

Abstract
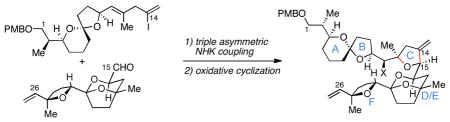
Synthesis of the C1-C26 hexacyclic subunit of pectenotoxin-2 (PTX-2) is described that features a stereoselective annulation to generate the C-ring by triple asymmetric Nozaki-Hiyama-Kishi coupling followed by oxidative cyclization. Preparation of the C1-C14 AB spriroketal-containing subunit employs a recently developed metallacycle-mediated reductive cross-coupling between a TMS-alkyne and a terminal alkene.
Pectenotoxin-2 (PTX2) is a rare marine-derived polyether natural product that displays rather profound anticancer properties (Figure 1A).1–3 Discovered in the digestive glands of the scallop Patinopecten yessoensis4 and traced back to the dinoflagellates Dinophysis fortii and D. acuminata,5 recent studies have described the isolation of PTX2 from a two-sponge association (Poecillastra sp. and Jaspis sp.).2 Initial biological evaluation of PTX2 established its substantial cytotoxic profile, with later studies concluding that this natural product is a unique actin depolymerizing agent. Binding to a site on G-actin that is distinct from other known marine toxins,6 recent studies have determined that PTX2 is selectively cytotoxic to p53-mutant and p53(–) cancers (representing approximately 50% of all human cancers).1,7
Figure 1.

Introduction.
A. Structure of PTX2
B. Convergent assembly of the C1-C26 subunit of PTX2.
C. C-ring annulation via asymmetric Nozaki-Hiyama-Kishi coupling/epoxidation/cyclization.
While no laboratory synthesis of PTX2 has been reported,8 a number of studies directed toward this goal have appeared.9 Here, we describe an efficient assembly of the C1-C26 ABCDEF hexacyclic subunit of PTX2 (2) by convergent union of the functionalized vinyliodide 3 with the tricyclic acetal-containing aldehyde 4 (Figure 1B). While these pursuits have led to the generation of a substantial subunit of pectenotoxin-2 (2), they have also defined an approach to stereodefined 2,2,5-trisubstituted THFs based on double- or triple asymmetric Nozaki-Hiyama-Kishi (NHK) coupling (8 + 9 → 7) and site and stereoselective oxidative cyclization via 5-exo ring closure (Figure 1C).
With initial focus on a more modest target than 2, and deriving inspiration from Professor Kishi’s approach to the synthesis of heterocyclic motifs present in the halichondrins,10 we targeted construction of the PTX2 CDEF heterocyclic system by double asymmetric NHK coupling11 between aldehyde 49o and vinyliodide 8 (Scheme 1A). Site- and stereoselective epoxidation of the diol product (10 → 11) followed by 5-exo ring closure (11 → 12) was then envisioned as a means to establish the complex C10-C26 tetracycle of PTX2.
Scheme 1.

C-ring annulation strategy and synthesis of 8.
A. Proposed annulation of the PTX2 C-ring:
B. Synthesis of vinyliodide 5:
As illustrated in Scheme 1B, vinyl iodide 8 was prepared by a simple six-step sequence from isoprene. First, conversion to the stereodefined vinylchloride 14 was accomplished by exposure to t-BuOCl in AcOH,12 and subsequent tranformation to enyne 15 was realized by sequential homologation with TMS-acetylene and desilylation.13 Conversion to the fully functionalized coupling partner 8 was then achieved by a simple three-step sequence: (1) silylation with TBDPSCl, (2) regioselective hydrostannylation,14 and (3) iodination.
Moving on to explore the basic steps of the annulation process on a model aldehyde, asymmetric NHK coupling11 with cyclohexane carboxaldehyde delivered allylic alcohol 17 in 54% yield with 87% ee (Scheme 2). Subsequent desilylation with TBAF provided diol 18 in 99% isolated yield — an intermediate that proved to be an ideal substrate for site- and stereoselective Sharpless asymmetric epoxidation15/ring closure. Exposure of 18 to reaction conditions for asymmetric epoxidation with (+)-diethyltartrate delivered the 2,2,5-cis trisubstituted THF 19 in 86% yield (dr = 10:1) by tandem asymmetric epoxidation/5-exo ring closure. While unrelated to the synthetic challenge associated with the C-ring of PTX2, use of (−)-diethyltartrate in this reaction process resulted in formation of the 2,2,5-trans trisubstituted product 20 in 86 % yield (dr = 6:1).16
Scheme 2.

NHK-based annulation for 2,2,5-trisubstituted THFs.
With confidence gained from the successful coupling of 8 with a simple aldehyde, we moved on to study the utility of this sequence for synthesis of the C10-C26 tetracyclic fragment of PTX2 12. As illustrated in Scheme 3, aldehyde 4 was prepared as previously described from linalool by an 11-step sequence.9o While single asymmetric NHK coupling between aldehyde 4 and vinyl iodide 8 proceeded without appreciable stereoselection (ds = 1.5:1), a double asymmetric variant of this process delivered the allylic alcohol 10 with exquisite levels of selectivity (dr ≥ 20:1) in 76% yield (after desilylation: TBAF, THF). Moving on, site-selective Sharpless asymmetric epoxidation and cyclization proved effective for advancing triene 10 to the tetracyclic target 12 in 83% yield.
Scheme 3.

Synthesis of a CDEF-containing subunit of PTX2.
With a sound foundation of preliminary data that supported the utility of NHK-coupling for establishment of the functionalized C-ring of the pectenotoxins, we then targeted synthesis of the fully functionalized C1-C26 subunit of PTX2. As illustrated in Scheme 4, synthesis of the AB spriroketal-containing subunit began with reductive cross-coupling between the stereodefined homoallylic alcohol 21 and TMS-alkyne 22.17 This Ti-mediated, hydroxyl-directed coupling process proceeded in 77% yield and delivered 23 as a single regio- and stereoisomer. Next, TBS deprotection (TBAF, THF) was followed by selective oxidation of the primary alcohol to the aldehyde (TEMPO, NCS, Bu4NCl, CH2Cl2, pH 8.6 buffer) and Carreira’s catalytic asymmetric acetylide addition18 [propyne, Zn(OTf)2, (–)-N-methylephedrine, Et3N, PhMe] to deliver the propargylic alcohol product 24 in 78% yield (dr ≥ 20:1). Protodesilylation of the vinylsilane (1 M HCl, THF, EtOH), oxidative cleavage of the alkene (O3, MeOH, then Me2S) and acid-promoted dehydration then delivered the AB spiroketal-containing subunit 25 as a mixture of C7-spiroketal isomers (dr = 14:1) in 84% yield. As expected, this spirocyclization provided the product containing the incorrect C7 stereochemistry for PTX2 – a structural feature that we plan to address in late stage acid-mediated equilibration once the fully functionalized macrocycle is in place. Finally, conversion to vinyliodide 3 was accomplished by regioselective hydrozirconation-iodination (Cp2ZrCl2, DIBAL, THF, then I2) and coupling with 2-iodo-allylbromide (n-BuLi, i-PrMgBr, CuCN•2LiCl).19
Scheme 4.

Synthesis of the AB spiroketal-containing subunit 3.
As illustratred in Scheme 5, triple asymmetric20 NHK coupling between vinyl iodide 3 and aldehyde 4 delivered the allylic alcohol product 26 in 79% yield (ds ≥ 20:1; Scheme 5). While we were delighted that this coupling proceeded with outstanding levels of stereochemical control and good yield, we were unable to identify reaction conditions for selective epoxidation of the C11-C12 trisubstituted alkene of 26. Standard reaction conditions for Shi epoxidation21 led to initial partial oxidation of the C14 1,1-disubstituted alkene, while prolonged exposure to the reaction conditions for this oxidation process was insufficient to oxidize both the C14 1,1-disubstituted and the C11-C12 trisubstituted alkene. In an attempt to advance substrate 10 to the desired product, mCPBA was also investigated as a potential oxidant but was similarly ineffective for accomplishing the desired epoxidation/cyclization sequence.
Scheme 5.

NHK coupling between 3 and 4, and attempted epoxidation/cyclization cascade.
In an attempt to overcome the unexpected difficulty in site-selective oxidation of the triene 26, we turned our attention to a substrate-controlled iodoetherification reaction to establish the 2,2,5-cis trisubstituted THF C-ring of PTX2. To our delight, treatment with N-iodosuccinimide in dichloromethane led to efficient cyclization of the C15 hydroxy group onto the trisubstituted alkene to deliver the polycyclic product containing the desired 2,2,5-cis trisubstituted THF 27 in 87% yield (dr = 12:1; Scheme 6). Stereochemical control in this process is quite interesting, as early studies of a related cyclization by Rychnovsky and Bartlett documented the preference of such ring-forming reactions to deliver 2,2,5-trans trisubstituted THFs with outstanding stereocontrol (ds = 20:1).23 While further study is required to understand the sense of stereoselection observed here, the transition state model A (Scheme 6) does not adequately support the selectivity observed in the conversion of 26 to 27. We propose that stereochemical control in this reaction is a result of an organized transition state that features minimization of A1,3 strain and stabilization by an intramolecular hydrogen bond (see B in Scheme 6).24
Scheme 6.

Closure of the C-ring via iodoetherification.
In conclusion, we report a synthesis of the C1-C26 hexacyclic subunit of PTX2 that proceeds by convergent establishment of the C-ring through sequential Nozaki-Hiyama-Kishi coupling and oxidative cyclization. Our model studies have confirmed that this general strategy is quite effective for generating either 2,2,5-cis or 2,2,5-trans trisubstituted tetrahydrofurans when employing substrates that are amenable to site- and stereoselective Sharpless epoxidation. This annulative process was initially employed to prepare the C10-C26 subunit of PTX2 (12), but proved problematic when challenged with a more complex coupling partner containing the C1-C14 northern hemisphere of PTX2 (3). In efforts to circumvent the unanticipated low reactivity of the C11-C12 trisubstituted alkene of 26 toward standard conditions for stereoselective epoxidation, we turned to iodoetherification as an alternative means of ring closure. The stereochemical control that we achieved in the conversion of 26 to the ABCDEF subunit of PTX2 27 is unique among iodoetherification reactions and may speak to the role that hydrogen bonding can play in dictating the stereochemical course of these cyclization reactions. Whether or not intermediate 27 will serve as a useful intermediate in efforts to prepare PTX2 is the subject of ongoing studies.
Supplementary Material
Acknowledgments
The authors are grateful for financial support of this work by the National Institutes of Health-NIGMS (GM080266), Scripps Research Institute and the Japan Society for the Promotion of Science (JSPS, postdoctoral fellowship to O.K.).
Footnotes
Supporting Information Available Experimental procedures and tabulated spectroscopic data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Chae H, Choi T, Kim B, Jung J, Bang Y, Shin DY. Oncogene. 2005;24:4813–4819. doi: 10.1038/sj.onc.1208640. [DOI] [PubMed] [Google Scholar]
- 2.Jung JH, Sim CJ, Lee C. J Nat Prod. 1995;58:1722–1726. doi: 10.1021/np50125a012. [DOI] [PubMed] [Google Scholar]
- 3.Spector I, Braet F, Shochet nR, Bubb MR. Micro Res And Tech. 1999;47:18–37. doi: 10.1002/(SICI)1097-0029(19991001)47:1<18::AID-JEMT3>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 4.(a) Yasumoto T, Murate M, Oshima Y, Sano M, Matsumoto GK, Clardy J. Tetrahedron. 1985;41:1019–1025. [Google Scholar]; (b) Sasaki K, Wright JLC, Yasumoto T. J Org Chem. 1998;63:2475–2480. doi: 10.1021/jo971310b. [DOI] [PubMed] [Google Scholar]; (c) Yasumoto T, Murata M. Chem Rev. 1993;93:1897–1909. [Google Scholar]
- 5.(a) Yasumoto T, Oshima Y, Sugawara W, Fukuyo Y, Oguri H, Igarashi T, Fujita N. Bull Jpn Soc Sci Fish. 1980;46:1405–1411. [Google Scholar]; (b) Lee J, Igarashi T, Fraga S, Dahl E, Hovgaard P, Yasumoto T. J Appl Phycol. 1989;1:147–152. [Google Scholar]; (c) Draisci R, Lucentini L, Giannetti L, Boria P, Poletti R. Toxicon. 1996;34:923–935. doi: 10.1016/0041-0101(96)00030-x. [DOI] [PubMed] [Google Scholar]
- 6.Allingham JS, Miles CO, Rayment I. J Mol Bio. 2007;371:959–970. doi: 10.1016/j.jmb.2007.05.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chae HD, Kim BM, Yun UJ, Shin DY. Oncogene. 2008;27:4115–4121. doi: 10.1038/onc.2008.46. [DOI] [PubMed] [Google Scholar]
- 8.For total syntheses of pectenotoxin-4 and -8, see: Evans DA, Rajapakse HA, Stenkamp D. Angew Chem Int Ed. 2002;41:4569–4573. doi: 10.1002/1521-3773(20021202)41:23<4569::AID-ANIE4569>3.0.CO;2-V.Evans DA, Rajapakse HA, Chiu A, Stenkamp D. Angew Chem Int Ed. 2002;41:4573–4576. doi: 10.1002/1521-3773(20021202)41:23<4573::AID-ANIE4573>3.0.CO;2-S.
- 9.For partial synthesis, and studies toward the syntheses of the pectenotoxins, see: Micalizio GC, Roush WR. Org Lett. 2001;3:1949–1952. doi: 10.1021/ol0160250.Paquette LA, Peng X, Bondar D. Org Lett. 2002;4:937–940. doi: 10.1021/ol010302l.Pihko PM, Aho JE. Org Lett. 2004;6:3849–3852. doi: 10.1021/ol048321t.Bondar D, Liu J, Müller T, Paquette LA. Org Lett. 2005;7:1813–1816. doi: 10.1021/ol0504291.Halim R, Brimble MA, Merten J. Org Lett. 2005;7:2659–2662. doi: 10.1021/ol0507975.Vellucci D, Rychnovsky SD. Org Lett. 2007;9:711–714. doi: 10.1021/ol0630447.O’Connor PD, Knight CK, Friedrich D, Peng X, Paquette LA. J Org Chem. 2007;72:1747–1754. doi: 10.1021/jo062513f.Lotesta SD, Hou Y, Williams LJ. Org Lett. 2007;9:869–872. doi: 10.1021/ol063087n.Heapy AM, Wagner TW, Brimble MM. Synlett. 2007:2359–2362.Fujiwara K, Aki Y, Yamamoto F, Kawamura M, Kobayashi M, Okano A, Awakura D, Shiga S, Murai A, Kawai H, Suzuki T. Tetrahedron Lett. 2007;48:4523–4527.Kolakowski RV, Williams LJ. Tetrahedron Lett. 2007;48:4761–4764.Aho JE, Salomäki E, Rissanen K, Pihko PM. Org Lett. 2008;10:4179–4182. doi: 10.1021/ol8015868.Carley S, Brimble MA. Org Lett. 2009;11:563–566. doi: 10.1021/ol8025457.Joyasawal S, Lotesta SD, Akhmedov NG, Williams LJ. Org Lett. 2010;12:988–991. doi: 10.1021/ol902984e.Canterbury DP, Micalizio GC. Org Lett. 2011;13:2384–2387. doi: 10.1021/ol200627d.
- 10.Choi H, Demeke D, Kang F, Kishi Y, Nakajima K, Nowak P, Wan Z, Xie C. Pure Appl Chem. 2003;75:1–17. [Google Scholar]
- 11.Guo H, Dong C, Kim D, Urabe D, Wang J, Kim JT, Liu X, Sasaki T, Kishi Y. J Am Chem Soc. 2009;131:15387–15393. doi: 10.1021/ja905843e. [DOI] [PubMed] [Google Scholar]
- 12.Bravo F, McDonald FE, Neiwert WA, Do B, Hardcastle KI. Org Lett. 2003;5:2123–2126. doi: 10.1021/ol034539o. [DOI] [PubMed] [Google Scholar]
- 13.Davies KA, Abel RC, Wulff JE. J Org Chem. 2009;74:3997–4000. doi: 10.1021/jo900444x. [DOI] [PubMed] [Google Scholar]
- 14.Fuwa H, Nakajima M, Shi J, Takeda Y, Saito T, Sasaki M. Org Lett. 2011;13:1106–1109. doi: 10.1021/ol1031409. [DOI] [PubMed] [Google Scholar]
- 15.Katsuki T, Martin VS. Org React. 1996;48:1–299. [Google Scholar]
- 16.Unfortunately, all attempts to employ an epoxide-containing vinyl iodide in the NHK coupling delivered a complex mixture of products.
- 17.(a) Reichard HA, Micalizio GC. Angew Chem Int Ed. 2007;46:1440–1443. doi: 10.1002/anie.200603515. [DOI] [PubMed] [Google Scholar]; (b) Canterbury DP, Micalizio GC. J Am Chem Soc. 2010;132:7602–7604. doi: 10.1021/ja102888f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Reichard HA, Micalizio GC. Chem Sci. 2011;2:573–589. doi: 10.1039/C0SC00394H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.(a) Frantz DE, Fässler R, Carreira EM. J Am Chem Soc. 2000;122:1806–1807. [Google Scholar]; (b) Sasaki H, Boyall D, Carreira EM. Helv Chim Acta. 2001;84:964–971. [Google Scholar]; (c) Boyall D, Frantz DE, Carreira EM. Org Lett. 2002;4:2605–2606. doi: 10.1021/ol026282k. [DOI] [PubMed] [Google Scholar]
- 19.For a review of synthetic applications of organochlorozirconocene complexes, see: Wipf P, Jahn H. Tetrahderon. 1996;52:12853–12910.For the preparation of 2-iodo-allylbromide, see: Kurosu M, Lin M, Kishi Y. J Am Chem Soc. 2004;126:12248–12249. doi: 10.1021/ja045557j.For a similar process for allylation of a vinyliodide, see: Kitagawa K, Inoue A, Shinokubo H, Oshima K. Angew Chem Int Ed. 2000;39:2481–2483. doi: 10.1002/1521-3773(20000717)39:14<2481::aid-anie2481>3.0.co;2-j.
- 20.Duplantier AJ, Nantz MH, Roberts JC, Short RP, Somfai P, Masamune S. Tetrahedron Lett. 1989;30:7357–7360. [Google Scholar]
- 21.(a) Wang Z, Tu Y, Frohn M, Zhang J, Shi Y. J Am Chem Soc. 1997;119:11224–11235. [Google Scholar]; (b) Wong OA, Shi Y. Chem Rev. 2008;108:3958–3987. doi: 10.1021/cr068367v. [DOI] [PubMed] [Google Scholar]; (c) Diffendal JM, Danheiser RL. Org Synth. 2009;coll.11:183–188. [Google Scholar]
- 22.Houk KN, Moses SR, Wu Y, Rondan NG, Jäger V, Schohe R, Fronczek FR. J Am Chem. 1984;106:3880–3882. [Google Scholar]
- 23.Rychnovsky SD, Bartlett PA. J Am Chem Soc. 1981;103:3963–3964. [Google Scholar]
- 24.When the iodoetherification is conducted in acetonitrile, a significant decrease in stereoselection is observed (2,2,5-cis:2,2,5-trans = 3:1).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.