4 in anaplastic large cell lymphoma and ALK-rearranged non-small cell lung cancer. Both 5A4 IHCs were detected with Leica automation. Magnification: ×200. Figure provided by Marie Mino-Kenudson.
Abbreviations: ALCL, anaplastic large cell lymphoma; NSCLC, non-small cell lung cancer.
Table 11.
Immunohistochemistry versus fluorescent in situ hybridization
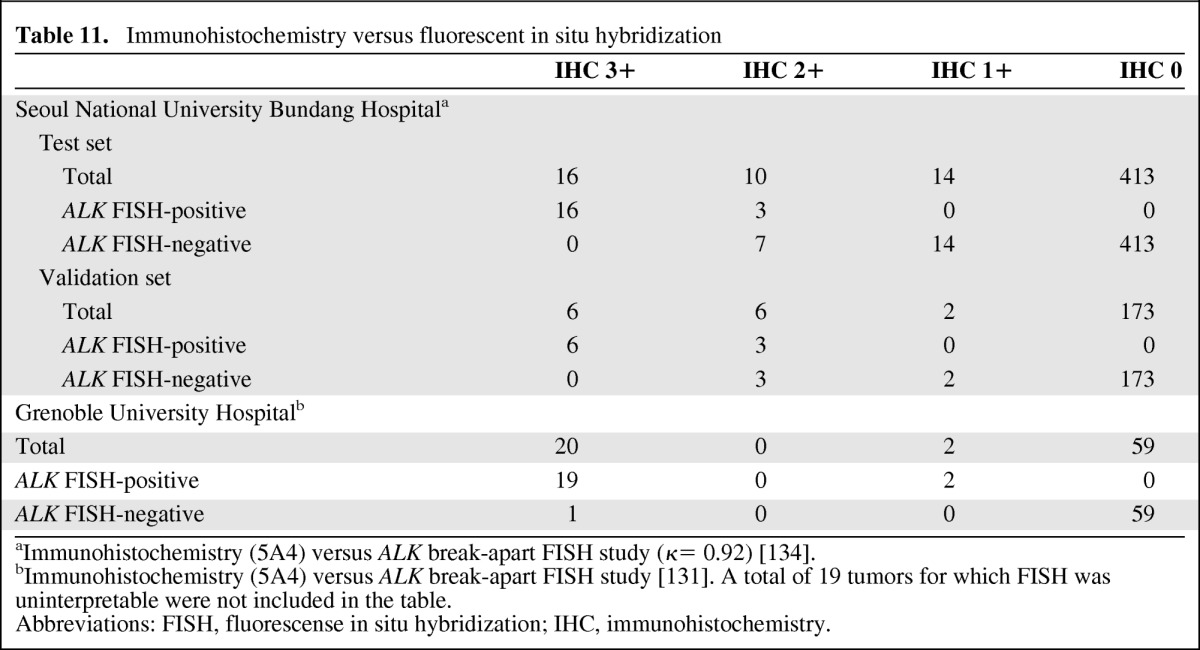
aImmunohistochemistry (5A4) versus ALK break-apart FISH study (κ= 0.92) [134].
bImmunohistochemistry (5A4) versus ALK break-apart FISH study [131]. A total of 19 tumors for which FISH was uninterpretable were not included in the table.
Abbreviations: FISH, fluorescense in situ hybridization; IHC, immunohistochemistry.
D5F3 is a promising rabbit monoclonal ALK antibody (developed by Cell Signaling Technology, Danvers, MA) for the detection of ALK rearrangement in NSCLC that has shown high sensitivity and specificity in a recent report [140]. However, the current impediments to ALK IHC being widely adopted in screening for ALK rearrangement include the lack of rigorous large-scale performance comparison with FISH and the lack of validation of clinical responses to crizotinib with ALK IHC. IHC is more likely to yield false-negative results, especially with low-ALK-expressing variants [59] or when specimens are small and/or poorly preserved. Unsuccessful FISH will be reported as “technical failure,” whereas unsuccessful IHC will be reported as a negative result.
If IHC methods (with or without amplification or scoring systems) with 5A4 and/or D5F3 are eventually proven to be accurate in detecting ALK rearrangements in larger-scale studies, IHC could become the primary screening modality for ALK-rearranged NSCLC, with only rare IHC-positive cases requiring confirmation with the FDA-approved ALK FISH test. In a research project, Sugawara et al. utilized 5A4 with an intercalated antibody-enhanced method and identified two ALK IHC-positive cases of non-clear cell renal cell carcinoma that were confirmed to harbor ALK rearrangements (EML4-ALK, TPM3-ALK) [81].
After FDA Approval of Crizotinib
The frontline PROFILE 1014 that compares crizotinib to platinum/pemetrexed is ongoing (Table 12). Recently, the second-line PROFILE 1007 study was reported as meeting its primary endpoint [100]. Both trials allow patients to receive crizotinib on disease progression if they are randomized to chemotherapy. Therefore, it is unlikely that significant improvement in OS with crizotinib will be shown in the first- or second-line setting based on these studies. Shaw et al. have performed a retrospective study and demonstrated that OS times in contemporaneously diagnosed patients with ALK-rearranged NSCLC who enrolled onto PROFILE 1001 were significantly improved compared with patients who did not enroll (Tables 3,4) [95]. Although there are certain limitations to this analysis, such retrospective analyses are likely to be the only evidence that crizotinib confers survival advantage in patients with ALK-rearranged NSCLC in the near future.
Table 12.
List of major ongoing trials with crizotinib

aClosed to accrual.
Abbreviations: ALCL, anaplastic large cell lymphoma; EORTC, European Organization for Research and Treatment of Cancer; MTD, maximum tolerated dose; NCT, National Clinical Trial; NSCLC, non-small cell lung cancer; PFS, progression-free survival; RR, response rate.
Screening for ALK-Rearranged NSCLC
The current draft version of the guidelines from CAP, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology recommends ALK testing in all patients with NSCLC exhibiting an adenocarcinoma component, regardless of age, ethnicity, sex, and smoking history [137]. The current National Comprehensive Cancer Network guidelines, developed by an expert opinion panel, recommend reflex testing of all adenocarcinoma, large cell, and NSCLC not otherwise specified for both EGFR mutations and ALK rearrangement and the use of crizotinib as a first-line treatment of ALK-rearranged NSCLC [141, 142]. Of the 24 ALK-rearranged patients who received crizotinib as a first-line treatment of their metastatic disease in PROFILE1001, 14 of 22 response-evaluable patients achieved an objective response (ORR, 64% with 95% CI, 40%–83%). The median PFS was 18.3 months (95% CI, 8.3–-not reached) [118].
The French National Cancer Institute recommends break-apart ALK FISH assay to screen all EGFR/KRAS-negative adenocarcinomas of the lung among the 28 French molecular genetics tumor laboratories [131]. Prior to the approval of crizotinib and the ALK companion diagnostic test in the USA and prior to the approval of crizotinib in Canada, a Canadian consensus report did not recommend routine screening for ALK-rearrangement in NSCLC with the argument that there was no approved treatment for ALK-rearranged NSCLC and no proven diagnostic test [143].
It is likely that individual countries will need to set up screening guidelines, especially in locations where crizotinib is approved. In regions (i.e., East Asia) where the incidence of activating EGFR mutations is high, sequential testing may be considered providing the overall turnaround time is within 14 days to allow the first-line use of crizotinib. However, double-positive mutant EGFR/ALK has been reported with increasing frequency from Asian tumor samples (Table 1), which could complicate formulation of a screening strategy.
Resistance to Crizotinib
Resistance to RTK inhibitors can be divided into primary (intrinsic) or secondary (acquired). Accelerated in vitro mutagenesis has identified mutations in six amino acid residues in ALK (C1156, I1171, F1174, G1269, L1196, S1206) that confer resistance at the highest level of crizotinib tested [41]. Mutations at three of these amino acid residues (L1196 in the gatekeeper region, S1206 near the ribose binding pocket, and G1269 in the DFG motif) confer the greatest resistance to crizotinib in vitro [41]. To date, multiple secondary point mutations in ALK that confer acquired resistance to crizotinib have been identified in patients with crizotinib-treated NSCLC (L1196M, C1156Y, L1152R, G1269A, S1206Y, G1202R, 1151Tins) [51, 71, 144–146] and inflammatory fibroblastic tumor (IMT; F1174L) [147] and they occurred in the same amino acid residues identified in the accelerated in vitro mutagenesis screen [41]. These secondary mutations accounted for approximately one-quarter of the resistance mechanisms. Furthermore, different ALK mutations can confer differential resistance according to the specific type of ALK inhibitor [145, 148].
Many “second-generation” ALK inhibitors are being developed with stronger ALK inhibitor properties than crizotinib, but also with the capability to inhibit to a varying degree these secondary acquired mutations, including the L1196M gatekeeper mutation [145, 149]. L1196M is considered a gatekeeper mutation similar to T351I, T670I, and T790M that developed in ABL, KIT, and EGFR, respectively [150]. L1196M is also a mutation that can be generated after exposing ALK-dependent NSCLC cell lines to increasing concentrations of crizotinib without the concomitant use of a mutagen [151]. Currently, at least four second-generation ALK inhibitors—AP26113 [41, 151], CH5424802 [152], AS3062 [145], and X396 [153]—have been reported to be able to inhibit the gatekeeper L1196M mutation. Other resistance mechanisms such as ALK gene copy number gain [71, 140], loss of the ALK fusion gene [71], and EGFR pathway activation [51, 140] including EGFR mutation [71], KRAS mutation [71], and KIT amplification [140] have so far been reported to confer resistance to crizotinib. Finally, in a substantial number of crizotinib resistance cases, the molecular mechanisms that underlie the resistance remain to be determined [71, 140].
Two heat shock protein (HSP) 90 inhibitors, retaspimycin (IPI-504) [154] and ganetespib (STA-9090) [155], have demonstrated clinical activity in patients with ALK-rearranged NSCLC. HSPs are a family of chaperone proteins that shepherd the aberrantly expressed EML4-ALK proteins to their subcellular location and substrates. In vitro, the addition of IPI-504 to an EML4-ALK-dependent cell line led to the rapid degradation of EML4-ALK [156], and this inhibition of EML4-ALK kinase activity occurs even in the presence of acquired crizotinib resistance [152, 156]. Therefore, HSP90 inhibitors alone or in combination with other therapy (ALK inhibitors, chemotherapy) have the potential to overcome acquired resistance to crizotinib independent of specific secondary ALK mutations and/or ALK fusion variants [78a]. Clinical trials testing this hypothesis have commenced.
In the PROFILE 1001 crizotinib study, 8 of 133 evaluable patients with ALK-rearranged NSCLC did not experience tumor shrinkage with crizotinib and 5 patients had disease progression as a best response, indicating the potential existence of primary resistance to crizotinib [118]. EML4-ALK is postulated to lead to antiapoptosis by decreasing the level of proapoptotic BIM through activation of ERK and increasing the level of antiapoptotic survivin through the activation of STAT3 [42]. Indeed, the pretreatment BIM level has been shown to predict response to a wide array of RTK inhibitors, with high pretreatment BIM level associated with a better response to RTK inhibitors and improved clinical outcome [157]. Another potential primary mechanism is the existence of bypass pathways. It has been demonstrated in vitro that activation of the EGFR pathway can lead to crizotinib resistance [51, 146]. In the future, it may be important to measure BIM levels and to ascertain the presence of EGFR mutations in ALK-rearranged NSCLC prior to crizotinib treatment [51, 71].
Crizotinib Activity in Other ALK-Dependent Tumors and MET-Amplified Tumors
Crizotinib was initially developed as a MET inhibitor, and the molecular enriched cohort of PROFILE 1001 was initially designed to screen for MET-amplified tumors (e.g., gastric carcinoma, non-Barrett's gastroesophogeal junction [GEJ] cancer) or MET-mutated tumors (e.g., squamous cell carcinoma of the head and neck, sporadic and hereditary papillary renal cell carcinoma). Other MET-dependent tumors, such as alveolar soft part sarcoma and alveolar rhabdomyosarcoma, were also eligible upon histologic confirmation.
The criteria for MET amplification in PROFILE 1001 followed the American Society of Clinical Oncology/CAP guidelines for definition of gene amplification (MET/CEP7 copy number ratio >2.2). As it turned out, true MET amplification in GEJ is an extremely rare event (2%), with aggressive tumor behavior that severely limited patient enrollment into the crizotinib clinical trial. To date, only two patients with MET-amplified GEJ derived clinical benefit on crizotinib, with time to progression being 105 and 112 days, respectively [158]. Similarly, de novo MET-amplified NSCLC is rare; no obvious clinicopathologic characteristics for this molecular subset of NSCLC have been identified.
Recently, the U.S. Lung Cancer Mutation Consortium reported a rate of 4.4% with MET/CEP7 cutoff at 2.2 and association with female sex [58]. To date, there is a report of one patient with a de novo highly MET-amplified NSCLC achieving confirmed partial response with crizotinib [159], with ongoing response for >19 months (S.-H.I. Ou, unpublished data). Additionally, one patient with MET-amplified glioblastoma achieved rapid radiographic response on crizotinib [160].
Promising antitumor activity has also been observed with crizotinib in patients with treatment-resistant ALK-positive ALCL [161]. The PROFILE 1013 study (NCT01121588) is enrolling patients with ALK-positive tumors other than NSCLC to better assess the activity of crizotinib in a wide range of malignancies. In addition, the European Organization for Research and Treatment of Cancer are in the process of initiating a study to explore crizotinib in patients with advanced tumors induced by casual alternations of ALK and MET (Table 12).
Currently, the U.S. Children's Oncology Group is conducting a phase I study in pediatric patients with refractory solid tumors and ALCL. A recent report showed that seven of eight enrolled patients with ALCL experienced a complete response. Among seven patients with ALK-positive IMT, three achieved partial response, one acheived prolonged stable disease (>24 months), and the other three were too early in treatment to be assessed. Two pediatric patients with ALK-positive NSCLC were enrolled. One achieved partial response and one achieved stable disease. Among the eight patients with neuroblastoma and a known ALK mutation, one experienced a complete response and two achieved stable disease. Of 19 patients with ALK-unknown neuroblastoma, one experienced a complete response and six had prolonged stable disease [162].
ROS1 Rearrangment in NSCLC
Rearrangements in ROS1, another human RTK, in NSCLC were discovered by Rikova et al. in 2007 with the same screening strategy for activated tyrosine kinases that identified ALK-rearranged NSCLC [19], with the initial incidence of ROS-rearranged NSCLC estimated to be <2% [163]. ROS1 is one of 58 human receptor tyrosine kinase RTKs [164] and is evolutionarily related to ALK [165]; the mouse ALK and ROS1 proteins share 52% amino acid homology within the kinase domain [20]. No biological ligand has been identified for ROS1 in humans, and its normal physiologic function in human is not well defined. Male ROS1 knockout mice are infertile due to disruption of the differentiation of the epididymal epithelium [166].
Rikova et al. identified two ROS1 fusion variants in NSCLC, including SLC34A2-ROS1 in HCC78 cell line. More recently, five additional fusion partners (TPM3, SDC4, EZR, LRIG3, FIG) to ROS1 have been identified in NSCLC [56, 68, 166a], representing 12 ROS1 fusion variants in NSCLC. More importantly, the identification of SCL34A2-ROS1 in the HCC78 cell line gave the first indication that an ALK inhibitor may also serve as a ROS1 inhibitor. The potential ROS1 inhibitory potential by ALK inhibitors was discovered by McDermott et al., who attempted to identify potential response mechanisms to ALK inhibition by screening the activity of TAE684, an ALK inhibitor in 603 different cell lines derived from a variety of human tumors [40]. They identified 10 cell lines derived from NSCLC, ACLC, and neuroblastoma that showed maximum inhibition by TAE684, including the HCC78 NSCLC cell line. No ALK abnormality or detectable ALK protein expression was found in HCC78 [40].
The discovery by Rikova et al. and the amino acid homology in the kinase domain between ALK and ROS1 led to the hypothesis that ALK inhibitors could act as ROS1 inhibitors. Consequently, ROS1-rearranged tumors based on a positive break-apart FISH assay were added as an additional eligibility criterion for molecularly enriched cohorts of PROFILE 1001 since May 2010. This modification provided the impetus to identify the clinicopathologic characteristics of patients with ROS1-rearranged NSCLC in order to implement a screening strategy to identify such patients.
Using a “home-grown” break-apart FISH assay at Massachusetts General Hospital, Bergethon et al. [167] screened 1,073 NSCLC tumor samples and identified 18 (1.7%) with ROS1-rearranged NSCLC and 31 (2.9%) with ALK-rearranged NSCLC, consistent with the previously reported low incidence of ROS1-rearranged NSCLC [163]. Perhaps not surprisingly, the clinicopathologic characteristics of ROS1-rearranged NSCLC are very similar to ALK-rearranged NSCLC: patients are young (a similar median age of diagnosis of approximately 50 years of age) never-smokers with adenocarcinoma [167]. These findings were independently demonstrated by Takeuchi et al. [68].
It seems that there is no difference in the prevalence of ROS1-rearranged NSCLC between Asian and non-Asian patients, with ROS1-rearranged NSCLC found in approximately 1% of East Asian patients with adenocarcinoma [56, 68, 168]. Among the first 15 patients (14 responses evaluable) enrolled in the ROS1 NSCLC cohort of A8081001, an ORR of 57% and disease control rate of 79% at 8 weeks suggest that crizotinib has marked antitumor activities in ROS1-rearranged NSCLC [169], similar to the activity of crizotinib initially reported for ALK-rearranged NSCLC [112]. Taken together, these data suggest that ROS1 rearrangement defines another unique molecularly defined subtype of lung cancer with heterogeneity, as well as a validated “driver mutation” and therapeutic target.
Whether the activity of crizotinib will be able to be extended to other tumors with ROS1 rearrangement remains to be determined [170, 171]. This will serve as a proof of principle demonstration that ROS1 rearrangement also function as a driver mutation in those tumors. In addition, optimized testing methods such as multiplexing are needed.
Summary
In 2011, we marked the beginning of the second decade of targeted therapy in oncology. The development and approval of crizotinib indicates that breathtaking advances are in store for the next 10 years. Already, novel chromosomal translocations involving receptor tyrosine kinases are now being discovered in other major epithelial malignancies [68, 81, 83–84, 135–136]. Although it took almost 20 years from the identification of the translocated ABL kinase in the Philadelphia chromosome in chronic myeloid leukemia [172] to the initial approval of imatinib as an ABL inhibitor in 2001 [173], it took only 4 years from the discovery of ALK rearrangement in NSCLC to the approval of crizotinib for the treatment of advanced ALK-positive NSCLC. The development of crizotinib put into focus several previously rare and relatively unknown molecular subsets of malignancy, such as ALK-rearranged NSCLC and IMT, in a similar way to the role of imatinib in defining gastrointestinal stromal tumor as a unique molecular subset of tumor. Similar to imatinib, crizotinib is also a multitargeted kinase inhibitor, and its emerging role as a MET and ROS1 inhibitor will likely spotlight further unique molecular tumor subsets underpinned by these driver mutations. The realization of the increasing incidence of chromosomal aberrations in solid tumors was actually predicted by Mitelman et al. [174].
Going forward, the emerging complexities of resistance mechanisms found in crizotinib-treated ALK-rearranged NSCLC will create a daunting task [71, 145–146]. To complicate the task further, progression on crizotinib occurred in diverse clinical scenarios, such as development of new metastasis, progression of pre-existing lesions, or both. Therefore, the importance of rebiopsying after treatment progression cannot be overemphasized [71, 144, 175]. Rebiopsying on treatment progression will likely be a major paradigm switch in solid tumor oncology as we go forward in the second decade of personalized therapy. Although rebiopsying in solid tumor malignancies will not be easy compared to repeated blood draws in chronic myeloid leukemia, it is only then that rational combination with targeted therapies can be implemented to delay or overcome resistance in order to prolong survival.
The approval of crizotinib was dependent on the simultaneous approval of a validated diagnostic assay. In contrast, when imatinib was conditionally approved for CD117-positive gastrointestinal stromal tumor in 2001, one of the post approval commitments was to develop a CD117 IHC assay in order to identify patients for imatinib treatment [176]. The codevelopment of drug with a companion diagnostic assay has spurred rapid development in the area of diagnostic assays for chromosomal translocation in solid tumors; it engenders an ongoing healthy and vigorous debate as to the most sensitive, specific, and cost-effective assay for the screening of chromosomal aberrations. With the advent of the second decade of molecular targeted therapy, the continual development of crizotinib points to more molecular subsets of solid tumors being discovered and defined, the increasing importance of repeat tissue acquisition and analysis during the clinical management of solid tumors, rapid advances in the diagnostic area of oncology, expanded translational research in oncology in the future, and the need for close collaboration between clinical oncologists, pathologists, and translational scientists.
Acknowledgments
We thank Keith Wilner (Pfizer Global Research, La Jolla, CA, USA) for shepherding the initial and ongoing clinical development of crizotinib. This work was supported by funding from Pfizer. Editorial assistance was provided by Martin Quinn at Acumed (Tytherington, U.K.).
Footnotes
- (C/A)
- Consulting/advisory relationship
- (RF)
- Research funding
- (E)
- Employment
- (H)
- Honoraria received
- (OI)
- Ownership interests
- (IP)
- Intellectual property rights/inventor/patent holder
- (SAB)
- Scientific advisory board
Author Contributions
Conception/Design: Sai-Hong Ignatius Ou, Cynthia Huang Bartlett
Collection and/or assembly of data: Sai-Hong Ignatius Ou, Cynthia Huang Barlett, Marie Mino-Kenudson, Jean Cui, A. John Iafrate
Data analysis and interpretation: Sai-Hong Ignatius Ou
Manuscript writing: Sai-Hong Ignatius Ou, Cynthia Huang Bartlett, Mari Mino-Kenudson, Jean Cui, A. John Iafrate
Final approval of manuscript: Sai-Hong Ignatius Ou, Cynthia Huang Bartlett, Mari Mino-Kenudson, Jean Cui, A. John Iafrate
References
- 1.Lynch TJ, Bell DW, Sordella R, et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N Engl J Med. 2004;350:2129–2139. doi: 10.1056/NEJMoa040938. [DOI] [PubMed] [Google Scholar]
- 2.Paez JG, Janne PA, Lee JC, et al. EGFR mutations in lung cancer: Correlation with clinical response to gefitinib therapy. Science. 2004;304:1497–1500. doi: 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- 3.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–13311. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cohen MH, Williams GA, Sridhara R, et al. United States Food and Drug Administration Drug Approval summary: Gefitinib (ZD1839; Iressa) tablets. Clin Cancer Res. 2004;10:1212–1218. doi: 10.1158/1078-0432.ccr-03-0564. [DOI] [PubMed] [Google Scholar]
- 5.Johnson JR, Cohen M, Sridhara R, et al. Approval summary for erlotinib for treatment of patients with locally advanced or metastatic non-small cell lung cancer after failure of at least one prior chemotherapy regimen. Clin Cancer Res. 2005;11:6414–6421. doi: 10.1158/1078-0432.CCR-05-0790. [DOI] [PubMed] [Google Scholar]
- 6.Maemondo M, Inoue A, Kobayashi K, et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 7.Mitsudomi T, Morita S, Yatabe Y, et al. Gefitinib versus cisplatin plus docetaxel in patients with non-small-cell lung cancer harbouring mutations of the epidermal growth factor receptor (WJTOG3405): An open label, randomised phase 3 trial. Lancet Oncol. 2010;11:121–128. doi: 10.1016/S1470-2045(09)70364-X. [DOI] [PubMed] [Google Scholar]
- 8.Rosell R, Carcereny E, Gervais R, et al. Erlotinib versus standard chemotherapy as first-line treatment for European patients with advanced EGFR mutation-positive non-small-cell lung cancer (EURTAC): A multicentre, open-label, randomised phase 3 trial. Lancet Oncol. 2012;13:239–246. doi: 10.1016/S1470-2045(11)70393-X. [DOI] [PubMed] [Google Scholar]
- 9.Zhou C, Wu YL, Chen G, et al. Erlotinib versus chemotherapy as first-line treatment for patients with advanced EGFR mutation-positive non-small-cell lung cancer (OPTIMAL, CTONG-0802): A multicentre, open-label, randomised, phase 3 study. Lancet Oncol. 2011;12:735–742. doi: 10.1016/S1470-2045(11)70184-X. [DOI] [PubMed] [Google Scholar]
- 10.Yang JC-H, Schuler MH, Yamamoto N, et al. LUX-Lung 3: A randomized, open-label, phase III study of afatinib versus pemetrexed and cisplatin as first-line treatment for patients with advanced adenocarcinoma of the lung harboring EGFR-activating mutations. J Clin Oncol. 2012;30(suppl):LBA7500. [Google Scholar]
- 11.Sun S, Schiller JH, Gazdar AF. Lung cancer in never smokers: A different disease. Nat Rev Cancer. 2007;7:778–790. doi: 10.1038/nrc2190. [DOI] [PubMed] [Google Scholar]
- 12.Toh CK, Gao F, Lim WT, et al. Never-smokers with lung cancer: Epidemiologic evidence of a distinct disease entity. J Clin Oncol. 2006;24:2245–2251. doi: 10.1200/JCO.2005.04.8033. [DOI] [PubMed] [Google Scholar]
- 13.Fukuoka M, Wu YL, Thongprasert S, et al. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS) J Clin Oncol. 2011;29:2866–2874. doi: 10.1200/JCO.2010.33.4235. [DOI] [PubMed] [Google Scholar]
- 14.Mok TS, Wu YL, Thongprasert S, et al. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 15.Han JY, Park K, Kim SW, et al. First-SIGNAL: First-line single-agent iressa versus gemcitabine and cisplatin trial in never-smokers with adenocarcinoma of the lung. J Clin Oncol. 2012;30:1122–1128. doi: 10.1200/JCO.2011.36.8456. [DOI] [PubMed] [Google Scholar]
- 16.Pao W, Girard N. New driver mutations in non-small-cell lung cancer. Lancet Oncol. 2011;12:175–180. doi: 10.1016/S1470-2045(10)70087-5. [DOI] [PubMed] [Google Scholar]
- 17.Soda M, Choi YL, Enomoto M, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature. 2007;448:561–566. doi: 10.1038/nature05945. [DOI] [PubMed] [Google Scholar]
- 18.Soda M, Takada S, Takeuchi K, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci U S A. 2008;105:19893–19897. doi: 10.1073/pnas.0805381105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rikova K, Guo A, Zeng Q, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell. 2007;131:1190–1203. doi: 10.1016/j.cell.2007.11.025. [DOI] [PubMed] [Google Scholar]
- 20.Morris SW, Naeve C, Mathew P, et al. ALK, the chromosome 2 gene locus altered by the t(2;5) in non-Hodgkin's lymphoma, encodes a novel neural receptor tyrosine kinase that is highly related to leukocyte tyrosine kinase (LTK) Oncogene. 1997;14:2175–2188. doi: 10.1038/sj.onc.1201062. [DOI] [PubMed] [Google Scholar]
- 21.Iwahara T, Fujimoto J, Wen D, et al. Molecular characterization of ALK, a receptor tyrosine kinase expressed specifically in the nervous system. Oncogene. 1997;14:439–449. doi: 10.1038/sj.onc.1200849. [DOI] [PubMed] [Google Scholar]
- 22.Morris SW, Kirstein MN, Valentine MB, et al. Fusion of a kinase gene, ALK, to a nucleolar protein gene, NPM, in non-Hodgkin's lymphoma. Science. 1994;263:1281–1284. doi: 10.1126/science.8122112. [DOI] [PubMed] [Google Scholar]
- 23.Barreca A, Lasorsa E, Riera L, et al. Anaplastic lymphoma kinase in human cancer. J Mol Endocrinol. 2011;47:R11–R23. doi: 10.1530/JME-11-0004. [DOI] [PubMed] [Google Scholar]
- 24.Chiarle R, Voena C, Ambrogio C, et al. The anaplastic lymphoma kinase in the pathogenesis of cancer. Nat Rev Cancer. 2008;8:11–23. doi: 10.1038/nrc2291. [DOI] [PubMed] [Google Scholar]
- 25.Lee HH, Norris A, Weiss JB, et al. Jelly belly protein activates the receptor tyrosine kinase Alk to specify visceral muscle pioneers. Nature. 2003;425:507–512. doi: 10.1038/nature01916. [DOI] [PubMed] [Google Scholar]
- 26.Stoica GE, Kuo A, Aigner A, et al. Identification of anaplastic lymphoma kinase as a receptor for the growth factor pleiotrophin. J Biol Chem. 2001;276:16772–16779. doi: 10.1074/jbc.M010660200. [DOI] [PubMed] [Google Scholar]
- 27.Stoica GE, Kuo A, Powers C, et al. Midkine binds to anaplastic lymphoma kinase (ALK) and acts as a growth factor for different cell types. J Biol Chem. 2002;277:35990–35998. doi: 10.1074/jbc.M205749200. [DOI] [PubMed] [Google Scholar]
- 28.Bowden ET, Stoica GE, Wellstein A. Anti-apoptotic signaling of pleiotrophin through its receptor, anaplastic lymphoma kinase. J Biol Chem. 2002;277:35862–35868. doi: 10.1074/jbc.M203963200. [DOI] [PubMed] [Google Scholar]
- 29.Motegi A, Fujimoto J, Kotani M, et al. ALK receptor tyrosine kinase promotes cell growth and neurite outgrowth. J Cell Sci. 2004;117:3319–3329. doi: 10.1242/jcs.01183. [DOI] [PubMed] [Google Scholar]
- 30.Perez-Pinera P, Zhang W, Chang Y, et al. Anaplastic lymphoma kinase is activated through the pleiotrophin/receptor protein-tyrosine phosphatase beta/zeta signaling pathway: An alternative mechanism of receptor tyrosine kinase activation. J Biol Chem. 2007;282:28683–28690. doi: 10.1074/jbc.M704505200. [DOI] [PubMed] [Google Scholar]
- 31.Reiff T, Huber L, Kramer M, et al. Midkine and Alk signaling in sympathetic neuron proliferation and neuroblastoma predisposition. Development. 2011;138:4699–4708. doi: 10.1242/dev.072157. [DOI] [PubMed] [Google Scholar]
- 32.Mehlen P, Bredesen DE. Dependence receptors: From basic research to drug development. Sci Signal. 2011;4:mr2. doi: 10.1126/scisignal.2001521. [DOI] [PubMed] [Google Scholar]
- 33.Mourali J, Benard A, Lourenco FC, et al. Anaplastic lymphoma kinase is a dependence receptor whose proapoptotic functions are activated by caspase cleavage. Mol Cell Biol. 2006;26:6209–6222. doi: 10.1128/MCB.01515-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bilsland JG, Wheeldon A, Mead A, et al. Behavioral and neurochemical alterations in mice deficient in anaplastic lymphoma kinase suggest therapeutic potential for psychiatric indications. Neuropsychopharmacology. 2008;33:685–700. doi: 10.1038/sj.npp.1301446. [DOI] [PubMed] [Google Scholar]
- 35.Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455:930–935. doi: 10.1038/nature07261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Y, Takita J, Choi YL, et al. Oncogenic mutations of ALK kinase in neuroblastoma. Nature. 2008;455:971–974. doi: 10.1038/nature07399. [DOI] [PubMed] [Google Scholar]
- 37.George RE, Sanda T, Hanna M, et al. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature. 2008;455:975–978. doi: 10.1038/nature07397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Janoueix-Lerosey I, Lequin D, Brugieres L, et al. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature. 2008;455:967–970. doi: 10.1038/nature07398. [DOI] [PubMed] [Google Scholar]
- 39.Koivunen JP, Mermel C, Zejnullahu K, et al. EML4-ALK fusion gene and efficacy of an ALK kinase inhibitor in lung cancer. Clin Cancer Res. 2008;14:4275–4283. doi: 10.1158/1078-0432.CCR-08-0168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.McDermott U, Iafrate AJ, Gray NS, et al. Genomic alterations of anaplastic lymphoma kinase may sensitize tumors to anaplastic lymphoma kinase inhibitors. Cancer Res. 2008;68:3389–3395. doi: 10.1158/0008-5472.CAN-07-6186. [DOI] [PubMed] [Google Scholar]
- 41.Zhang S, Wang F, Keats J, et al. Crizotinib-resistant mutants of EML4-ALK identified through an accelerated mutagenesis screen. Chem Biol Drug Des. 2011;78:999–1005. doi: 10.1111/j.1747-0285.2011.01239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Takezawa K, Okamoto I, Nishio K, et al. Role of ERK-BIM and STAT3-survivin signaling pathways in ALK inhibitor-induced apoptosis in EML4-ALK-positive lung cancer. Clin Cancer Res. 2011;17:2140–2148. doi: 10.1158/1078-0432.CCR-10-2798. [DOI] [PubMed] [Google Scholar]
- 43.Sequist LV, Heist RS, Shaw AT, et al. Implementing multiplexed genotyping of non-small-cell lung cancers into routine clinical practice. Ann Oncol. 2011;22:2616–2624. doi: 10.1093/annonc/mdr489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Solomon B, Varella-Garcia M, Camidge DR. ALK gene rearrangements: A new therapeutic target in a molecularly defined subset of non-small cell lung cancer. J Thorac Oncol. 2009;4:1450–1454. doi: 10.1097/JTO.0b013e3181c4dedb. [DOI] [PubMed] [Google Scholar]
- 45.Tiseo M, Gelsomino F, Bartolotti M, et al. Anaplastic lymphoma kinase as a new target for the treatment of non-small-cell lung cancer. Expert Rev Anticancer Ther. 2011;11:1677–1687. doi: 10.1586/era.11.157. [DOI] [PubMed] [Google Scholar]
- 46.Doebele RC, Lu X, Sumey C, et al. Oncogene status predicts patterns of metastatic spread in treatment-naive nonsmall cell lung cancer. Cancer. 2012;118:4502–4511. doi: 10.1002/cncr.27409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kris MG, Johnson BE, Kwiatkowski DJ, et al. Identification of driver mutations in tumor specimens from 1,000 patients with lung adenocarcinoma: The NCI's Lung Cancer Mutation Consortium (LCMC) J Clin Oncol. 2011;29(suppl):7506. [Google Scholar]
- 48.Kuo YW, Wu SG, Ho CC, et al. Good response to gefitinib in lung adenocarcinoma harboring coexisting EML4-ALK fusion gene and EGFR mutation. J Thorac Oncol. 2010;5:2039–2040. doi: 10.1097/JTO.0b013e3181f43274. [DOI] [PubMed] [Google Scholar]
- 49.Martelli MP, Sozzi G, Hernandez L, et al. EML4-ALK rearrangement in non-small cell lung cancer and non-tumor lung tissues. Am J Pathol. 2009;174:661–670. doi: 10.2353/ajpath.2009.080755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Popat S, Vieira de AA, Min T, et al. Lung adenocarcinoma with concurrent exon 19 EGFR mutation and ALK rearrangement responding to erlotinib. J Thorac Oncol. 2011;6:1962–1963. doi: 10.1097/JTO.0b013e31822eec5e. [DOI] [PubMed] [Google Scholar]
- 51.Sasaki T, Koivunen J, Ogino A, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71:6051–6060. doi: 10.1158/0008-5472.CAN-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tiseo M, Gelsomino F, Boggiani D, et al. EGFR and EML4-ALK gene mutations in NSCLC: A case report of erlotinib-resistant patient with both concomitant mutations. Lung Cancer. 2011;71:241–243. doi: 10.1016/j.lungcan.2010.11.014. [DOI] [PubMed] [Google Scholar]
- 53.Yang J, Zhang X, Su J, et al. Concomitant EGFR mutation and EML4-ALK gene fusion in non-small cell lung cancer. J Clin Oncol. 2011;29(suppl):10517. [Google Scholar]
- 54.Zhang X, Zhang S, Yang X, et al. Fusion of EML4 and ALK is associated with development of lung adenocarcinomas lacking EGFR and KRAS mutations and is correlated with ALK expression. Mol Cancer. 2010;9:188. doi: 10.1186/1476-4598-9-188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee JK, Kim TM, Koh Y, et al. Differential sensitivities to tyrosine kinase inhibitors in NSCLC harboring EGFR mutation and ALK translocation. Lung Cancer. 2012;77:460–463. doi: 10.1016/j.lungcan.2012.04.012. [DOI] [PubMed] [Google Scholar]
- 56.Rimkunas VM, Crosby K, Kelly M, et al. Analysis of receptor tyrosine kinase ROS1 positive tumors in non-small cell lung cancer: Identification of a FIG-ROS1 fusion. Clin Cancer Res. 2012;18:4449–4457. doi: 10.1158/1078-0432.CCR-11-3351. [DOI] [PubMed] [Google Scholar]
- 57.Peters S, Blackhall F, Boffetta P, et al. Building a comprehensive database for the LUNGSCAPE project: A way to bridge genomics and clinicial practice in the European Thoracic Oncology Platform (ETOP) J Thorac Oncol. 2011;6:S994. [Google Scholar]
- 58.Varella-Garcia M, Berry LD, Su P-F, et al. ALK and MET genes in advanced lung adenocarcinomas: The Lung Cancer Mutation Consortium experience. J Clin Oncol. 2012;30(suppl):7589. [Google Scholar]
- 59.Li T, Huang E, Desai S, et al. Update on the large-scale screening of ALK fusion oncogene transcripts in archival NSCLC tumor specimens using multiplexed RT-PCR assays. J Clin Oncol. 2012;30(suppl):7594. [Google Scholar]
- 59a.Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012;18:1–9. doi: 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- 60.Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK fusion is linked to histological characteristics in a subset of lung cancers. J Thorac Oncol. 2008;3:13–17. doi: 10.1097/JTO.0b013e31815e8b60. [DOI] [PubMed] [Google Scholar]
- 61.Inamura K, Takeuchi K, Togashi Y, et al. EML4-ALK lung cancers are characterized by rare other mutations, a TTF-1 cell lineage, an acinar histology, and young onset. Mod Pathol. 2009;22:508–515. doi: 10.1038/modpathol.2009.2. [DOI] [PubMed] [Google Scholar]
- 62.Jokoji R, Yamasaki T, Minami S, et al. Combination of morphological feature analysis and immunohistochemistry is useful for screening of EML4-ALK-positive lung adenocarcinoma. J Clin Pathol. 2010;63:1066–1070. doi: 10.1136/jcp.2010.081166. [DOI] [PubMed] [Google Scholar]
- 63.Wong DW, Leung EL, So KK, et al. The EML4-ALK fusion gene is involved in various histologic types of lung cancers from nonsmokers with wild-type EGFR and KRAS. Cancer. 2009;115:1723–1733. doi: 10.1002/cncr.24181. [DOI] [PubMed] [Google Scholar]
- 64.Rodig SJ, Mino-Kenudson M, Dacic S, et al. Unique clinicopathologic features characterize ALK-rearranged lung adenocarcinoma in the western population. Clin Cancer Res. 2009;15:5216–5223. doi: 10.1158/1078-0432.CCR-09-0802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shaw AT, Yeap BY, Mino-Kenudson M, et al. Clinical features and outcome of patients with non-small-cell lung cancer who harbor EML4-ALK. J Clin Oncol. 2009;27:4247–4253. doi: 10.1200/JCO.2009.22.6993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yoshida A, Tsuta K, Watanabe SI, et al. Frequent ALK rearrangement and TTF-1/p63 co-expression in lung adenocarcinoma with signet-ring cell component. Lung Cancer. 2011;72:309–315. doi: 10.1016/j.lungcan.2010.09.013. [DOI] [PubMed] [Google Scholar]
- 67.Yoshida A, Tsuta K, Nakamura H, et al. Comprehensive histologic analysis of ALK-rearranged lung carcinomas. Am J Surg Pathol. 2011;35:1226–1234. doi: 10.1097/PAS.0b013e3182233e06. [DOI] [PubMed] [Google Scholar]
- 68.Takeuchi K, Soda M, Togashi Y, et al. RET, ROS1, and ALK fusions in lung cancer. Nat Med. 2012;18:378–381. doi: 10.1038/nm.2658. [DOI] [PubMed] [Google Scholar]
- 69.Nishino M, Klepeis VE, Bergethon K, et al. Histologic and cytomorphologic features of ALK-rearranged lung adenocarcinomas. Mod Pathol. 2012;25(suppl):285A. doi: 10.1038/modpathol.2012.109. [DOI] [PubMed] [Google Scholar]
- 70.Choi YL, Takeuchi K, Soda M, et al. Identification of novel isoforms of the EML4-ALK transforming gene in non-small cell lung cancer. Cancer Res. 2008;68:4971–4976. doi: 10.1158/0008-5472.CAN-07-6158. [DOI] [PubMed] [Google Scholar]
- 71.Doebele RC, Pilling AB, Aisner DL, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18:1472–1482. doi: 10.1158/1078-0432.CCR-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sanders HR, Li HR, Bruey JM, et al. Exon scanning by reverse transcriptase-polymerase chain reaction for detection of known and novel EML4-ALK fusion variants in non-small cell lung cancer. Cancer Genet. 2011;204:45–52. doi: 10.1016/j.cancergencyto.2010.08.024. [DOI] [PubMed] [Google Scholar]
- 73.Takahashi T, Sonobe M, Kobayashi M, et al. Clinicopathologic features of non-small-cell lung cancer with EML4-ALK fusion gene. Ann Surg Oncol. 2010;17:889–897. doi: 10.1245/s10434-009-0808-7. [DOI] [PubMed] [Google Scholar]
- 74.Takeuchi K, Choi YL, Soda M, et al. Multiplex reverse transcription-PCR screening for EML4-ALK fusion transcripts. Clin Cancer Res. 2008;14:6618–6624. doi: 10.1158/1078-0432.CCR-08-1018. [DOI] [PubMed] [Google Scholar]
- 75.Takeuchi K, Choi YL, Togashi Y, et al. KIF5B-ALK, a novel fusion oncokinase identified by an immunohistochemistry-based diagnostic system for ALK-positive lung cancer. Clin Cancer Res. 2009;15:3143–3149. doi: 10.1158/1078-0432.CCR-08-3248. [DOI] [PubMed] [Google Scholar]
- 75a.Wang R, Pan Y, Li C, et al. The use of quantitative real-time reverse transcriptase PCR for 5′ and 3′ protions of ALK transcripts to detect ALK rearrangements in lung cancers. Clin Cancer Res. 2012;18:4725–4732. doi: 10.1158/1078-0432.CCR-12-0677. [DOI] [PubMed] [Google Scholar]
- 75b.Penzel R, Schirmacher P, Warth A. A novel EML4-ALK variant. Exon 6 of EML4 fused to exon 19 of ALK. J Thorac Oncol. 2012;7:1198–1199. doi: 10.1097/JTO.0b013e3182598af3. [DOI] [PubMed] [Google Scholar]
- 76.Wong DW, Leung EL, Wong SK, et al. A novel KIF5B-ALK variant in nonsmall cell lung cancer. Cancer. 2011;117:2709–2718. doi: 10.1002/cncr.25843. [DOI] [PubMed] [Google Scholar]
- 77.Togashi Y, Soda M, Sakata S, et al. KLC1-ALK: A novel fusion in lung cancer identified using a formalin-fixed paraffin-embedded tissue only. PLoS One. 2012;7:e31323. doi: 10.1371/journal.pone.0031323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jung Y, Kim P, Jung Y, et al. Discovery of ALK-PTPN3 gene fusion from human non-small cell lung carcinoma cell line using next generation RNA sequencing. Genes Chromosomes Cancer. 2012;51:590–597. doi: 10.1002/gcc.21945. [DOI] [PubMed] [Google Scholar]
- 78a.Heuckmann JM, Balke-Want H, Malchers F, et al. Differential protein stability and ALK inhibitor sensitivity of EML4-ALK fusion variants. Clin Cancer Res. 2012;18:4682–4690. doi: 10.1158/1078-0432.CCR-11-3260. [DOI] [PubMed] [Google Scholar]
- 78b.Takeda M, Okamoto I, Sakai K, et al. Clinical outcome for EML4-ALK-positive patients with advanced non-small-cell lung cancer treated with first-line platinum-based chemotherapy. Ann Oncol. 2012 Jul 5; doi: 10.1093/annonc/mds124. doi: 10.1093/annonc/mds124 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 79.Fukuyoshi Y, Inoue H, Kita Y, et al. EML4-ALK fusion transcript is not found in gastrointestinal and breast cancers. Br J Cancer. 2008;98:1536–1539. doi: 10.1038/sj.bjc.6604341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lin E, Li L, Guan Y, et al. Exon array profiling detects EML4-ALK fusion in breast, colorectal, and non-small cell lung cancers. Mol Cancer Res. 2009;7:1466–1476. doi: 10.1158/1541-7786.MCR-08-0522. [DOI] [PubMed] [Google Scholar]
- 81.Sugawara E, Togashi Y, Kuroda N, et al. Identification of anaplastic lymphoma kinase fusions in renal cancer: Large-scale immunohistochemical screening by the intercalated antibody-enhanced polymer method. Cancer. 2012;118:4427–4436. doi: 10.1002/cncr.27391. [DOI] [PubMed] [Google Scholar]
- 82.Hirokawa N, Takemura R. Molecular motors and mechanisms of directional transport in neurons. Nat Rev Neurosci. 2005;6:201–214. doi: 10.1038/nrn1624. [DOI] [PubMed] [Google Scholar]
- 83.Kohno T, Ichikawa H, Totoki Y, et al. KIF5B-RET fusions in lung adenocarcinoma. Nat Med. 2012;18:375–377. doi: 10.1038/nm.2644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Lipson D, Capelletti M, Yelensky R, et al. Identification of new ALK and RET gene fusions from colorectal and lung cancer biopsies. Nat Med. 2012;18:382–384. doi: 10.1038/nm.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ju YS, Lee WC, Shin JY, et al. A transforming KIF5B and RET gene fusion in lung adenocarcinoma revealed from whole-genome and transcriptome sequencing. Genome Res. 2012;22:436–445. doi: 10.1101/gr.133645.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pollmann M, Parwaresch R, dam-Klages S, et al. Human EML4, a novel member of the EMAP family, is essential for microtubule formation. Exp Cell Res. 2006;312:3241–3251. doi: 10.1016/j.yexcr.2006.06.035. [DOI] [PubMed] [Google Scholar]
- 89.Sasaki T, Rodig SJ, Chirieac LR, et al. The biology and treatment of EML4-ALK non-small cell lung cancer. Eur J Cancer. 2010;46:1773–1780. doi: 10.1016/j.ejca.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Kwak EL, Bang YJ, Camidge DR, et al. Anaplastic lymphoma kinase inhibition in non-small-cell lung cancer. N Engl J Med. 2010;363:1693–1703. doi: 10.1056/NEJMoa1006448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nakajima T, Kimura H, Takeuchi K, et al. Treatment of lung cancer with an ALK inhibitor after EML4-ALK fusion gene detection using endobronchial ultrasound-guided transbronchial needle aspiration. J Thorac Oncol. 2010;5:2041–2043. doi: 10.1097/JTO.0b013e3181f77b58. [DOI] [PubMed] [Google Scholar]
- 92.Perner S, Wagner PL, Demichelis F, et al. EML4-ALK fusion lung cancer: A rare acquired event. Neoplasia. 2008;10:298–302. doi: 10.1593/neo.07878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shinmura K, Kageyama S, Tao H, et al. EML4-ALK fusion transcripts, but no NPM-, TPM3-, CLTC-, ATIC-, or TFG-ALK fusion transcripts, in non-small cell lung carcinomas. Lung Cancer. 2008;61:163–169. doi: 10.1016/j.lungcan.2007.12.013. [DOI] [PubMed] [Google Scholar]
- 94.Wu SG, Kuo YW, Chang YL, et al. EML4-ALK translocation predicts better outcome in lung adenocarcinoma patients with wild-type EGFR. J Thorac Oncol. 2012;7:98–104. doi: 10.1097/JTO.0b013e3182370e30. [DOI] [PubMed] [Google Scholar]
- 95.Shaw AT, Yeap BY, Solomon BJ, et al. Effect of crizotinib on overall survival in advanced NSCLC harboring anaplastic lymphoma kinase gene rearrangement: A retrospective analysis. Lancet Oncol. 2011;11:1004–1012. doi: 10.1016/S1470-2045(11)70232-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Altavilla G, Santarpia M, Arrigo C, et al. EML4-ALK fusion gene in lung adenocarcinoma: A retrospective analysis of the outcome of cisplatin plus pemetrexed treated patients. J Clin Oncol. 2010;28(suppl):7610. [Google Scholar]
- 97.Camidge DR, Kono SA, Lu X, et al. Anaplastic lymphoma kinase gene rearrangements in non-small cell lung cancer are associated with prolonged progression-free survival on pemetrexed. J Thorac Oncol. 2011;6:774–780. doi: 10.1097/JTO.0b013e31820cf053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee JO, Kim TM, Lee SH, et al. Anaplastic lymphoma kinase translocation: A predictive biomarker of pemetrexed in patients with non-small cell lung cancer. J Thorac Oncol. 2011;6:1474–1480. doi: 10.1097/JTO.0b013e3182208fc2. [DOI] [PubMed] [Google Scholar]
- 98a.Shaw AT, Varghese AM, Solomon BJ, et al. Pemetrexed-based chemotherapy in patients with advanced, ALK-positive non-small cell lung cancer. Ann Oncol. 2012 Aug 10; doi: 10.1093/annonc/mds242. doi: 10.1093/annonc/mds242 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Scagliotti G, Kim DW, Shaw AT, et al. A large retrospective analysis of the activity of pemetrexed (PEM) in patients (pts) with ALK-positive (ALK+) non-small cell lung cancer (NSCLC) prior to crizotinib (CRIZ) J Clin Oncol. 2012;30(suppl):7599. [Google Scholar]
- 100.Pfizer Inc. Pfizer announces positive results from phase 3 study PROFILE 1007 evaluating Xalkori (crizotinib) in previously treated patients with ALK-positive advanced non-small cell lung cancer. [Accessed June 19, 2012]. Available at http://www.marketwatch.com/story/pfizer-announces-positive-results-from-phase-3-study-profile-1007-evaluating-xalkorir-crizotinib-in-previously-treated-patients-with-alk-positive-advanced-non-small-cell-lung-cancer-2012–06-19.
- 101.Zhang XC, Blanckmeister C, Yang J, et al. Retrospective study of clinicopathologic factors associated with ALK rearrangement and survival outcome in Chinese patients with NSCLC. Abstract presented at: American Association for Cancer Research Annual Meeting; March 31-April 4, 2012; Chicago, IL. [Google Scholar]
- 102.Lee JK, Park HS, Kim DW, et al. Comparative analyses of overall survival in patients with anaplastic lymphoma kinase-positive and matched wild-type advanced nonsmall cell lung cancer. Cancer. 2012;118:3579–3586. doi: 10.1002/cncr.26668. [DOI] [PubMed] [Google Scholar]
- 103.Kim HR, Shim HS, Chung JH, et al. Distinct clinical features and outcomes in never-smokers with nonsmall cell lung cancer who harbor EGFR or KRAS mutations or ALK rearrangement. Cancer. 2012;118:729–739. doi: 10.1002/cncr.26311. [DOI] [PubMed] [Google Scholar]
- 104.Yang P, Kulig K, Boland JM, et al. Worse disease-free survival in never-smokers with ALK+ lung adenocarcinoma. J Thorac Oncol. 2012;7:90–97. doi: 10.1097/JTO.0b013e31823c5c32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Timofeevski SL, McTigue MA, Ryan K, et al. Enzymatic characterization of c-Met receptor tyrosine kinase oncogenic mutants and kinetic studies with aminopyridine and triazolopyrazine inhibitors. Biochemistry. 2009;48:5339–5349. doi: 10.1021/bi900438w. [DOI] [PubMed] [Google Scholar]
- 106.Zou HY, Li Q, Lee JH, et al. An orally available small-molecule inhibitor of c-Met, PF-2341066, exhibits cytoreductive antitumor efficacy through antiproliferative and antiangiogenic mechanisms. Cancer Res. 2007;67:4408–4417. doi: 10.1158/0008-5472.CAN-06-4443. [DOI] [PubMed] [Google Scholar]
- 107.Cui JJ, Tran-Dube M, Shen H, et al. Structure based drug design of crizotinib (PF-02341066), a potent and selective dual inhibitor of mesenchymal-epithelial transition factor (c-MET) kinase and anaplastic lymphoma kinase (ALK) J Med Chem. 2011;54:6342–6363. doi: 10.1021/jm2007613. [DOI] [PubMed] [Google Scholar]
- 108.RCSB Protein Database. X-ray structure of PF-02341066 bound to the kinase domain of c-MET. [Accessed June 27, 2012]. Available at http://www.rcsb.org/pdb/explore.do?structureId=2wgj.
- 109.RCSB Protein Database. Structure of the human anaplastic lymphoma kinase in complex with crizotinib (PF-02341066) [Accessed June 27, 2012]. Available at http://www.rcsb.org/pdb/explore/explore.do?pdbId=2xp2.
- 110.Christensen JG, Zou HY, Arango ME, et al. Cytoreductive antitumor activity of PF-2341066, a novel inhibitor of anaplastic lymphoma kinase and c-Met, in experimental models of anaplastic large-cell lymphoma. Mol Cancer Ther. 2007;6:3314–3322. doi: 10.1158/1535-7163.MCT-07-0365. [DOI] [PubMed] [Google Scholar]
- 111.Cullinane C, Dorow DS, Jackson S, et al. Differential (18)F-FDG and 3′-deoxy-3′-(18)F-fluorothymidine PET responses to pharmacologic inhibition of the c-MET receptor in preclinical tumor models. J Nucl Med. 2011;52:1261–1267. doi: 10.2967/jnumed.110.086967. [DOI] [PubMed] [Google Scholar]
- 112.Kwak EL, Camidge DR, Clark J, et al. Clinical activity observed in a phase I dose escalation trial of an oral c-met and ALK inhibitor, PF-02341066. J Clin Oncol. 2009;27(suppl):3509. [Google Scholar]
- 113.Pfizer, Inc. Xalkori prescribing information. [Accessed October 7, 2011]. Available at http://www.accessdata.fda.gov/drugsatfda_docs/label/2011/202570s000lbl.pdf.
- 114.Tan W, Wilner KD, Bang Y, et al. Pharmacokinetics (PK) of PF-02341066, a dual ALK/MET inhibitor after multiple oral doses to advanced cancer patients. J Clin Oncol. 2010;28(suppl):2596. [Google Scholar]
- 115.Ou SI, Salgia R, Clark J, et al. Comparison of crizotinib (PF-02341066) pharmacokinetics between Asian and non-Asian patients with advanced malignancies. J Thorac Oncol. 2010;5:S382. [Google Scholar]
- 116.French J, Tan W, Kang D, et al. Preliminary exposure-response analysis of crizotinib in patients with ALK-positive advanced non-small cell lung cancer. Poster presented at: American Society for Clinical Pharmacology and Therapeutics Annual Meeting; March 12–17, 2012; National Harbor, MD. [Google Scholar]
- 117.Camidge DR, Bang Y, Kwak EL, et al. Progression-free survival (PFS) from a phase I study of crizotinib (PF-02341066) in patients with ALK-positive non-small cell lung cancer (NSCLC). Presented at: 47th Annual Meeting of the American Society of Clinical Oncology; June 3–7, 2011; Chicago, IL. [Google Scholar]
- 118.Camidge DR, Bang Y, Kwak EL, et al. Efficacy and safety of crizotinib (PF-02341066) in patients with ALK-positive non-small cell lung cancer: Updated results from the molecularly defined cohort of this first-in-man phase I study. Lancet Oncol. 2012 Sep 3; doi: 10.1016/S1470-2045(12)70344-3. doi: 10.1016/S1470204512703443 [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kim D-W, Ahn M-J, Shi Y, et al. Results of a global phase II study with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC). Poster presented at: 48th Annual Meeting of the American Society of Clinical Oncology; June 1–5, 2012; Chicago, IL. [Google Scholar]
- 120.Kim DW, Blackhall F, Soria JC, et al. A global phase 2 study including efficacy, safety and patient-reported outcomes (PROs) with crizotinib in patients (Pts) with ALK-positive non-small cell lung cancer (NSCLC) Eur J Cancer. 2011;47:S617. [Google Scholar]
- 121.Besse B, Salgia R, Solomon B, et al. Visual disturbances in patients (pts) with anaplastic lymphoma kinase (ALK)-positive advanced non-small cell lung cancer (NSCLC) treated with crizotinib. Poster presented at: European Society of Medical Oncology Annual Meeting; September 28-October 2, 2012; Vienna, Austria. [Google Scholar]
- 122.Schnell P, Safferman AZ, Bartlett CH, Tang Y, Wilner KD. Clinical presentation of hepatotoxicity-associated crizotinib in ALK-positive (ALK+) advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2012;30(suppl):7598. [Google Scholar]
- 123.Weickhardt AJ, Rothman MS, Salian-Mehta S, et al. Rapid-onset hypogonadism secondary to crizotinib use in men with metastatic nonsmall cell lung cancer; Cancer; 2012. Apr 4, doi: 10.1002/cncr.27450 [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 124.Costa DB, Kobayashi S, Pandya SS, et al. CSF concentration of the anaplastic lymphoma kinase inhibitor crizotinib. J Clin Oncol. 2011;29:e443–e445. doi: 10.1200/JCO.2010.34.1313. [DOI] [PubMed] [Google Scholar]
- 125.Otterson GA, Riely GJ, Shaw AT, et al. Clinical characteristics of ALK+ NSCLC patients (pts) treated with crizotinib beyond disease progression (PD): Potential implications for management. J Clin Oncol. 2012;30(suppl):7600. [Google Scholar]
- 126.Weickhardt JA, Scheier B, Burke JM, et al. Continuation of EGFR/ALK inhibition after local therapy of oligoprogressive disease in EGFR mutant (Mt) and ALK+ non-small cell lung cancer (NSCLC) J Clin Oncol. 2012;30(suppl):7526. [Google Scholar]
- 127.Crinò L, Kim D-W, Riely G, et al. Initial phase 2 results with crizotinib in advanced ALK-positive non-small cell lung cancer (NSCLC): PROFILE 1005. J Clin Oncol. 2011;29(suppl):7514. [Google Scholar]
- 128.Tomlins SA, Rhodes DR, Perner S, et al. Recurrent fusion of TMPRSS2 and ETS transcription factor genes in prostate cancer. Science. 2005;310:644–648. doi: 10.1126/science.1117679. [DOI] [PubMed] [Google Scholar]
- 129.Boland JM, Erdogan S, Vasmatzis G, et al. Anaplastic lymphoma kinase immunoreactivity correlates with ALK gene rearrangement and transcriptional up-regulation in non-small cell lung carcinomas. Hum Pathol. 2009;40:1152–1158. doi: 10.1016/j.humpath.2009.01.012. [DOI] [PubMed] [Google Scholar]
- 130.Camidge DR, Kono SA, Flacco A, et al. Optimizing the detection of lung cancer patients harboring anaplastic lymphoma kinase (ALK) gene rearrangements potentially suitable for ALK inhibitor treatment. Clin Cancer Res. 2010;16:5581–5590. doi: 10.1158/1078-0432.CCR-10-0851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.McLeer-Florin A, Moro-Sibilot D, Melis A, et al. Dual IHC and FISH testing for alk gene rearrangement in lung adenocarcinomas in a routine practice: A French study. J Thorac Oncol. 2012;7:348–354. doi: 10.1097/JTO.0b013e3182381535. [DOI] [PubMed] [Google Scholar]
- 132.Yoshida A, Tsuta K, Nitta H, et al. Bright-field dual-color chromogenic in situ hybridization for diagnosing echinoderm microtubule-associated protein-like 4-anaplastic lymphoma kinase-positive lung adenocarcinomas. J Thorac Oncol. 2011;6:1677–1686. doi: 10.1097/JTO.0b013e3182286d25. [DOI] [PubMed] [Google Scholar]
- 133.Camidge DR, Theodoro M, Maxson DA, et al. Correlations between the percentage of tumor cells showing an ALK (anaplastic lymphoma kinase) gene rearrangement, ALK signal copy number, and response to crizotinib therapy in ALK fluorescence in situ hybridization-positive nonsmall cell lung cancer. Cancer. 2012;118:4486–4494. doi: 10.1002/cncr.27411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Paik JH, Choe G, Kim H, et al. Screening of anaplastic lymphoma kinase rearrangement by immunohistochemistry in non-small cell lung cancer: Correlation with fluorescence in situ hybridization. J Thorac Oncol. 2011;6:466–472. doi: 10.1097/JTO.0b013e31820b82e8. [DOI] [PubMed] [Google Scholar]
- 135.Debelenko LV, Raimondi SC, Daw N, et al. Renal cell carcinoma with novel VCL-ALK fusion: New representative of ALK-associated tumor spectrum. Mod Pathol. 2011;24:430–442. doi: 10.1038/modpathol.2010.213. [DOI] [PubMed] [Google Scholar]
- 136.Marino-Enriquez A, Ou WB, Weldon CB, et al. ALK rearrangement in sickle cell trait-associated renal medullary carcinoma. Genes Chromosomes Cancer. 2011;50:146–153. doi: 10.1002/gcc.20839. [DOI] [PubMed] [Google Scholar]
- 137.College of American Pathologists, International Association for the Study of Lung Cancer, Association for Molecular Pathology. Lung cancer biomarkers guideline draft recommendations. [Accessed March 6, 2012]. Available at http://www.cap.org/apps/docs/membership/transformation/new/lung_public_comment_supporting_materials.pdf.
- 138.Garcia-Caballero T, Grabau D, Green AR, et al. Determination of HER2 amplification in primary breast cancer using dual-colour chromogenic in situ hybridization is comparable to fluorescence in situ hybridization: A European multicentre study involving 168 specimens. Histopathology. 2010;56:472–480. doi: 10.1111/j.1365-2559.2010.03503.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kim H, Yoo SB, Choe JY, et al. Detection of ALK gene rearrangement in non-small cell lung cancer: A comparison of fluorescence in situ hybridization and chromogenic in situ hybridization with correlation of ALK protein expression. J Thorac Oncol. 2011;6:1359–1366. doi: 10.1097/JTO.0b013e31821cfc73. [DOI] [PubMed] [Google Scholar]
- 140.Mino-Kenudson M, Chirieac LR, Law K, et al. A novel, highly sensitive antibody allows for the routine detection of ALK-rearranged lung adenocarcinomas by standard immunohistochemistry. Clin Cancer Res. 2010;16:1561–1571. doi: 10.1158/1078-0432.CCR-09-2845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.National Comprehensive Cancer Network (NCCN) Fort Washington, PA, USA: National Comprehensive Cancer Network®; Guidelines for Management of Stage IV NSCLC. NCCN guidelines version 2, 2012. [Google Scholar]
- 142.Riely GJ, Chaft JE, Ladanyi M, et al. Incorporation of crizotinib into the NCCN guidelines. J Natl Compr Canc Netw. 2011;9:1328–1330. doi: 10.6004/jnccn.2011.0113. [DOI] [PubMed] [Google Scholar]
- 143.Ellis PM, Blais N, Soulieres D, et al. A systematic review and Canadian consensus recommendations on the use of biomarkers in the treatment of non-small cell lung cancer. J Thorac Oncol. 2011;6:1379–1391. doi: 10.1097/JTO.0b013e318220cb8e. [DOI] [PubMed] [Google Scholar]
- 144.Choi YL, Soda M, Yamashita Y, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363:1734–1739. doi: 10.1056/NEJMoa1007478. [DOI] [PubMed] [Google Scholar]
- 145.Katayama R, Shaw AT, Khan TM, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung cancers. Sci Transl Med. 2012;4:120ra17. doi: 10.1126/scitranslmed.3003316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lovly CM, Pao W. Escaping ALK inhibition: Mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4:120ps2. doi: 10.1126/scitranslmed.3003728. [DOI] [PubMed] [Google Scholar]
- 147.Sasaki T, Okuda K, Zheng W, et al. The neuroblastoma-associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK-translocated cancers. Cancer Res. 2010;70:10038–10043. doi: 10.1158/0008-5472.CAN-10-2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Heuckmann JM, Holzel M, Sos ML, et al. ALK mutations conferring differential resistance to structurally diverse ALK inhibitors. Clin Cancer Res. 2011;17:7394–7401. doi: 10.1158/1078-0432.CCR-11-1648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Sasaki T, Janne PA. New strategies for treatment of ALK rearranged non-small cell lung cancers. Clin Cancer Res. 2011;17:7213–7218. doi: 10.1158/1078-0432.CCR-11-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Pao W, Chmielecki J. Rational, biologically based treatment of EGFR-mutant non-small-cell lung cancer. Nat Rev Cancer. 2010;10:760–774. doi: 10.1038/nrc2947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Katayama R, Khan TM, Benes C, et al. Therapeutic strategies to overcome crizotinib resistance in non-small cell lung cancers harboring the fusion oncogene EML4-ALK. Proc Natl Acad Sci U S A. 2011;108:7535–7540. doi: 10.1073/pnas.1019559108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sakamoto H, Tsukaguchi T, Hiroshima S, et al. CH5424802, a selective ALK inhibitor capable of blocking the resistant gatekeeper mutant. Cancer Cell. 2011;19:679–690. doi: 10.1016/j.ccr.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 153.Lovly CM, Heuckmann JM, de SE, et al. Insights into ALK-driven cancers revealed through development of novel ALK tyrosine kinase inhibitors. Cancer Res. 2011;71:4920–4931. doi: 10.1158/0008-5472.CAN-10-3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Sequist LV, Gettinger S, Senzer NN, et al. Activity of IPI-504, a novel heat-shock protein 90 inhibitor, in patients with molecularly defined non-small-cell lung cancer. J Clin Oncol. 2010;28:4953–4960. doi: 10.1200/JCO.2010.30.8338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Wong K, Koczywas M, Goldman JW, et al. An open-label phase II study of the Hsp90 inhibitor ganetespib (STA-9090) as monotherapy in patients with advanced non-small cell lung cancer (NSCLC) J Clin Oncol. 2011;29(suppl) abstr 7500. [Google Scholar]
- 156.Normant E, Paez G, West KA, et al. The Hsp90 inhibitor IPI-504 rapidly lowers EML4-ALK levels and induces tumor regression in ALK-driven NSCLC models. Oncogene. 2011;30:2581–2586. doi: 10.1038/onc.2010.625. [DOI] [PubMed] [Google Scholar]
- 157.Faber A, Corcoran RB, Ebi H, et al. BIM expression in treatment naive cancers predicts responsiveness to kinase inhibitors. Cancer Discov. 2011;1:352–365. doi: 10.1158/2159-8290.CD-11-0106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lennerz JK, Kwak EL, Ackerman A, et al. MET Amplification Identifies a Small and Aggressive Subgroup of Esophagogastric Adenocarcinoma With Evidence of Responsiveness to Crizotinib. J Clin Oncol. 2011;29:4803–4810. doi: 10.1200/JCO.2011.35.4928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Ou SH, Kwak EL, Siwak-Tapp C, et al. Activity of crizotinib (PF02341066), a dual mesenchymal-epithelial transition (MET) and anaplastic lymphoma kinase (ALK) inhibitor, in a non-small cell lung cancer patient with de novo MET amplification. J Thorac Oncol. 2011;6:942–946. doi: 10.1097/JTO.0b013e31821528d3. [DOI] [PubMed] [Google Scholar]
- 160.Chi AS, Kwak EL, Clark JW, et al. Clinical improvement and rapid radiographic regression induced by a MET inhibitor in a patient with MET-amplified glioblastoma. J Clin Oncol. 2011;29(suppl):2072. [Google Scholar]
- 161.Pogliani EM, Dilda I, Villa F, et al. High response rate to crizotinib in advanced, chemoresistant ALK+ lymphoma patients. J Clin Oncol. 2011;29(suppl):e18507. [Google Scholar]
- 162.Mossé YP, Balis FM, Lim MS, et al. Efficacy of crizotinib in children with relapsed/refractory ALK-driven tumors including anaplastic large cell lymphoma and neuroblastoma: A Children's Oncology Group phase I consortium study. J Clin Oncol. 2012;30(suppl):9500. [Google Scholar]
- 163.Rimkunas V, Crosby K, Kelly M, et al. Frequencies of ALK and ROS in NSCLC FFPE tumor samples utilizing a highly specific immunohistochemistry-based assay and FISH analysis. J Clin Oncol. 2010;28(suppl):10536. [Google Scholar]
- 164.Blume-Jensen P, Hunter T. Oncogenic kinase signalling. Nature. 2001;411:355–365. doi: 10.1038/35077225. [DOI] [PubMed] [Google Scholar]
- 165.Robinson DR, Wu YM, Lin SF. The protein tyrosine kinase family of the human genome. Oncogene. 2000;19:5548–5557. doi: 10.1038/sj.onc.1203957. [DOI] [PubMed] [Google Scholar]
- 166.Sonnenberg-Riethmacher E, Walter B, Riethmacher D, et al. The c-ros tyrosine kinase receptor controls regionalization and differentiation of epithelial cells in the epididymis. Genes Dev. 1996;10:1184–1193. doi: 10.1101/gad.10.10.1184. [DOI] [PubMed] [Google Scholar]
- 166a.Davies KD, Le AT, Theodoro MF, et al. Identifying and targeting ROS1 gene fusions in non-small cell lung cancer. Clinc Cancer Res. 2012;18:4570–4579. doi: 10.1158/1078-0432.CCR-12-0550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Bergethon K, Shaw AT, Ignatius Ou SH, et al. ROS1 Rearrangements define a unique molecular class of lung cancers. J Clin Oncol. 2012;30:863–870. doi: 10.1200/JCO.2011.35.6345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Li C, Fang R, Sun Y, et al. Spectrum of oncogenic driver mutations in lung adenocarcinomas from East Asian never smokers. PLoS One. 2011;6:e28204. doi: 10.1371/journal.pone.0028204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Shaw AT, Camidge DR, Engelman JA, et al. Clinical activity of crizotinib in advanced non-small cell lung cancer (NSCLC) harboring ROS1 gene rearrangement. Presented at: 48th Annual Meeting of the American Society of Clinical Oncology; June 1–5, 2012; Chicago, IL. [Google Scholar]
- 170.Charest A, Lane K, McMahon K, et al. Fusion of FIG to the receptor tyrosine kinase ROS in a glioblastoma with an interstitial del(6)(q21q21) Genes Chromosomes Cancer. 2003;37:58–71. doi: 10.1002/gcc.10207. [DOI] [PubMed] [Google Scholar]
- 171.Gu TL, Deng X, Huang F, et al. Survey of tyrosine kinase signaling reveals ROS kinase fusions in human cholangiocarcinoma. PLoS One. 2011;6:e15640. doi: 10.1371/journal.pone.0015640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.de KA, van Kessel AG, Grosveld G, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature. 1982;300:765–767. doi: 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 173.Cohen MH, Williams G, Johnson JR, et al. Approval summary for imatinib mesylate capsules in the treatment of chronic myelogenous leukemia. Clin Cancer Res. 2002;8:935–942. [PubMed] [Google Scholar]
- 174.Mitelman F, Johansson B, Mertens F. Fusion genes and rearranged genes as a linear function of chromosome aberrations in cancer. Nat Genet. 2004;36:331–334. doi: 10.1038/ng1335. [DOI] [PubMed] [Google Scholar]
- 175.Sequist LV, Waltman BA, as-Santagata D, et al. Genotypic and histological evolution of lung cancers ac-quiring resistance to EGFR inhibitors. Sci Transl Med. 2011;3:75ra26. doi: 10.1126/scitranslmed.3002003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Dagher R, Cohen M, Williams G, et al. Approval summary: Imatinib mesylate in the treatment of metastatic and/or unresectable malignant gastrointestinal stromal tumors. Clin Cancer Res. 2002;8:3034–3038. [PubMed] [Google Scholar]