

Abstract
Purpose
Genetic alterations of KRAS, CDKN2A, TP53 and SMAD4 are the most frequent events in pancreatic cancer. We determined the extent to which these four alterations are coexistent in the same carcinoma, and their impact on patient outcome.
Experimental Design
Pancreatic cancer patients who underwent an autopsy were studied (n=79). Matched primary and metastasis tissues were evaluated for intragenic mutations in KRAS, CDKN2A and TP53 and immunolabeled for CDKN2A, TP53 and SMAD4 protein products. The number of altered driver genes in each carcinoma was correlated to clinicopathologic features. Kaplan-Meier estimates were used to determine median disease free and overall survival, and a Cox proportional hazards model used to compare risk factors.
Results
The number of genetically altered driver genes in a carcinoma was variable, with only 29 patients (37%) having an alteration in all four genes analyzed. The number of altered driver genes was significantly correlated with disease free survival (p=0.008), overall survival (p=0.041) and metastatic burden at autopsy (p=0.002). On multivariate analysis, the number of driver gene alterations in a pancreatic carcinoma remained independently associated with overall survival (p=0.046). Carcinomas with only one to two driver alterations were enriched for those patients with the longest survival (median 23 months, range 1–53).
Conclusions
Determinations of the status of the four major driver genes in pancreatic cancer, and specifically the extent to which they are coexistent in an individual patients cancer, provides distinct information regarding disease progression and survival that is independent of clinical stage and treatment status.
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is one of the most lethal solid malignancies and a major cause of cancer-related deaths in developed countries (1), with a > 95% mortality rate. Most patients present with locally advanced or metastatic disease at initial diagnosis leaving relatively few as candidates for a potentially curative resection. Unfortunately, even in patients who undergo pancreatic resection, both local and systemic recurrences are common with a median post-resection survival of less than 18 months (2).
The recent completion of the pancreatic cancer exome marked a notable milestone (3). The coding regions of 20,661 genes were sequenced in 24 PDACs indicating that these neoplasms contain an average of 63 genomic alterations, the majority of which are point mutations. Moreover, the genetic landscape of the PDAC genomes is notable for four frequently mutated genes, designated “mountains”, including KRAS, CDKN2A (p16), TP53 and SMAD4 (DPC4). Numerous candidate cancer genes altered at low frequency, designated “hills”, were also identified such as MLL3 and ARID1A (3, 4). These four mountain genes are well recognized as contributing to pancreatic carcinogenesis (5), and are thus classifiable as “driver” genes for this tumor type. Furthermore, based on comparative lesion sequencing these four genes are also classifiable as “founder” mutations in that they are present in the original parental clone that gave rise to the infiltrating carcinoma (6). While additional genetic alterations accumulate during the ongoing clonal evolution of the carcinoma (“progressor” mutations), the constellation of founder mutations contained within the parental clone presumably constitutes the major characteristics for that carcinoma (6, 7).
The relationship between the genetic status of these four genes and clinicopathological features, including survival, have been previously studied. However, until now this work has focused on individual genes and has yielded conflicting results (8–14). Furthermore, although genetically engineered mouse models indicate that the concomitant expression of these mutated genes is crucial to progress to invasion and metastasis in PDACs (15–19), the extent to which the coexistence of three or more of these altered genes in the same PDAC influence the biological behavior and survival outcome is unknown.
The objective of the current study was to clarify the clinical significance of the genetic landscape of pancreatic cancer, specifically the genetic status of the KRAS, CDKN2A, TP53 and SMAD4 driver genes in a large series of nonfamilial advanced stage PDACs with known outcomes including patterns of failure and in a second set of xenografted PDACs. We now show that there are distinct patterns and prevalences to which these driver genes occur in the same carcinoma, and that these patterns are highly correlated with clinical features of patients.
PATIENTS AND METHODS
Patients and tissue samples
Paraffin-embedded and snap-frozen tissue samples from 79 patients collected in association with the Gastrointestinal Cancer Rapid Medical Donation Program (GICRMDP) were used. This program was previously reported in detail (20). Among these 79 patients, 20 initially underwent surgical resection and the remaining 59 patients were initially diagnosed with Stage III/IV unresectable disease. Based on autopsy findings and clinical chart review, all patients died of causes directly related to their disease. The Johns Hopkins Institutional Review Board approved use of all patient samples for this study.
Sanger sequencing
Snap frozen tissue samples were embedded in OCT compound (Sakura Finetek, Tokyo, Japan), sectioned by a cryostat and stained by hematoxylin and eosin. Tumor tissues were dissected macroscopically if the neoplastic cellularity was at least 50%, or microscopically using a PALM MicroLaser System (Carl Zeiss MicroImaging, Oberkochen, Germany) for cases with low neoplastic cellularity. Genomic DNA from dissected tissues was extracted using phenol-chloroform, or QIAmp DNA Micro Kits if microdissected (Qiagen, Valencia, CA). Genomic DNA from microdissected tissues was quantified by calculating long interspersed nuclear elements (LINE) by real-time PCR as described previously (6) and whole genome amplification (WGA) was performed using 10 ng total template gDNA and an illustra GenomiPhi V2 DNA Amplification Kit (GE Healthcare, Buckinghamshire, UK). PCR amplification was performed using 20 ng of gDNA for KRAS exons 1 and 2, TP53 exons 5–9 and CDKN2A exons 1 and 2 using intronic primers flanking these exons (Supplemental Table 1). PCR products were sequenced by use of a M13F primer (5′-GTAAAACGACGGCCAGT-3′) or M13R primer (5′-CAGGAAACAGCTATGACC-3′) that was incorporated into the forward and reverse primer of each primer pair, respectively (Beckman Coulter Genomics, Danvers, MA). Sequencing data were analyzed with Sequencher 4.10 software (Gene Codes, Ann Arbor, MI). Mutation analysis, confirmation and determination of somatic status were carried out using matched normal tissues from the same patient.
Immunohistochemistry
Paraffin-embedded samples of the primary carcinoma and matched metastases were immunolabeled for Cdkn2A, Tp53 and Smad4 as an adjunct to sequencing. At least five different distinct regions of the primary carcinoma were immunolabeled for each case to evaluate for potential heterogeneity. In the event of positive immunolabeling for Cdkn2A or Smad4 in the primary carcinoma, at least five different matched metastases, and local recurrences if available, were also labeled to assess for gene inactivation during disease progression. Immunohistochemical labeling was performed using antibodies to Cdkn2A protein (ready-to-use, clone E6H4, MTM Laboratories), Tp53 protein (ready-to-use, Bp-53-11, Ventana) and Smad4 protein (clone B8, Santa Cruz Biotechnology) as reported (21). Nuclear labeling of Cdkn2A was scored as intact (positive, indicating the presence of an intact gene) or lost (negative, indicating a deletion, inactivating mutation or promoter hypermethylation) (22, 23). As previously described (21), Tp53 immunolabeling was considered abnormal when it showed robust nuclear accumulation of immunolabeled protein in ≥ 30% of the neoplastic cells compared to adjacent normal cells, or if the neoplastic cells showed a virtual absence of immunolabeling compared to immediately adjacent normal cells suggesting the presence of an intragenic deletion, nonsense or frameshift mutation (24–26). In all instances p53 labeling was evaluated within sections cut from at least two different paraffin blocks of the same carcinoma. Nuclear labeling of Smad4 was scored as intact (positive, indicating the presence of an intact gene) or lost (negative, indicating a deletion or inactivating mutation of the gene has occurred) (27). Normal islets for Cdkn2A and normal acinar cells, islets, lymphocytes and stromal cells for TP53 and Smad4 were regarded as internal positive controls for each case. Negative controls for each of the antibodies were performed using nonimmune serum instead of the primary antibody. Slides were scored by two of the authors (S.Y and C.I.D.).
Statistics
Dichotomous variables were compared using Fisher’s exact test or the chi-square test, and continuous variables were compared using the Student’s t-test or the Mann-Whitney U-test, where appropriate. Multiple groups were compared by the Kruskal-Wallis test or the chi-square test, where appropriate. Survival analyses were performed by the Kaplan-Meier method or Cox regression and survival curves were compared with the logrank test. P values ≤ 0.05 was considered statistically significant. Statistical analyses were performed using SPSS 20.0 software (SPSS, Chicago, IL).
RESULTS
Clinicopathologic Features of Autopsied Patients
The clinicopathologic features of all 79 patients with lethal pancreatic ductal adenocarcinoma whose tissues were collected in association with the GICRMDP are summarized in Table 1. Detailed findings at autopsy of 60 of these patients were previously described (28). Among all 79 patients, 56% were male and 81% of the primary carcinomas developed in the head or body of the pancreas. Most patients (75%) had advanced stage disease at diagnosis (Stage III or IV), and this corresponded to a median overall survival of 10 months for all 79 patients. Nonetheless, when stratified by stage at diagnosis the median overall survival was 24 months for Stage I/II, 11.5 months for Stage III, and 6.5 months for Stage IV patients. At autopsy, 17 (85%) of Stage I/II patients had a local recurrence although for three of these it was the only site of disease found. The liver was the most common site of metastatic disease among all patients and was found in 76% of patients. However, the extent of metastatic disease burden among all patients varied greatly (less than 10 to >100), a reflection of the inherent “metastatic efficiency” of each patient’s pancreatic cancer (28).
Table 1.
Clinicopathologic Features of Patients
| Characteristic | Autopsy Patients (n=79) |
|---|---|
|
| |
| Age at Diagnosis, years (Mean ± SD) | 62.2±11.4 |
|
| |
| Gender (%) | |
| Male | 44 (56%) |
| Female | 35 (44%) |
|
| |
| Tumor location (%) | |
| Head/Body | 64 (81%) |
| Tail | 14 (18%) |
| NA | 1 (1%) |
|
| |
| Stage at Diagnosis (%) | |
| I/II | 20 (26%) |
| III | 19 (24%) |
| IV | 40 (50%) |
|
| |
| Tumor Differentiationd (%) | |
| Well/Moderate | 27 (34%) |
| Poor | 52 (66%) |
|
| |
| Treatment (%) | |
| Chemoradiation | 32 (41%) |
| Chemotherapy | 34 (43%) |
| None | 13 (16%) |
|
| |
| Median overall survival, months (range) | 10 (0.75 – 58) |
|
| |
| Major Sites Involved by Metastatic Disease at Autopsya (%) | |
| Liver (n=79) | 60 (76%) |
| Lung (n=65)b | 31 (48%) |
| Peritoneum (n=69)c | 41 (59%) |
|
| |
| Number of Sites Involved by Metastatic Disease (%)b,c | |
| 0 | 8 (10%) |
| 1 | 26 (33%) |
| 2 | 29 (37%) |
| ≥3 | 16 (20%) |
|
| |
| Metastatic burden (%) | |
| 0–10 (oligometastatic) | 22 (28%) |
| 11–100 (moderate) | 27 (34%) |
| >100 (widely metastatic) | 29 (37%) |
Refers to frequency at each site independently.
Data regarding presence of lung metastasis not available for 14 patients.
Data regarding presence of peritoneal metastasis not available for 10 patients. na, not available.
Genetic Features of Pancreatic Cancers Obtained from Autopsy
DNA was extracted from snap frozen samples of normal tissue, primary infiltrating ductal adenocarcinomas and multiple matched metastases for all patients and sequenced for KRAS, CDKN2A and TP53. Multiple samples taken from distinct regions of each primary carcinoma were analyzed (mean 5.9 samples per carcinoma), as well as multiple different metastases (mean 6.3 matched metastases per patient) corresponding to a total of 884 individual samples and greater than 2.5 million bases of sequencing data analyzed.
Activating mutations in KRAS were identified in 73 (92%) of 79 carcinomas analyzed (Supplemental Table 2). Mutations at codon 12 were most common (66 of 73 mutations, 90%), with G12D accounting for 38 (52%) of 73 carcinomas. For six carcinomas without a detectable KRAS mutation of codons 12,13 or 61 we also analyzed for mutations of codon 146 (29), but no mutations were found.
High quality sequencing data were obtained for CDKN2A in 76 of 79 patient’s carcinomas (Supplemental Table 2). Intragenic mutations were identified in 21 (28%) of 76 carcinomas analyzed, corresponding to eight (38%) missense mutations, seven (33%) nonsense mutations, and six (29%) frameshift mutations. All but one carcinoma with an intragenic mutation had loss of Cdkn2A protein expression. Because CDKN2A may undergo homozygous deletion or hypermethylation-induced silencing that would not be detected by sequencing (30), we also immunolabeled all 55 carcinomas in which no intragenic mutations were found. Of these, 48 (87%) had loss of Cdkn2A labeling. In total, loss of Cdkn2A secondary to any potential mechanism was detected in 72 of 79 (91%) carcinomas analyzed.
Inactivating mutations in TP53 were identified in 58 of 79 (73%) carcinomas, of which 28 (48%) were missense mutations, 11 (19%) were frameshift mutations, nine (16%) were nonsense mutations, six (10%) were intragenic deletions, and four (7%) were splice-site mutations (Supplemental Table 2). Carcinomas found to be TP53 wild type by sequencing were also immunolabeled for Tp53 protein to assess for potential large homozygous deletions or mutations outside of the analyzed region. Of these, three had robust nuclear accumulation of Tp53 and five of 21 had complete absence of Tp53 protein. Overall, TP53 was altered in 66 of 79 (84%) carcinomas.
Finally, we also determined Smad4 immunolabeling patterns, which is a strong marker of SMAD4 genetic status (27, 31). Of 79 carcinomas analyzed, 39 (49%) showed loss of Smad4 immunolabeling consistent with inactivation of the SMAD4 gene (Supplemental Table 2).
In 73 patients analyzed (92%), there was complete concordance for genetic status and/or immunolabeling patterns of all genes in the primary carcinoma and the matched metastases. Of the remaining six patients, one showed intact Smad4 labeling in the primary carcinoma and peritoneal metastases, whereas the matched liver metastases in this patient showed loss of labeling, indicating genetic inactivation of SMAD4 occurred during subclonal evolution and metastatic progression (Figure 1A,1B). In an additional five patients intratumoral heterogeneity for Cdkn2A labeling was observed in the primary carcinoma in that regions of both strong positive and complete loss of labeling were seen (Figure 1C–1E). True heterogeneity versus a labeling artifact was confirmed by use of a second antibody to Cdkn2A raised against a different epitope of the protein that showed the identical pattern of labeling in these five carcinomas. One of these carcinomas contained a 6 bp in-frame deletion of the Cdkn2A gene, and the matched liver metastases showed complete loss of Cdkn2A labeling. In the remaining four carcinomas no mutations were found, and the matched liver metastases also had loss of Cdkn2A labeling.
Figure 1.
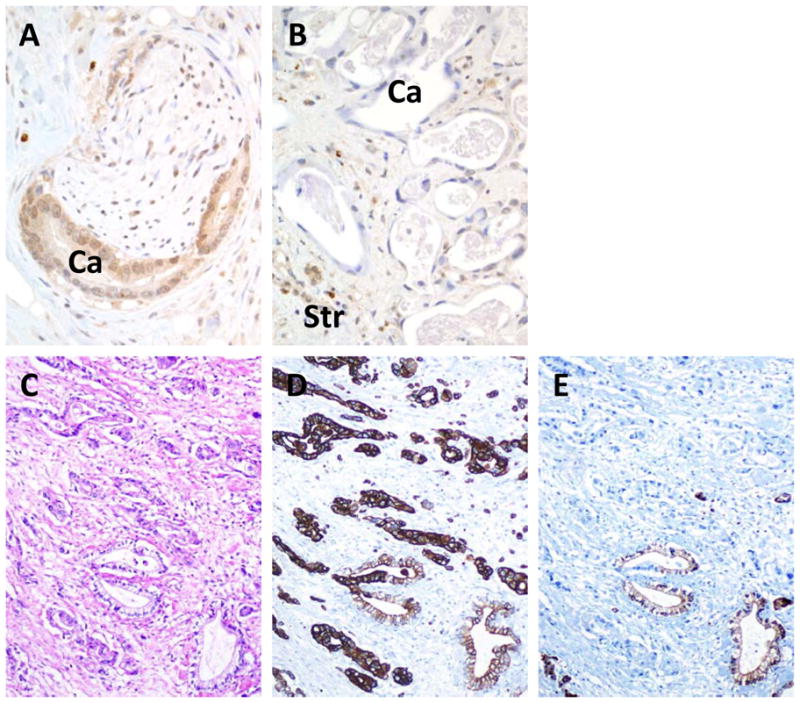
Deviant SMAD4 and CDKN2A immunohistochemical labeling patterns in pancreatic cancer tissues. A. Intact Smad4 immunolabeling in a primary carcinoma. Both nuclear and cytoplasmic labeling for SMAD4 is present within the neoplastic glands (Ca) in an area of perineural invasion. B. Loss of SMAD4 immunolabeling in a liver metastasis derived from the carcinoma shown in A. In this example, no labeling of SMAD4 is seen within the neoplastic glands (Ca). By contrast, positive labeling of surrounding stromal cells (Str) is present. C. Hematoxylin and eosin stained section of infiltrating pancreatic carcinoma. D. CK19 labeling of the carcinoma shown in C indicating strong positive labeling throughout the neoplastic epithelium. E. Example of focal loss of CDKN2A immunolabeling in the carcinoma shown in C. No labeling of CDKN2A is seen in the neoplastic epithelium within the upper half of the shown section, whereas strong positive labeling is seen within scattered neoplastic glands in the lower half.
Coexistent Genetic Alterations in Pancreatic Cancer
We next determined the specific genes altered in pancreatic cancers with one, two and three total genetic alterations (Table 2), as well as the type of alterations for these genes in each category. Of interest, for one aggressive carcinoma (patient A68) only a KRAS mutation was found, despite analysis of 8 different microdissected samples of the primary carcinoma and 24 different matched metastases. Among carcinomas with two genetic alterations, all 14 had a KRAS or CDKN2A alteration and nine of 14 (64%) harbored an alteration in both KRAS and CDKN2A. The remaining five carcinomas had either a KRAS or CDKN2A alteration in combination with a TP53 alteration. Among the 35 carcinomas with three genetic alterations, all 35 had a KRAS or CDKN2A alteration and for 28 of 35 (80%) carcinomas KRAS and CDKN2A were coexistent. Moreover, 25 of these 28 carcinomas (89%) contained TP53 as the third genetic alteration, and the remaining three carcinomas contained loss of SMAD4 as the third genetic alteration. The remaining seven of 35 (20%) carcinomas had a KRAS or CDKN2A alteration in association with both TP53 and SMAD4 alterations.
Table 2.
Coexistence of KRAS, CDKN2A, TP53 and SMAD4 Alterations in Pancreatic Cancer
| Category | Autopsy Patients (n=79) | Xenografts (n=84) |
|---|---|---|
|
| ||
| One Gene | ||
| KRAS | 1 (100%) | - |
|
| ||
| Two Genes | ||
| KRAS/CDKN2A | 9 (64%) | 9 (75%) |
| KRAS/TP53 | 2 (14%) | 2 (17%) |
| CDKN2A/TP53 | 3 (21%) | 1 (8%) |
|
| ||
| Three Genes | ||
| KRAS/CDKN2A/TP53 | 25 (71%) | 33 (85%) |
| KRAS/CDKN2A/SMAD4 | 3 (9%) | 5 (13%) |
| KRAS/TP53/SMAD4 | 4 (11%) | 1 (2%) |
| CDKN2A/TP53/SMAD4 | 3 (9%) | 0 |
|
| ||
| Four Genes | ||
| KRAS/CDKN2A/TP53/SMAD4 | 29 (100%) | 33 (100%) |
Given the observations made in autopsied patients, we further explored the extent to which these driver gene alterations are coexistent in a second and more uniform set of xenografts derived from 84 pancreatic cancer patients with Stage I/II disease seen at our institution. The specific genetic features of KRAS, CDKN2A, TP53 and SMAD4 in these xenografts have previously been reported in association with whole exome sequencing of a large series of pancreatic cancers (3). These xenografts were also previously analyzed as part of a larger series of xenografted carcinomas evaluating the relationship of each of these genes to overall survival (8). However as the frequency and prevalence of coexistent mutations in xenografts from these patients were not addressed, we focused specifically on that aspect.
The genetic features of KRAS, CDKN2A, TP53 and SMAD4 in these xenografts were similar to that found for the autopsy cohort. All but one carcinoma (99%) had a mutation in KRAS with G12D the most common mutation identified in 40 of 84 (48%) carcinomas analyzed. Inactivating mutations or homozygous deletions of CDKN2A were found in 81 of 84 carcinomas (96%), and of TP53 in 71 of 84 (83%) of these same cases. Inactivation of SMAD4 by mutation or homozygous deletion was identified in 39 of 84 (46%) carcinomas and was most often seen in association with TP53 mutation (34 of 39, 87%). The frequency at which these driver gene alterations were coexistent in a single pancreatic cancer was also similar to the autopsy cohort, with the majority of carcinomas also having three (46%) or four (39%) coexistent alterations. Thus, our findings of the frequency and coexistence of driver genes in autopsied patients is likely correct and not an underestimate due to our sample type analyzed.
Given that SMAD4 loss was commonly seen in association with TP53 inactivation, we further explored this relationship. SMAD4 inactivation always occurred in association with two or three coexistent driver gene alterations, and the vast majority of SMAD4 inactive carcinomas had coexistent TP53 mutations (36 of 39, 92%). By contrast, TP53 alterations were equally likely to be found independent of SMAD4 inactivation with 35 of 66 (53%) in SMAD4 wild type carcinomas versus 31 of 66 (47%) in association with SMAD4 loss. SMAD4 status alone was significantly correlated with high metastatic burden (p=0.008), as was TP53 status (p=0.039). However, as these two gene alterations are commonly coexistent we compared the features among pancreatic cancers with TP53 alterations only, with SMAD4 alterations only, with alterations in both genes and in neither gene. To our surprise, TP53 alterations were similarly correlated with high metastatic burden disease when they occurred with or without coexistent SMAD4 alterations (p=0.170), and differed from carcinomas without TP53 and SMAD4 alterations in which metastatic burden was more commonly oligometastatic (p=0.008). To determine if the types of TP53 alterations differ among these groups to explain this observation, we assessed the frequency of TP53 missense versus null mutations (nonsense, deletion or frameshift) in the 58 carcinomas with complete sequencing data available. Of interest, null mutations were significantly more common in SMAD4 intact carcinomas (18 of 28, 64%) than in carcinomas with SMAD4 loss (7 of 22, 38%, p=0.046). Collectively, this suggests that pancreatic cancers with high metastatic efficiency may be represented by at least two genetic subtypes, i.e. TP53 null mutant and TP53 missense mutant in association with SMAD4 loss.
Relationships of Genetic Features to Clinical Features in Pancreatic Cancer Patients
Among all 79 carcinomas analyzed, one (1%) had a single detectable gene alteration, 14 (18%) had two gene alterations, 35 (44%) had three gene alterations and 29 (37%) had an alteration in all four genes analyzed (Table 2). Carcinomas with one or two alterations only were combined into a single group, as were carcinomas with three or four alterations, and the relationships of the number of genetic alterations to clinical features of each patient’s carcinoma was analyzed (Table 3). There were no differences in mean age or gender distribution among patients in relation to number of gene alterations, nor were there differences in tumor size, location or differentiation at initial diagnosis. No relationship was found either with clinical stage at diagnosis, although 1–2 gene mutant carcinomas were twice more commonly observed in association with Stage I/II disease (30% of patients, versus 15% of Stage III and 15% of Stage IV). By univariate analysis the number of altered genes was significantly correlated with both median disease free survival (p=0.008) in patients with Stage I/II disease, and median overall survival (p=0.041) (Figure 2) among all patients although this was not maintained when separated out by stage. However, a greater number of altered genes was also significantly correlated with high metastatic burden at autopsy with 10 of 15 (66%) patients with 1–2 altered genes having oligometastatic failure compared to 2 of 29 (14%) of patients with widespread metastatic failure (p=0.002) (Table 4). This relationship was also maintained when patients were stratified by tumor stage. Of interest, when controlling for clinical stage at diagnosis the number of altered genes remained significantly correlated to patient survival (p=0.046) (Table 5).
Table 3.
Relationship of Number of Genetic Alterations to Clinical Features in 79 Autopsied Pancreatic Cancer Patients
| Feature | Number of Altered Genes
|
P Value | |
|---|---|---|---|
| 1–2 (n=15) | 3–4 (n=64) | ||
|
| |||
| Age (yrs) | 66.1±9.0 | 61.3±11.7 | 0.147 |
|
| |||
| Gender | 0.469 | ||
| Male | 9 (20%) | 35 (80%) | |
| Female | 6 (17%) | 29 (83%) | |
|
| |||
| Clinical Stage at Diagnosis | 0.347 | ||
| I/II | 6 (30%) | 14 (70%) | |
| III | 3 (15%) | 16 (85%) | |
| IV | 6 (15%) | 34 (85%) | |
|
| |||
| Tumor Size at Diagnosis (cm) | |||
| I/II | 2.7±0.8 | 3.2±1.5 | 0.468 |
| III | 4.7±2.8 | 3.6±1.0 | 0.195 |
| IV | 4.9±2.0 | 4.3±1.5 | 0.429 |
|
| |||
| Tumor Locationa | 0.865 | ||
| Head/Body | 12 (80%) | 52 (81%) | |
| Tail | 3 (20%) | 11 (19%) | |
|
| |||
| Tumor Differentiation | 0.404 | ||
| Well/Moderate | 6 (40%) | 43 (67%) | |
| Poor | 9 (60%) | 21 (33%) | |
|
| |||
| Median Disease Free Survival, Stage I/II (mo) | 20 | 7 | 0.008 |
|
| |||
| Median Overall Survival (mo) | |||
| All stages | 23 | 9 | 0.041 |
| I/II only | 24 | 24 | 0.448 |
| III only | 18 | 10 | 0.134 |
| IV only | 2 | 6 | 0.428 |
info on one patient not available.
Figure 2.

Kaplan-Meier survival curves demonstrating the relationship of number of driver gene alterations (1–2 versus 3–4) to disease free survival in 20 Stage I/II patients specifically (A) and overall survival among all 79 patients (B). Survival curves were compared by a log rank test. The percent of patients alive at interval time points are also indicated for each arm.
Table 4.
Relationship of Number of Genetic Alterations to Metastatic Burden in Autopsied Pancreatic Cancer Patients
| Feature | Number of Altered Genes
|
P Value | |
|---|---|---|---|
| 1–2 (n=15) | 3–4 (n=64) | ||
|
All Patients (n=79)
| |||
| Metastatic Burden (All Patients) | 0.002 | ||
| Oligometastatic (≤10) | 10 (43%) | 13 (52%) | |
| Moderate (11–100) | 3 (11%) | 24 (89%) | |
| Widely metastatic (>100) | 2 (7%) | 27 (93%) | |
|
| |||
|
Stage I/II Patients Only (n=20)
| |||
| Metastatic Burden | 0.019 | ||
| Oligometastatic (≤10) | 4 (80%) | 1 (20%) | |
| Moderate (11–100) | 1 (13%) | 7 (87%) | |
| Widely metastatic (>100) | 1 (14%) | 6 (86%) | |
|
| |||
|
Stage III/IV Patients Only (n=59)
| |||
| Metastatic Burden | 0.033 | ||
| Oligometastatic (≤10) | 6 (33%) | 12 (66%) | |
| Moderate (11–100) | 2 (11%) | 17 (89%) | |
| Widely metastatic (>100) | 1 (5%) | 21 (95%) | |
Table 5.
Cox Regression Analysis of Driver Genes versus Clinical Stage
| Hazard Ratio | 95.0% CI | P value | |
|---|---|---|---|
| Clinical Stage at Diagnosis (I/II versus III versus IV) | .211 | .114–.390 | .000 |
| Number of Driver Genes (1/2 versus 3 versus 4) | 1.392 | 1.006–1.927 | .046 |
DISCUSSION
The pancreatic cancer progression model illustrates the approximate timing of accumulation of genetic alterations during PanIN progression (32). KRAS mutations are an early event and are followed by inactivating mutations in CDKN2A, whereas TP53 and SMAD4 alterations occur relatively later during PanIN-3. While our data are in agreement with this model, they also suggests that this mode of genetic progression likely occurs for only a subset of patients in that only 37–39% of carcinomas contain alterations in all genes. Thus, a more complete understanding of the extent to which alterations of these genes are coexistent in pancreatic cancer should not only provide insight into the dynamics by which they occur during pancreatic carcinogenesis, but also the biologic features of the infiltrating carcinomas that developed from those precursors.
The major clinical implication of this work is that knowledge of the gene status of the four major driver genes in pancreatic cancer, and specifically the extent to which they are coexistent in an individual patients cancer, provides distinct information regarding patterns of disease progression, metastatic failure and survival outcome. It is important to emphasize that other genes also play an important role in the biology of pancreatic cancer, for example inactivating BRCA2, PALB2 or FANC gene mutations that may confer susceptibility to cisplatin or PARP inhibitors (33, 34). However, because mutations in those genes are relatively uncommon our rationale was to identify genetic factors that influence outcomes for a greater number of patients. For example, among Stage I/II patients’ carcinomas with two driver gene alterations were associated with relatively longer median disease-free survival, and carcinomas with two driver gene alterations were significantly more likely to develop oligometastatic failure. Ultimately, while the demographics of these patients are entirely in keeping with the epidemiology and clinical features of larger cohorts of patients in well-controlled studies, additional validations in a controlled setting will be necessary.
The most common initiating genetic events in pancreatic cancer are oncogenic mutations in KRAS and inactivating mutations, deletions or methylation of CDKN2A (30), and the sole identification of these two driver genes accounted for many of these cases. However, in other carcinomas the two driver gene alterations corresponded to alternative combinations, for example KRAS and TP53, but importantly never included SMAD4. Overall, these carcinomas with “two” driver genes had significantly longer disease free and median overall survival, suggesting the subset of patients whose carcinomas have these genetic features may be enriched for long-term survivors. Of note, it is highly likely that additional genes may be mutated in the TP53 (apoptotic) and TGFβ pathways in these carcinomas that were not evaluated by our approach. For example, Jones et al proposed that the significance of genetic alterations in pancreatic cancer were largely for their indication of the core signaling pathways they occurred in, and that while more than one gene may be targeted in a pathway only one gene of the pathway is targeted per carcinoma (3). Moreover, it is conceivable that these alternative genetic alterations may not have the same effects on survival or progression as for TP53 and SMAD4 that are the most frequent genetic targets in their respective pathways. Consistent with this notion, Blackford et al found that among all members of the TGFβ signaling pathway that may be genetically inactivated in pancreatic cancer, only SMAD4 loss is associated with worse overall survival (8). By contrast, in one patient in our study only a KRAS mutation was found despite careful methodology, and this patient had widespread metastatic disease at autopsy following a mere 5 month overall survival, suggesting relatively rare genetic events occurred during carcinogenesis leading to a particularly aggressive phenotype (35).
We have previously shown that SMAD4 status of the primary carcinoma correlates with patterns of failure in pancreatic cancer (28), and now extend this observation by illustrating that SMAD4 loss is most often seen in the setting of coexistent mutations in TP53. In this regard, SMAD4 loss is a marker of genetically complex pancreatic cancers (i.e. those with all four driver gene mutations). These data also clarify prior observations that not all patients with widespread metastatic disease at autopsy have SMAD4 loss, and provide evidence that mutations that specifically abolish TP53 gene expression may also promote widespread metastatic failure independently of SMAD4 loss in some patients. Thus, determinations of both SMAD4 and TP53 status may have value in identifying patients at risk for widespread metastatic failure. Furthermore, as additional genes are functionally validated as drivers in this tumor type (3, 4), it is conceivable that they will provide added information regarding prognosis and risk of metastatic failure for pancreatic cancer patients.
KRAS mutations in normal cells leads to replicative senescence (36), and it has been suggested that CDKN2A inactivation provides a selective advantage to KRAS mutant cells by allowing cell division to proceed unhampered through the G1 checkpoint (37). That the vast majority of pancreatic cancers in this study have coexistent KRAS and CDKN2A mutations (89%) provides support to this concept. Beyond KRAS and CDKN2A, the frequencies by which alterations in TP53 or SMAD4 occur are relatively lower. SMAD4 loss most often occurred in a background of TP53 mutations yet TP53 mutations occurred at similar frequency in the presence or absence of SMAD4 loss, suggesting SMAD4 inactivation follows TP53 during the genetic progression of PanINs. In this context SMAD4 loss may provide a selective advantage to cells with coexistent KRAS, CDKN2A and TP53 mutations. In support of this hypothesis, we noted that TP53 null mutations were less commonly found in association with SMAD4 inactivation suggesting that TP53 null mutations select against SMAD4 loss. Alternatively, TP53 null mutations may have similar “potency” in progressing to an infiltrating carcinoma as coexistent TP53 missense mutations and SMAD4 loss. Consistent with this concept the metastatic burden of patients whose carcinomas corresponded to these two genetic categories (KRAS/CDKN2A/TP53-null versus KRAS/CDKN2A/TP53-missense/SMAD4) were similar to each other and significantly different from carcinomas that did not have TP53 or SMAD4 mutations.
The significance of exomic sequencing can only be realized by translational studies that include well-annotated patient data. We now demonstrate the clinical significance of such data for patients with pancreatic cancer. In time, these data may also have value for personalized approaches to management of pancreatic cancer patients.
Supplementary Material
STATEMENT OF TRANSLATIONAL RELEVENCE.
Irrespective of clinical stage at diagnosis, most patients with pancreatic cancer will die of their disease. Although genomic efforts have now clarified the genetic basis for pancreatic cancer, the relationship of the genetic landscape to an individual patients’ outcome is unknown. This study shows that there are distinct patterns and prevelences of the number of genetically altered driver genes in pancreatic cancer, a concept of significance for screening efforts based on identification of mutated alleles in body fluids. We also show that the number of altered driver genes is independently correlated with patient outcome, and that specific subsets of coexistent genes correspond to a greater incidence of metastatic failure. Finally, we show that carcinomas with one or two driver gene alterations identify a subset of patients with relatively more indolent disease, a finding of significance for early identification of long-term survivors.
Acknowledgments
Supported by National Institutes of Health grants CA140599, CA101955, CA62924 and CA121113, The Uehara Memorial Foundation, The Alfredo Scatena Memorial, The George Rubis Endowment for Pancreatic Cancer Research, The Michael Rolfe Pancreatic Cancer Foundation, Sigma Beta Sorority, The Joseph C. Monastra Foundation, The Gloria Swan Pancreatic Cancer Foundation, The Skip Viragh Pancreatic Cancer Center, The Patty Boshell Pancreas Cancer Foundation and an AACR Stand Up To Cancer-Dream Team Translational Cancer Research Grant.
Footnotes
The authors have no financial conflicts of interest related to this work.
References
- 1.Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62(1):10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- 2.Mayo SC, Gilson MM, Herman JM, Cameron JL, Nathan H, Edil BH, et al. Management of patients with pancreatic adenocarcinoma: national trends in patient selection, operative management, and use of adjuvant therapy. J Am Coll Surg. 2012;214(1):33–45. doi: 10.1016/j.jamcollsurg.2011.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jones S, Zhang X, Parsons DW, Lin JC, Leary RJ, Angenendt P, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Balakrishnan A, Bleeker FE, Lamba S, Rodolfo M, Daniotti M, Scarpa A, et al. Novel somatic and germline mutations in cancer candidate genes in glioblastoma, melanoma, and pancreatic carcinoma. Cancer Res. 2007;67(8):3545–50. doi: 10.1158/0008-5472.CAN-07-0065. [DOI] [PubMed] [Google Scholar]
- 5.Iacobuzio-Donahue CA. Genetic evolution of pancreatic cancer: lessons learnt from the pancreatic cancer genome sequencing project. Gut. 2011 Jul 11; doi: 10.1136/gut.2010.236026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Yachida S, Jones S, Bozic I, Antal T, Leary R, Fu B, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467(7319):1114–7. doi: 10.1038/nature09515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Campbell PJ, Yachida S, Mudie LJ, Stephens PJ, Pleasance ED, Stebbings LA, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467(7319):1109–13. doi: 10.1038/nature09460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Blackford A, Serrano OK, Wolfgang CL, Parmigiani G, Jones S, Zhang X, et al. SMAD4 gene mutations are associated with poor prognosis in pancreatic cancer. Clin Cancer Res. 2009;15(14):4674–9. doi: 10.1158/1078-0432.CCR-09-0227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nakamori S, Yashima K, Murakami Y, Ishikawa O, Ohigashi H, Imaoka S, et al. Association of p53 gene mutations with short survival in pancreatic adenocarcinoma. Jpn J Cancer Res. 1995;86(2):174–81. doi: 10.1111/j.1349-7006.1995.tb03036.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Smith RA, Tang J, Tudur-Smith C, Neoptolemos JP, Ghaneh P. Meta-analysis of immunohistochemical prognostic markers in resected pancreatic cancer. Br J Cancer. 2011;104(9):1440–51. doi: 10.1038/bjc.2011.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Salek C, Minarikova P, Benesova L, Nosek V, Strnad R, Zavoral M, et al. Mutation status of K-ras, p53 and allelic losses at 9p and 18q are not prognostic markers in patients with pancreatic cancer. Anticancer Res. 2009;29(5):1803–10. [PubMed] [Google Scholar]
- 12.Garcea G, Neal CP, Pattenden CJ, Steward WP, Berry DP. Molecular prognostic markers in pancreatic cancer: a systematic review. Eur J Cancer. 2005;41(15):2213–36. doi: 10.1016/j.ejca.2005.04.044. [DOI] [PubMed] [Google Scholar]
- 13.Biankin AV, Morey AL, Lee CS, Kench JG, Biankin SA, Hook HC, et al. DPC4/Smad4 expression and outcome in pancreatic ductal adenocarcinoma. J Clin Oncol. 2002;20(23):4531–42. doi: 10.1200/JCO.2002.12.063. [DOI] [PubMed] [Google Scholar]
- 14.Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJ, et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res. 2001;7(12):4115–21. [PubMed] [Google Scholar]
- 15.Hingorani SR, Wang L, Multani AS, Combs C, Deramaudt TB, Hruban RH, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7(5):469–83. doi: 10.1016/j.ccr.2005.04.023. [DOI] [PubMed] [Google Scholar]
- 16.Aguirre AJ, Bardeesy N, Sinha M, Lopez L, Tuveson DA, Horner J, et al. Activated Kras and Ink4a/Arf deficiency cooperate to produce metastatic pancreatic ductal adenocarcinoma. Genes Dev. 2003;17(24):3112–26. doi: 10.1101/gad.1158703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P, et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev. 2006;20(22):3130–46. doi: 10.1101/gad.1478706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kojima K, Vickers SM, Adsay NV, Jhala NC, Kim HG, Schoeb TR, et al. Inactivation of Smad4 accelerates Kras(G12D)-mediated pancreatic neoplasia. Cancer Res. 2007;67(17):8121–30. doi: 10.1158/0008-5472.CAN-06-4167. [DOI] [PubMed] [Google Scholar]
- 19.Qiu W, Sahin F, Iacobuzio-Donahue CA, Garcia-Carracedo D, Wang WM, Kuo CY, et al. Disruption of p16 and activation of Kras in pancreas increase ductal adenocarcinoma formation and metastasis in vivo. Oncotarget. 2011;2(11):862–73. doi: 10.18632/oncotarget.357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Embuscado EE, Laheru D, Ricci F, Yun KJ, de Boom Witzel S, Seigel A, et al. Immortalizing the complexity of cancer metastasis: genetic features of lethal metastatic pancreatic cancer obtained from rapid autopsy. Cancer Biol Ther. 2005;4(5):548–54. doi: 10.4161/cbt.4.5.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yachida S, Vakiani E, White CM, Zhong Y, Saunders T, Morgan R, et al. Small cell and large cell neuroendocrine carcinomas of the pancreas are genetically similar and distinct from well-differentiated pancreatic neuroendocrine tumors. Am J Surg Pathol. 2012;36(2):173–84. doi: 10.1097/PAS.0b013e3182417d36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wilentz RE, Geradts J, Maynard R, Offerhaus GJ, Kang M, Goggins M, et al. Inactivation of the p16 (INK4A) tumor-suppressor gene in pancreatic duct lesions: loss of intranuclear expression. Cancer Res. 1998;58(20):4740–4. [PubMed] [Google Scholar]
- 23.Geradts J, Hruban RH, Schutte M, Kern SE, Maynard R. Immunohistochemical p16INK4a analysis of archival tumors with deletion, hypermethylation, or mutation of the CDKN2/MTS1 gene. A comparison of four commercial antibodies. Appl Immunohistochem Mol Morphol. 2000;8(1):71–9. doi: 10.1097/00129039-200003000-00011. [DOI] [PubMed] [Google Scholar]
- 24.Melhem MF, Law JC, el-Ashmawy L, Johnson JT, Landreneau RJ, Srivastava S, et al. Assessment of sensitivity and specificity of immunohistochemical staining of p53 in lung and head and neck cancers. Am J Pathol. 1995;146(5):1170–7. [PMC free article] [PubMed] [Google Scholar]
- 25.Obata A, Eura M, Sasaki J, Saya H, Chikamatsu K, Tada M, et al. Clinical significance of p53 functional loss in squamous cell carcinoma of the oropharynx. Int J Cancer. 2000;89(2):187–93. doi: 10.1002/(sici)1097-0215(20000320)89:2<187::aid-ijc14>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 26.Sjogren S, Inganas M, Norberg T, Lindgren A, Nordgren H, Holmberg L, et al. The p53 gene in breast cancer: prognostic value of complementary DNA sequencing versus immunohistochemistry. J Natl Cancer Inst. 1996;88(3–4):173–82. doi: 10.1093/jnci/88.3-4.173. [DOI] [PubMed] [Google Scholar]
- 27.Wilentz RE, Su GH, Dai JL, Sparks AB, Argani P, Sohn TA, et al. Immunohistochemical labeling for Dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol. 2000;156(1):37–43. doi: 10.1016/S0002-9440(10)64703-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Iacobuzio-Donahue CA, Fu B, Yachida S, Luo M, Abe H, Henderson CM, et al. DPC4 gene status of the primary carcinoma correlates with patterns of failure in patients with pancreatic cancer. J Clin Oncol. 2009;27(11):1806–13. doi: 10.1200/JCO.2008.17.7188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Edkins S, O’Meara S, Parker A, Stevens C, Reis M, Jones S, et al. Recurrent KRAS codon 146 mutations in human colorectal cancer. Cancer Biol Ther. 2006;5(8):928–32. doi: 10.4161/cbt.5.8.3251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Schutte M, Hruban RH, Geradts J, Maynard R, Hilgers W, Rabindran SK, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–30. [PubMed] [Google Scholar]
- 31.Iacobuzio-Donahue CA, Song J, Parmiagiani G, Yeo CJ, Hruban RH, Kern SE. Missense mutations of MADH4: characterization of the mutational hot spot and functional consequences in human tumors. Clin Cancer Res. 2004;10(5):1597–604. doi: 10.1158/1078-0432.ccr-1121-3. [DOI] [PubMed] [Google Scholar]
- 32.Hruban RH, Goggins M, Parsons J, Kern SE. Progression model for pancreatic cancer. Clin Cancer Res. 2000;6(8):2969–72. [PubMed] [Google Scholar]
- 33.Villarroel MC, Rajeshkumar NV, Garrido-Laguna I, De Jesus-Acosta A, Jones S, Maitra A, et al. Personalizing cancer treatment in the age of global genomic analyses: PALB2 gene mutations and the response to DNA damaging agents in pancreatic cancer. Mol Cancer Ther. 2011;10(1):3–8. doi: 10.1158/1535-7163.MCT-10-0893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fogelman DR, Wolff RA, Kopetz S, Javle M, Bradley C, Mok I, et al. Evidence for the efficacy of Iniparib, a PARP-1 inhibitor, in BRCA2-associated pancreatic cancer. Anticancer Res. 2011;31(4):1417–20. [PubMed] [Google Scholar]
- 35.Stephens PJ, Greenman CD, Fu B, Yang F, Bignell GR, Mudie LJ, et al. Massive genomic rearrangement acquired in a single catastrophic event during cancer development. Cell. 2011;144(1):27–40. doi: 10.1016/j.cell.2010.11.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Schubbert S, Shannon K, Bollag G. Hyperactive Ras in developmental disorders and cancer. Nat Rev Cancer. 2007;7(4):295–308. doi: 10.1038/nrc2109. [DOI] [PubMed] [Google Scholar]
- 37.Caldas C, Hahn SA, da Costa LT, Redston MS, Schutte M, Seymour AB, et al. Frequent somatic mutations and homozygous deletions of the p16 (MTS1) gene in pancreatic adenocarcinoma. Nat Genet. 1994;8(1):27–32. doi: 10.1038/ng0994-27. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.