

Abstract
We identified Bub1b as an essential element for the growth and survival of rhabdomyosarcoma (RMS) cells using a bar-coded, tetracycline-inducible shRNA library screen. Knockdown of Bub1b resulted in suppression of tumor growth in vivo, including the regression of established tumors. The mechanism by which this occurs is via post-mitotic endoreduplication checkpoint and mitotic catastrophe. Furthermore, using a chromatin immunoprecipitation (ChIP) assay we found that Bub1b is a direct transcriptional target of Forkhead Box M1 (FoxM1). Suppression of FoxM1 either by shRNA or the inhibitor siomycin A resulted in reduction of Bub1b expression and inhibition of cell growth and survival. These results demonstrate the important role of the Bub1b/FoxM1 pathway in RMS and provide potential therapeutic targets.
Keywords: Bub1b, FoxM1, RMS, shRNA library screen, endoreduplication
Introduction
The spindle assembly checkpoint (SAC), also known as the mitotic checkpoint, is an important surveillance mechanism that prevents chromosome missegregation during mitotic cell division by delaying the metaphase to anaphase transition. SAC defects, such as deletion or inactivation of several components of the spindle checkpoint machinery, have been shown to contribute to chromosomal instability, aneuploidy and cancer (1, 2). Bub1b, known also as BubR1, is an essential SAC protein that interacts with Bub3, Mad2 and CDC20 to form the mitotic checkpoint complex (MCC). Activation of MCC results in inhibition of anaphase-promoting complex or cyclosome (APC/C) activity in the kinetochore, APC/C-mediated ubiquitination of cyclin B and securin, and the initiation of anaphase, thereby preventing premature chromosome segregation (3–5).
Bub1b is the mammalian homolog of yeast Mad3, but differs significantly since Bub1b has a kinase domain that is absent in Mad3 (6). Complete deletion of Bub1B in the mouse germline results in early embryonic death (7). Bub1b+/− (haploinsufficient) mice display increased megakaryopoiesis and increased chromosome instability, as well as susceptibility to cancer (8, 9). Reducing the levels of Bub1b or inhibition of its kinase activity in human cancer cells results in massive chromosome loss and apoptotic cell death (10). Somatic mutations in Bub1b have been found in various tumor samples including colorectal, lung, breast and hematopoietic malignancies (3). Biallelic mutations of the Bub1B gene have been detected in mosaic variegated aneuploidy (MVA) syndrome, also referred to as premature chromatid separation (PCS) syndrome (11, 12). MVA is rare autosomal recessive disorder, 37 cases of MVA have been reported worldwide (13). MVA is characterized by constitutional aneuploidy and early onset childhood cancer predisposition to rhabdomyosarcoma (RMS), Wilms tumor and leukemia. The high incidence of childhood cancer in MVA patients suggested that mitotic spindle dysfunction related to a Bub1b mutation might contribute to the development of these childhood cancers.
RMS is the most common pediatric soft tissue sarcoma and consists of two major subtypes, alveolar (ARMS) and embryonal (ERMS), which are associated with distinct genetic alterations. ARMS is characterized by non-random translocations involving the DNA binding domain of either PAX3 on the long arm of chromosome 2 or PAX7 on the short arm of chromosome 1 and the transactivation domain of the FOXOA1 gene [t(2;13) (PAX3-FOXOA1) or t(1;13) (PAX7-FOXOA1)] (14). Loss of heterozygosity (LOH) at chromosome 11p15.5 has been identified in ERMS (15). Despite increasing cure rates for RMS, children with high-risk tumors including metastatic disease, recurrent tumor, and certain histologies carry a poor prognosis and require identification of new molecular therapeutic targets for recurrent and metastatic RMS.
In this study, we determined that Bub1b is necessary for the growth and survival of both ARMS and ERMS cells using a loss of function high-throughput shRNA screen. Knockdown of Bub1b also resulted in significant suppression of Rh30 and RD xenograft growth in vivo. Mechanistically, flow cytometry analysis of Bub1b knockdown cells showed an increase in >4N DNA content. Live cell time-lapse microscopy studies provided direct evidence that knockdown of Bub1b promotes endoreduplication, eventually causing mitotic catastrophe. Further, ChIP assay demonstrated that Forkhead Box M1 (FoxM1), an essential transcriptional factor for G1/S transition and mitotic progression, directly binds to the Bub1b promoter. Suppression of FoxM1, either by shRNA or the inhibitor siomycin A, led to reduction of Bub1b expression and inhibition of cell growth and survival in vitro. These findings indicate that the FoxM1-Bub1b axis is important for the growth and survival of RMS cells. Further evaluation of components in this pathway may identify potential therapeutic targets in RMS
Materials and methods
Cell lines
Alveolar RMS cell lines Rh30 and Rh4, and embryonal RMS cell line RD have been previously described (16) and authenticated by Genetica DNA Laboratories Inc July 2012 (Cincinnati, OH). RD and Rh30 doxycycline-inducible Cell lines were engineered as described (17). Human fibroblast cell lines N1 and N2 were obtained from Dr. SB Lee (Genetics of Development and Disease Branch, NIDDK, NIH). These cells were immortalized by transfection with human papillomaviruses E6 and E7 (18). Neuroblastoma cell lines AS and KCNR were obtained from Dr. C Thiele (Pediatric Oncology Branch, CCR, NCI, NIH). Breast cancer cell lines MCF-7, MDA-MB231 and ovarian cancer cell line SKOV3 have been described previously (19).
Preparation of doxycycline-inducible cell line and bar-code shRNA screen
Rh30 and RD cells used in the bar-code screen were first infected with a feline endogenous virus expressing the ecotropic retroviral receptor and then second infected with an ecotropic retrovirus expressing the bacterial tetracycline repressor (TETR). Bar-code screen using inducible shRNA retrviral expressing library was performed as described (17).
Establishment of doxycycline-inducible shBub1b and shFoxM1 cell lines
shBub1b and shFoxM1 cell lines were generated by infection of RD and Rh30 with retrovirus encoding the shRNA targeting Bub1b at bp3099 of NM_001211 or FoxM1 at bp1778 of NM_202002. Both RD and Rh30 control cell lines were generated by infection of retrovirus encoding an shRNA targeting a region of GFP at bp288. Cell lines were batch selected in puromycin (1 µg/mL) for 3 weeks.
Western Blot Analysis
Western blot analysis was done as published previously (20). Antibodies to Bub1b, Bub3, CDC20 Cyclin B1 CDC2 PLK1, Aurora A, Aurora B, and Aurora C were purchased from Cell Signaling Technology Inc. (Beverly, MA). Anti-Securin antibody was purchased from Epitomics Inc (Burlingame, CA). Anti-actin antibody was purchased from Abcam Inc. (Cambridge, MA).
Cell proliferation and survival assays
Cell proliferation and survival was determined using MTT assay or stained with Neat Stain Helatologystain kit (Astral Diagnostics Inc, West Deptford, NJ).
Lentivirus transduction
Lentivirus-expressing shRNA against Bub1b, FoxM1 and control were purchased from Sigma (St. Louis, MO) and infection carried out according to the manufacturer's procedure. Puromycin (1 µg/mL) was used for lentivirus-infected cell selection.
In vivo study
Animal studies were performed in accordance with guidelines of the National Institutes of Health Animal Care and Use Committee. Four- to 6-week-old female Cohort Nude-SCID mice were purchased from Charles River Laboratories (Wilmington, MA). Three million cells of each cell line (Rh30-Bub1bshRNA3099 and RD-Bub1bshRNA3099) were injected orthotopically into the gastrocnemius muscle in the left hind leg.
For Bub1b-off groups, mice were fed with doxycycline diet (Bio-Serv, Frenchtown, NJ) before one week of injection. For Bub1b-delayed off group, mice were fed with doxycycline diet after tumor was established. For control groups, mice were fed with regular diet.
Cell synchronization and flow cytometry
Cells were treated with 2 mM thymidine (Sigma) for 18 h. After cells were washed three times with phosphate-buffered saline (PBS) and released into fresh medium for 8 h. These cells were then treated with thymidine for an additional 18 h. After double thymidine block, cells were treated 50ng/ml of doxycycline (Sigma) at indicated times and were subjected to flow cytometry as previously described (21).
Spectral Karyotyping
SKY was carried out as described previously (22). The chromosome paints used was the 24-color Human SKY paint kit from Applied Spectral Imaging followed manufacturer’s protocol. Spectral images of the hybridized metaphases were acquired using a SD301 SpectraCubeTM system (Applied Spectral Imaging Inc., Vista, CA) mounted on top of an epifluorescence microscope Axioplan 2 (Zeiss). Images were analyzed using Spectral Imaging 6.0 acquisition software (Applied Spectral Imaging Inc., Vista, CA). G banding was simulated by electronic inversion of DAPI-counterstaining.
Time-lapse imaging
Cells were treated with or without doxycycline for 4 days, and then collected mitotic cells by shake-off, spread these cells to 6-well plate at very density, and incubated 24 hours, and started to monitor the cells using a Zeiss AxioObserver Z1 microscope (Zeiss Inc., Thornwood, NY, USA) captured at 3-minute intervals for 72 hours.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) assays were performed using the ChIP-IT Express Enzymatic kit from Active Motif Inc. (Carlsbad, CA) followed manufacture’s instruction. Anti-FoxM1 antibody (Bethyl, TX) and control IgG (Santa Cruz, CA) were used for immunoprecipitation. ChIP samples including input and negative control were assayed by quantitative real-time PCR using a SYBR green kit (Applied Biosystems, Carlsbad, CA) to amplify a 135-bp region (−350 to −216 bp) of the Bub1b promoter. The nucleotide sequences of the quantitative real-time PCR were as follows: sense primer 5’-taagcctgctgcacttccac-3’ and antisense primer 5’-ctcctccgtgctctc gcgtct-3’.
Luciferase assays of Bub1b promoter-luciferase constructs
We amplified the −350/−216 and −545/−397 regions of the human Bub1b promoter from RD genomic DNA by PCR using the primers containing the restriction sites Xho1 and HindIII respectively. These nucleotide sequences of primers were as follows: sense primer 5’-ctcgagtaagcctgctgcacttccac-3’ and antisense primer 5’-aagcttctcctccgtgctctcgcgtct-3’ for −350/−216 fragment, and sense primer 5’-ctcgagccggacgggtgagatttg ggg-3’ and antisense primer 5’-aagcttccg agtctctgagccgagcga-3’ for −545/−397 fragment. These fragments were ligated into a TOPO-TA vector (Invitrogen) and then cloned into a pGL4.18 firefly luciferase reporter vector (Promega, Madison, WI) after digestion with Xho1 and HindIII. PCR-based site-directed mutagenesis was performed to generate a single point mutation in FoxM1 binding sites of Bub1b promoter region by using Quickchange II XL Site-directed Mutagenesis Kit (Agilent Technologies, La Jolla, CA). Sequences were verified by DNA sequencing. . RD-FoxM1 cells were transfected via Lipofectamine™ 2000 with 1 µg of empty vector, or −350/−216 or −545/−397 Bub1b promoter luciferase reporter and cultured with 500 mg/L of G418 for two weeks. RD cells were transiently transfected via the Amaxa Nucleofector with 3 µg of wild-type Bub1b promoter or its mutants or empty vector. Luciferase assays were performed using Bright-Glo reagent (Promega) on the Victor3 counter (Perkin Elmer, Walthan, MA).
Quantitative RT-PCR
RNA was isolated using an RNeasy kit (Qiagen, Valencia, CA) and 500 ng of total RNA was reverse transcribed using a high-capacity reverse transcriptase kit (Applied Biosystems). The quantitative real-time PCR was performed using a SYBR green kit (Applied Biosystems). The following primers were used: FoxM1 sense primer 5’-AAGCGAGTCCGCATTGCCCC-3’, FoxM1 antisense primer 5’-CGGGAGGGCCACTTCCA-3’, Bub1b sense primer 5’-GCGGCGGTGAAGAAGGAAGGG-3’, Bub1b antisense 5’-TGCCAGTGCTCCCTGAAGCG-3’, glyceraldehyde-3-phosphate dehydrogenase (GAPDH) sense primer 5’-AGGTGACACTATAGAATAAAGGTGAAGGTCGGAGTCAA-3’, GAPDH antisense 5’-GTACGACTCACTATAGGGAGATCTCGCTCCTGGAAGATG-3’. Quantitation was done using the formula 2−ddCT (23).
Statistical analysis
Statistical analyses were performed in Prism version 4.0 (GraphPad Software) using a nonparametric t-test. Statistical significance was defined as P <0.05.
Results
A large scale shRNA barcode screen identifies Bub1b as necessary for RMS cell growth and survival
To identify new molecular targets as key regulators of RMS cell proliferation and survival, we performed a large-scale shRNA screen using Rh30 and RD. Each cell line was engineered to express a doxycycline-inducible, bar-coded shRNA library against approximately 5000 human genes with 3 different shRNAs per gene (17). As shown in Fig. 1A, following subtraction of genes that have been identified to be required for proliferation and survival of lymphoma cells using the same shRNA library screen (17), we identified 330 genes that significantly decreased proliferation and survival in RD cells and 53 genes in Rh30 cells. 38 genes were common to the two distinct cell lines. We focused on Bub1b because of mutations of the gene in MVA are associated with RMS as described above. We examined the expression of Bub1b protein by Western blot analysis in Rh30 and RD, in addition to 2 human fibroblast cell lines (N1 and N2), 2 breast cancer lines (MCF-7 and MDA-MB231), and 1 ovarian cancer cell line (SKOV3)(Fig. 1B). Bub1b was highly expressed in RMS cell lines, but nearly undetectable in human fibroblast cell lines, and expressed at much lower levels in the other cancer cell lines. To examine the function of Bub1b in mitotic checkpoint control, both Rh30 and RD cells were synchronized by thymidine block, released from G1/S arrest, and arrested by nocodazole treatment, which activates the mitotic checkpoint. We collected cell samples at 0, 1, 3, and 6 h after release from nocodazole and analyzed expression of Bub1b and various mitotic checkpoint proteins such as Cyclin B1, CDC2, PIK1, FoxM1, and aurora B in thymidine-nocodazole synchronized cells. Thymidine treatment alone induced cell arrest at the G1/S phase boundary and thus serves as a control as indicated by high expression of CyclinE. As shown in Fig.1C, Bub1b, Cyclin B1, PIK1, and FoxM1 showed similar behavior, in that they were markedly increased in nocodazole-arrested cells and declined as the cells exited mitosis. No changes in Bub3 and CDC2 and only a minor increase in Aurora B were detected in nocodazole-arrested cells.
Figure 1.

A, A large-scale, barcoded, inducible shRNA library screen identified a total of 38 genes that were necessary for growth and survival of both ARMS and ERMS cell lines. B, Western blot analysis revealed that RMS cell lines expressed high levels of Bub1b, compared to other cancer cell lines and normal fibroblasts. Beta actin was used as a loading control. C, Rh30 and RD cells were released from thymidine (2mM) synchrony and then exposed to nocodazole (100 ng/ml) for 18 h. Cells were collected at 0, 1, 3, and 6 h after release from nocodazole and analyzed for expression of Bub1b, Bub3, Cyclin B1, CDC2, PIK1, FoxM1, aurora B, and Cyclin E. Thymidine treatment alone served as a control.
Verification of Bub1b knockdown mediated growth suppression
To verify the shRNA screening data, Rh30 and RD cells were infected with the specific Bub1b shRNA-3099 clone from the library screen that targets the kinase domain of Bub1b. Western blot demonstrates that both RD and Rh30 cells treated with doxycycline have markedly decreased protein expression of Bub1b only, without affecting the expression of other MCC proteins including Bub3 and CDC20, the mitosis regulatory kinases Aurora kinase A, B, C and Polo-like kinase (PIK)(Fig. 2A). APC/C is a critical target of MCC, and Bub1b is the only MCC protein that contains a kinase domain with enzymatic activity. Knockdown of Bub1b is expected to activate APC/C and to increase APC/C-mediated ubiquitination of cyclin B1 and securin. However, we did not detect changes of cyclin B1 and securin in either asynchronous or synchronous Bub1b-knockdown cells (Fig. 2A and 2B). p16INK4a is a important cell cycle regulator and mediator of irreversible senescence growth arrest (24). It has been reported that the levels of p16INK4a were significantly elevated in Bub1b insufficient mice tissues including adipose tissue, skeletal muscle and eye (25). To determine whether cellular senescence is related to Bub1b-knockdown induced suppression of RMS growth, we examined the protein levels of p16INK4a, a biomarker for cellular senescence, in Bub1b-knockdown cells. However, knockdown of Bub1b did not affect p16INK4a expression in either asynchronous cells (data not shown) or synchronous cells (Fig. 2B). Consistent with the shRNA library screening data, knockdown of Bub1b resulted in significant inhibition of both Rh30 and RD cell growth and survival as measured by MTT assay and cell staining (Fig. 2C and 2D). These results show that we attain specific inhibition of Bub1b using the Bub1b shRNA-3099 clone, resulting in decreased cell proliferation and survival of both ARMS and ERMS cell lines.
Figure 2.

Knockdown of Bub1b inhibits RD and Rh30 cell growth and survival. To verify the shRNA library screening data, Rh30 and RD cells were infected with the Bub1b-specific clone shRNA3099, followed by 2 weeks of batch selection in medium containing puromycin. A, cells were divided into two aliquots and half were treated with doxycycline at indicated days, followed by protein lysate harvest. The Western blot demonstrates that both RD and Rh30 cells treated with doxycycline have markedly decreased Bub1b protein expression. B, cells released from double thymidine (2mM) synchrony were treated with or without doxycycline at indicated times and then harvested for Western blot analysis. C, cells were plated in 96 well plates at a density of 2000 cells per well and half were treated with doxycycline, followed by MTT measurement at days 3, 5, and 7. Data are mean ± SE (n =5). D, cells were plated in 6-well plates and half were treated with doxycycline for 12 days, followed by Neat Stain kit (astraldiagnostics) staining. The results are representative of 3 independent experiments. E and F, cells were infected with different Bub1b lentivirus shRNAs. After selection for10 days with puromycin, cells were read by MTT assay. Data are mean ± SE (n =4). G, Bub1b expression was examined in RD and SKOV3 cell lines after 6 days of selection.
To provide additional evidence that Bub1b is necessary for growth and survival of RMS, we introduced additional lentivirus Bub1b shRNA constructs that targeted different sequences of Bub1b into RMS cell lines Rh30 and RD, ovarian cancer cell line SKOV3, and human fibroblast NI and N2 cell lines. Bub1b shRNA 464 and 465 displayed a significant inhibition of cell growth and survival in Rh30 and RD cells (Fig. 2E). Furthermore, human RMS cells were more sensitive to Bub1b knockdown compared to SKOV3 which express less Bub1b or human fibroblast N1 and N2 cells which express almost no Bub1b as shown in Fig. 1B (Fig. 2E). Western blot analysis confirmed that Bub1b expression was markedly inhibited by both Bub1b shRNA 464 and 465 in RD and SKOV3 cells (Fig. 2F). Taken together, we conclude that Bub1b expression is necessary for the growth and survival of RMS cells.
Knockdown of Bub1b resulted in significant suppression of Rh30 and RD xenograft growth in vivo
To study the consequence of loss of Bub1b for tumor formation in vivo, we performed xenograft experiments using both Rh30 and RD cells that express inducible Bub1b shRNA-3099. We injected cells orthotopically into the gastrocnemius musculature of mice, which were fed either a normal diet (Dox−, Bub1b-on) or a diet containing doxycycline (Dox+, Bub1b-off). As shown in Fig. 3, knockdown of Bub1b resulted in significant suppression of both Rh30 and RD xenograft growth. For Rh30 xenografts, all nine mice in the control group (DOX−) developed tumors 40 days after injection of cells, compared to none in the Bub1b-knockdown group (p=0.0002)(Fig. 3A). We continued to monitor the mice in the Bub1b-knockdown group for 87 days and five of ten (50%) mice remained free of tumor (Fig. 3B). Six mice in the Rh30 control group (Bub1b-on) who had developed tumors were then changed to a diet containing doxycycline (Bub1b-off) beginning at day 40, and 2 of the 6 mice then had tumor regression (Fig. 3C). For RD xenografts, all twenty mice in the control group developed tumor by day 58, compared to only one of ten mice in the Bub1b-knockdown group (p<0.0001)(Fig. 3D). Two of ten (20%) mice in the Bub1b-knockdown group remained free of tumor at 87 days (Fig. 3E). Half of the mice in the control cohort began a diet containing doxycycline at 58 days, and one of ten mice had tumor regression while another mouse had tumor stabilization (data not shown). In addition, mice given a diet containing doxycycline but injected with parental Rh30 and RD cells without the Bub1b inducible construct all developed tumors (data not shown). To determine whether Bub1b was still suppressed in the tumors of mice that received doxycycline, we examined Bub1b expression in 4 control tumor samples (2 from Rh30 control and 2 from RD control) and 4 Bub1b-knockdown tumor samples (2 from Rh30-Bub1bshRNA3099 and 2 from RD-Bub1bshRNA3099) by Western blot. Bub1b is re-expressed in all 4 Bub1b-knockdown tumor samples at levels that were comparable to control tumor samples (Fig. 3F). The mechanism by which these tumors re-express Bub1b remains unclear, but includes the possibility that some cells failed to respond to doxycycline.
Figure 3.

Knockdown of Bub1b inhibits Rh30 (A–C) and RD (D, E) xenografts growth in vivo. Three million cells were injected orthotopically into the gastrocnemius musculature of the hind leg of Nude Scid-Beige mice who were fed either normal diet (Bub1b shRNA not induced) or doxycycline diet (Bub1b shRNA induced). F, Bub1b expression was analyzed by Western blot analysis in these Dox− and Dox+ tumor samples.
Knockdown of Bub1b resulted in endoreduplication and mitotic catastrophe
To explore the mechanisms by which knockdown of Bub1b inhibited RMS cell growth and survival, we performed cell cycle analyses of Bub1b-knockdown cells. Flow cytometry analysis of Bub1b-knockdown cells showed a significant increase in the percentage of polyploid (>4N) cells from an average of 11% in control cells to 21% in knockdown cells for Rh30 (p <0.02) and from 9% to 22% for RD (p <0.02) following 5 days of doxycycline treatment to induce the shRNA constructs (Fig. 4A). There was a concomitant reduction in the number of cells in the G1 phase (2N) from an average of 44% to 36% for Rh30 (p <0.05) and from 49% to 37% for RD cells (p <0.05). A significant decrease in S phase was seen only in the RD cell line from an average of 9.7% to 6.7% for RD (p <0.05)(Fig. 4A). Similar data was obtained for each cell phase, even at 10 days of treatment with doxycycline. The accumulation of >4N DNA content with knockdown of Bub1b suggested that these cells might be undergoing endoreduplication, the phenomenon by which cells exit the cell cycle and replicate their genomic DNA but do not divide, resulting in genome size augmentation (26). Numerical metaphase chromosomal analysis revealed that both RD and Rh30 cell lines showed similar chromosome distribution with a majority (78% for RD and 88% for Rh30) of cells containing over 70 chromosomes (Fig. 4B). However, knockdown of Bub1b resulted an increase to over 80 chromosomes (from 40% to 48% for RD and 60% to 84% for Rh30)(Fig. 4B).
Figure 4.
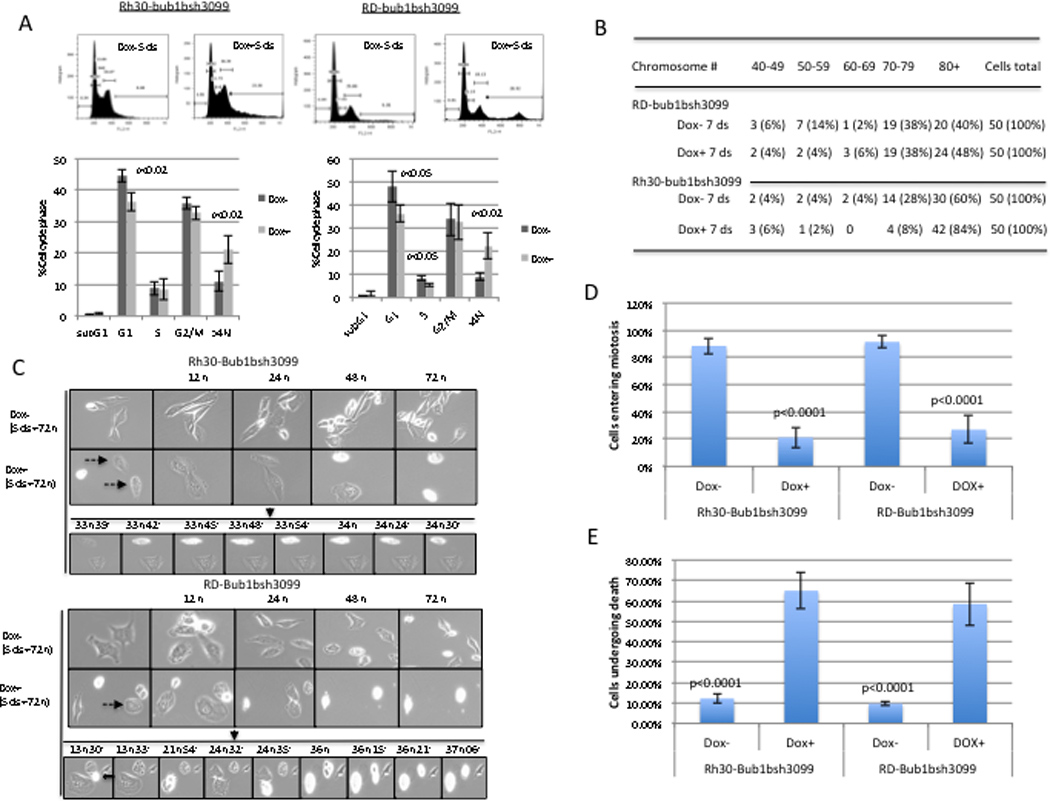
A, the percentages of cells at each stage of the cell cycle were analyzed by flow cytometry after PI staining. Bub1b-knockdown cells showed a decrease in G1 phase cells and an increase in >4N DNA content in both Rh30 and RD cell lines at day 5 after treatment with doxycycline to induce shRNA constructs. Data are mean ± SE. The results were representative of three independent experiments. B, chromosome numbers were quantified in Bub1b knockdown or control cells (n=50). C, Live-cell time-lapse microscopy studies demonstrated that knockdown of Bub1b promotes endoreduplication and mitotic catastrophe. Cells were treated with doxycycline for 4 days to induce the siRNA constructs, and then collected mitotic cells by shake-off, added these cells to a 6-well plate at very low density, incubated overnight, and started to monitor the cells with time-lapse microscopy captured at 3-minute intervals for 72 hours. The dashed arrows point to cells that underwent endoreduplication, eventually causing mitotic catastrophe. An open arrow points to cell that underwent mitotic catastrophe. D, mitotic cells were scored by their transition to a small rounded morphology, followed by cytokinesis and cell duplication. E, dead cells were counted from undivided DOX− and DOX+ cells. C and D data represent four independent experiments. Data are mean ± SE (n >100).
We utilized time-lapse live-cell imaging of Bub1b-knockdown cells and control cells. As shown in Fig. 4C and D, imaging over a 3-day period showed that 90% of control cells went through at least one round of mitosis, compared to only around 20% of Bub1b-knockdown cells. This means that around 80% of Bub1b-knockdown cells failed to completely divide. Further, 60% of undivided cells eventually underwent death by mitotic catastrophe, which is a type of cell death that occurs during mitosis (Fig. 4C and E). In total, our data indicated that knockdown of Bub1b suppresses RMS cell growth and survival via promotion of endoreduplication and mitotic catastrophe.
FoxM1 directly binds to the Bub1b promoter
FoxM1 is a member of the Forkhead Box (Fox) family of transcription factors that play a key role in cell proliferation by controlling the G1/S transition and mitotic progression (27, 28). How FoxM1 regulates mitotic progression is not completely understood. We found that both Bub1b and FoxM1 expression are increased following nocodazol-induced mitotic arrest in RD and Rh30 cells (Fig. 1C). Our Western blot analysis showed that Bub1b expression (Fig. 1B) correlates with FoxM1 expression (Fig. 5A). Thus, we hypothesized that FoxM1 may regulate Bub1b expression. To determine whether Bub1b was a direct transcriptional target of FoxM1, we scanned the 3-kb promoter region of Bub1b for the FoxM1 binding consensus sequence (TAAACA)(29). We identified this consensus FoxM1 binding sequence between −278 and −273 bp of the human Bub1b promoter (Fig. 5B). To determine if FoxM1 binds directly to the TAAACA sequence of the Bub1b promoter, we performed ChIP assay. The cross-linked chromatin fragments from control RD (dox−) and FoxM1-knockdown RD cells (dox+) were immunoprecipitated with either anti-FoxM1 or control anti-IgG antibodies. FoxM1-bound Bub1b promoter DNA was quantified by quantitative real-time PCR analysis with primers specific for the putative FoxM1 binding sites. As shown in Fig. 5C, ChIP assays showed that FoxM1 directly binds to the Bub1b promoter. In addition, knockdown of FoxM1 by shRNA obliterated FoxM1-binding to the Bub1b promoter. These data demonstrate that the Bub1b promoter region is a direct transcriptional target of FoxM1.
Figure 5.

Bub1b is a direct transcriptional target of FoxM1. A, FoxM1 expression was analyzed by Western blot analysis in indicated cell lines. B, schematic drawing of the 3-kb promoter region of the human Bub1b. Sequence and position of FoxM1-binding sites on Bub1b promoter are underlined (-278/−273). The two luciferase reporter constructs with or without FoxM1-binding sites are also shown. C, the direct binding of FoxM1 to promoter region of Bub1b was confirmed by ChIP analysis. Cross-linked chromatin from either control RD cells (Dox−) or FoxM1-knockdown RD cells (Dox+ 4 days) was immunoprecipitated with either anti-FoxM1 antibody or IgG control. After immunopecipitation, genomic DNA was analyzed using quantitative real-time PCR with primers specific for FoxM1-binding region. Data are shown as mean ± SE (n+3) and represent three independent experiments. D, luciferase reporter constructs with or without FoxM1-binding sites or empty vector were stablly transfected into doxycycline-inducible RD-FoxM1shRNA cells. These cells were treated with or without doxycycline for 4 days and then collected for analysis of luciferase activity. E, RD cells were transiently transfected with expression plasmids for the wild-type Bub1b promoter or its mutants or empty vector (3 µg). Luciferase activity was then determined. D and E data are shown as mean ± SE (n+5) and represent three independent experiments.
FoxM1-binding region is critical for the activation of the Bub1b promoter
To further assess the functional role of the FoxM1-binding region in Bub1b transcription, we performed a luciferase reporter assay with constructs driven by a human Bub1b promoter with or without the FoxM1-binding sites (Fig. 5B). As shown in Fig. 5D, −350/−216 construct containing FoxM1-binding sites drove greater luciferase activity compared to −545/−397 construct without FoxM1-binding sites (p <0.0001) or empty vector construct (p <0.0001). Furthermore, shRNA-mediated-knockdown of FoxM1 (Dox+) resulted in a marked decrease in transcription activity of FoxM1-binding region (p <0.0001). In addition, disruption of one of the FoxM1-binding sites significantly attenuated Bub1b promoter activity in RD cells (Fig. 5E). These data suggest that the FoxM1-binding region was critical for Bub1b promoter activation in RMS cells.
FoxM1 regulates Bub1b expression and is required for growth and survival of RMS Cells
To determine the functional consequence of the interaction between FoxM1 and Bub1b, we examined Bub1b expression in FoxM1-knockdown cells. As shown in Fig. 6A, knockdown of FoxM1 by retrovirus shRNA in RD cells resulted in a reduction of Bub1b expression at the protein level. Real-time PCR analysis of these FoxM1 knocked-down RD cells also revealed a substantial decrease in Bub1b mRNA level (Fig. 6B). To further confirm that FoxM1 indeed regulates Bub1b expression, we introduced additional lentivirus FoxM1 shRNA544 that targeted different sequences of FoxM1 into RMS cell lines Rh30, RD and Rh4. Western blot analysis demonstrated that FoxM1 shRNA544 significantly knocked down FoxM1 expression, which in turn also resulted in a downregulation of Bub1b in all three infected RMS cell lines (Fig. 6C). Since Bub1b expression is required for RMS cell growth and survival and FoxM1 is an upstream regulator of Bub1b, we hypothesized that targeting FoxM1 should also impact RMS cell growth and survival. By searching our shRNA library screen data, we found that targeting FoxM1 resulted in an average 9.4 fold decrease (p=0.017) for RD cells and an average 2.5 fold decrease (p=0.84) for Rh30 in hybridization intensity. To further confirm that targeting of FoxM1 is critical for growth and survival of RMS cells, we measured cell growth and survival in FoxM1-shRNA544 infected Rh30, RD, and Rh4 cell lines. It displayed a significant inhibition of cell growth and survival in all three RMS cell lines (Fig. 6D). We next followed FoxM1 knockdown cells by cell cycle analysis. Knockdown of FoxM1 resulted in significant accumulation of subG1, G1, and polyploidy cells (>4N) as well a significant reduction in cells in S and G2/M (Fig. 6E). We found that FoxM1 knockdown induced less polyploidy compared to Bub1b knockdown cells. This was, at least in part, due to a large proportion of FoxM1 knockdown cells remained arrested in G1.
Figure 6.

Knockdown of FoxM1 either by different shRNA sequences or by Siomycin A resulted in suppression of Bub1b expression and cell growth and survival in tested cancer cell lines. A and B, knockdown of FoxM1 by shRNA lead to decreased Bub1b expression at the level of both protein and RNA. Data are mean ± SE (n=4). C and D, cells were infected with different FoxM1 lentivirus shRNA. After selection with puromycin, cells were harvested for Western blot analysis (C) and read by MTT assay (D). E, RD cells released from double thymidine (2mM) synchrony were treated with or without doxycycline for 4 days and then analyzed by flow cytometry. F, cells were treated with Siomycin A (5 µM) at indicated time points and then harvested for Western blot analysis. G, cells were treated with Siomycin A for 72 hours and then read by MTT. Data are mean ± SE (n=5).
A recent study has found that the antibiotic thiazole compound Siomycin A inhibits FoxM1 transcriptional activity (30). Based on this finding, we performed a time course experiment to examine the effects of Siomycin A at 5 uM on FoxM1 and Bub1b expression in RMS cell lines. As shown in Fig. 6F, treatment of RD and Rh30 cells with Siomycin A at 5 µM resulted in suppression of both FoxM1 and Bub1b in a time dependent manner. In contrast, Siomycin A treatment had no effect on Cyclin B1 and PIK1 expression. These results suggested that Bub1b is a direct downstream target of FoxM1.Treatment Rh30 and RD cells with Siomycin A also led to a significant inhibition of cell growth and survival (Fig. 6G). These data support our findings that FoxM1 regulates Bub1b expression and is also required for the growth and survival of RMS cells.
Discussion
Our data in vitro and in vivo demonstrate that Bub1b is a critical protein for growth and survival of RMS. This finding is consistent with a previous report that reduction of Bub1b caused cell death in the human cervical cancer cell line Hela and glioblastoma line T98G (10). Biallelic and heterozygous mutations of the in Bub1b have been found in MVA that has been associated with RMS (11). Our findings that knockdown of Bub1b leads to loss of RMS cell growth and previous reports indicating an association of Bub1b mutations seen in patients with MVA and its association with the development of RMS appear to provide conflicting data. There are several possible explanations for this apparent discrepancy. First, certain mutations of Bub1b do not completely inactivate Bub1b function. Indeed, no homozygous mutation in Bub1b has found in MVA patient. Homozygous deletions of Bub1b resulted in embryonic lethality accompanied by enhanced apoptosis (7, 8). Second, this might reflect potential difference in rates of chromosome missegregation between Bub1b knockdown in RMS cell lines and Bub1b mutations in MVA patients. It is posited that low rates of chromosome missegregation can promote tumor development, whereas higher levels might promote cell death and suppress tumorigenesis (31). Finally, it is of note that in APCmin/+ mice with knockdown of Bub1b, there in an increased the incidence of colon tumors observed, but a reduced incidence of small intestinal tumors, with a corresponding increase in apoptosis in the small intestinal tumors (32). Thus the context in which Bub1b functions appears to be very important, and may also explain why mitotic catastrophe in rarely seen in Bub1b knockdown Hela cells and MVA patient derived cells (33–35). APC/C –mediated ubiquitination and degradation of anaphase inhibitors cyclin B and securin represents one of the key steps in the metaphase to anaphase transition (3, 4). Bub1b inhibits APC/C activity during metaphase as part of MCC (3, 4). Knockdown of Bub1b would be expected to increase APC/C activity and consequently to drive cells into anaphase by degradation of cyclin B1 and securin leading to inactivation of Cdk1 (CDC2) and activation of separase. However, knockdown of Bub1b failed to decrease expression of cyclin B1 and securin in both Rh30 and RD cells (Fig. 2A and 2B). This suggests that knockdown of Bub1b induced suppression of RMS cell growth and survival via an APC/C-cyclin B1/securin independent mechanism.
Endoreduplication is the replication of genome during cell cycle without the subsequent completion of mitosis and/or cytokinesis (26). A wide variety of agents including microtubule inhibitors, topoisomerase II inhibitors and DNA damaging agents have been reported as able to induce endoreduplication (36). The physiological significance and the molecular mechanism of endoreduplication are still not well understood. Endoreduplication can be associated with cell differentiation but also frequently occurs in malignant cells and may play a role in maintaining cell fate (37, 38). Recently, several cell cycle related proteins including p21, Aurora B, cyclin B1 and B2, CDK1, have been found to regulate endoreduplication (39–42). Our data demonstrate that knockdown of Bub1b resulted in endoreduplication (Fig. 4C & D). Our live cell imaging data also showed that around 80% Bub1b knockdown cells are unable to complete cell division and most of undivided cells finally die through mitotic catastrophe (Fig. 4C and E). These data suggest that Bub1b knockdown induced endoreduplication and mitotic catastrophe can exist together. A previous report in BRCA1 mutant breast cancer cells found that G2 arrest induced by genistein led to endoreduplication and cell death through activation of DNA damage response mechanisms (43). Another study evaluated the effect of FoxM1 knockdown in breast cancer cells and showed both endoreduplication and or mitotic catastrophe (44). Our study showing Bub1b knockdown induced endoreduplication and mitotic catastrophe thus appears to be consistent with these studies in breast cancer cells. It is still unclear how Bub1b knockdown precisely leads to endoreduplication and mitotic catastrophe.
FoxM1 is a typical proliferation-associated transcription factor. However, the transcriptional mechanisms downstream of FoxM1 in cell cycle progression remain to be determined. In this study, we demonstrate that FoxM1 is essential for Bub1b expression. To our knowledge, this is the first report to show the Bub1b is a direct transcriptional target of FoxM1. Specifically, we show that FoxM1 expression drives a Bub1b promoter construct containing a consensus FoxM1 binding site. Second, we show direct binding of FoxM1 to this promoter using ChiP assays. Furthermore, a strong correlation between the expression levels of FoxM1 and Bub1b was observed not only in RMS but also in other pediatric malignant and normal tissue samples by microarray expression analysis (data not shown). Recently, the thiazole antibiotic Siomycin A has been identified as a potent inhibitor of FoxM1 (30). Consistent with our shRNA data, Siomycin A downregulates both FoxM1 and Bub1b expression, and inhibits RMS cell growth and survival. Taken together, disruption of this FoxM1-Bub1b signaling cascade, both genetically and pharmacologically, results in suppression of RMS cell growth and survival. These findings could be interesting because this is the first report that implicates the FoxM1-Bub1b signaling pathway in RMS tumorigenesis and presents potential targets for therapy.
Footnotes
Disclosure of potential conflicts of interest: none
References
- 1.Jallepalli PV, Lengauer C. Chromosome segregation and cancer: cutting through the mystery. Nat Rev Cancer. 2001;1:109–117. doi: 10.1038/35101065. [DOI] [PubMed] [Google Scholar]
- 2.Rajagopalan H, Lengauer C. Aneuploidy and cancer. Nature. 2004;432:338–341. doi: 10.1038/nature03099. [DOI] [PubMed] [Google Scholar]
- 3.Kops GJ, Weaver BA, Cleveland DW. On the road to cancer: aneuploidy and the mitotic checkpoint. Nat Rev Cancer. 2005;5:773–785. doi: 10.1038/nrc1714. [DOI] [PubMed] [Google Scholar]
- 4.Bharadwaj R, Yu H. The spindle checkpoint, aneuploidy, and cancer. Oncogene. 2004;23:2016–2027. doi: 10.1038/sj.onc.1207374. [DOI] [PubMed] [Google Scholar]
- 5.Musacchio A, Salmon ED. The spindle-assembly checkpoint in space and time. Nat Rev Mol Cell Biol. 2007;8:379–393. doi: 10.1038/nrm2163. [DOI] [PubMed] [Google Scholar]
- 6.Taylor SS, Ha E, McKeon F. The Human Homologue of Bub3 Is Required for Kinetochore Localization of Bub1 and a Mad3/Bub1-related Protein Kinase. J Cell Biol. 1998;142:1–11. doi: 10.1083/jcb.142.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Baker DJ, Jeganathan KB, Cameron JD, Thompson M, Juneja S, Kopecka A, et al. BubR1 insufficiency causes early onset of aging-associated phenotypes and infertility in mice. Nature Genetics. 2004;36:744–749. doi: 10.1038/ng1382. [DOI] [PubMed] [Google Scholar]
- 8.Wang Q, Liu T, Fang Y, Xie S, Huang X, Mahmood R, et al. BUBR1 deficiency results in abnormal megakaryopoiesis. Blood. 2004;103:1278–1285. doi: 10.1182/blood-2003-06-2158. [DOI] [PubMed] [Google Scholar]
- 9.Dai W, Wang Q, Liu T, Swamy M, Fang Y, Xie S, et al. Slippage of Mitotic Arrest and Enhanced Tumor Development in Mice with BubR1 Haploinsufficiency. Cancer Research. 2004;64:440–445. doi: 10.1158/0008-5472.can-03-3119. [DOI] [PubMed] [Google Scholar]
- 10.Kops PL, Foltz DR, Cleveland DW. Lethality to human cancer cells through massive chromosome loss by inhibition of the mitotic checkpoint. Proc Natl Acad Sci USA. 2004;101:8699–8704. doi: 10.1073/pnas.0401142101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hanks S, Coleman K, Reid S, Plaja A, Firth H, Fitzpatrick D, et al. Constitutional aneuploidy and cancer predisposition caused by biallelic mutations in BUB1B. Nat Genet. 2004;36:1159–1161. doi: 10.1038/ng1449. [DOI] [PubMed] [Google Scholar]
- 12.Matsuura S, Matsumoto Y, Morishima K, Izumi H, Matsumoto H, Ito E, et al. Monoallelic BUB1B mutations and defective mitotic-spindle checkpoint in seven families with premature chromatid separation (PCS) syndrome. Am J Med Genet A. 2006;140:358–367. doi: 10.1002/ajmg.a.31069. [DOI] [PubMed] [Google Scholar]
- 13.Rio Frio T, Lavoie J, Hamel N, Geyer FC, Kushner YB, Novak DJ, et al. Homozygous BUB1B mutation and susceptibility to gastrointestinal neoplasia. N Engl J Med. 2010;363:2628–2637. doi: 10.1056/NEJMoa1006565. [DOI] [PubMed] [Google Scholar]
- 14.Merlino G, Helman L. Rhabdomyosarcoma--working out the pathways. Oncogene. 1999;18:5340–5348. doi: 10.1038/sj.onc.1203038. [DOI] [PubMed] [Google Scholar]
- 15.Scrable H, Cavenee W, Ghavimi F, Lovell M, Morgan K, Sapienza C. A model for embryonal rhabdomyosarcoma tumorigenesis that involves genome imprinting. Proc Natl Acad Sci USA. 1989;86:1117–1121. doi: 10.1073/pnas.86.19.7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wan X, Helman LJ. Levels of PTEN protein modulate Akt phos phorylation on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcomas cells. Oncogene. 2003;22:8205–8211. doi: 10.1038/sj.onc.1206878. [DOI] [PubMed] [Google Scholar]
- 17.Ngo VN, Young RM, Schmitz R, Jhavar S, Xiao W, Lim KH, et al. A loss-of-function RNA interference screen for molecular targets in cancer. Nature. 2006;441:106–110. doi: 10.1038/nature04687. [DOI] [PubMed] [Google Scholar]
- 18.Oram JF, Mendez AJ, Lymp J, Kavanagh TJ, Halbert CL. Reduction in apolipoprotein-mediated removal of cellular lipids by immortalization of human fibroblasts and its reversion by cAMP: lack of effect with Tangier disease cells. J Lipid Res. 1999;40:1769–1781. [PubMed] [Google Scholar]
- 19.Grohar PJ, Woldemichael GM, Griffin LB, Mendoza A, Chen QR, Yeung C, et al. Identification of an Inhibitor of the EWS-FLI1 Oncogenic Transcription Factor by High-Throughput Screening. J Natl Cancer Inst. 2011;103:962–978. doi: 10.1093/jnci/djr156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wan X, Mendoza A, Khanna C, Helman LJ. Rapamycin inhibits ezrin-mediated metastatic behavior in a murine model of osteosarcoma. Cancer Res. 2005;65:2406–2411. doi: 10.1158/0008-5472.CAN-04-3135. [DOI] [PubMed] [Google Scholar]
- 21.Schrock E, du Manoir S, Veldman T, Schoell B, Wienberg J, Ferguson MA, et al. Multicolor spectral karyotyping of human chromosomes. Science. 1996;273:494–497. doi: 10.1126/science.273.5274.494. [DOI] [PubMed] [Google Scholar]
- 22.Wan X, Helman LJ. Effect of insulin-like growth factor II on protecting myoblast cells against cisplatin-induced apoptosis through p70 S6 kinase pathway. Neoplasia. 2002;4:400–408. doi: 10.1038/sj.neo.7900242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schmittgen TD, Livak KJ. Analyzing real-time PCR data by the comparative C(T) method. Nat Protoc. 2008;3:1101–1108. doi: 10.1038/nprot.2008.73. [DOI] [PubMed] [Google Scholar]
- 24.Gil J, Peters G. Regulation of the INK4b-ARF-INK4a tumour suppressor locus: all for one or one for all. Nat Rev Mol Cell Biol. 2006;7:667–677. doi: 10.1038/nrm1987. [DOI] [PubMed] [Google Scholar]
- 25.Baker DJ, Perez-Terzic C, Jin F, Pitel KS, Niederländer NJ, Jeganathan K, et al. Opposing roles for p16Ink4a and p19Arf in senescence and ageing caused by BubR1 insufficiency. Nat Cell Biol. 2008;10:825–836. doi: 10.1038/ncb1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Edgar BA, Orr-Weaver TL. Endoreplication cell cycles: more for less. Cell. 2001;105:297–306. doi: 10.1016/s0092-8674(01)00334-8. [DOI] [PubMed] [Google Scholar]
- 27.Kaestne KH, Knochel W, Martinez DE. Unified nomenclature for the winged helix/forkhead transcription factors. Gene Dev. 2000;14:142–146. [PubMed] [Google Scholar]
- 28.Laoukili J, Kooistra MR, Brás A, Kauw J, Kerkhoven RM, Morrison A, et al. FoxM1 is required for execution of the mitotic programme and chromosome stability. Nat Cell Biol. 2005;7:126–136. doi: 10.1038/ncb1217. [DOI] [PubMed] [Google Scholar]
- 29.Littler DR, Alvarez-Fernández M, Stein A, Hibbert RG, Heidebrecht T, Aloy P, et al. Structure of the FoxM1 DNA-recognition domain bound to a promoter sequence. Nucleic Acids Res. 2010;38:4527–4538. doi: 10.1093/nar/gkq194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Radhakrishnan SK, Bhat UG, Hughes DE, Wang IC, Costa RH, Gartel AL. Identification of a chemical inhibitor of the oncogenic transcription factor forkhead box M1. Cancer Res. 2006;66:9731–9735. doi: 10.1158/0008-5472.CAN-06-1576. [DOI] [PubMed] [Google Scholar]
- 31.Holland AJ, Cleveland DW. Losing balance: the origin and impact of aneuploidy in cancer. EMBO reports. 2012;13:501–514. doi: 10.1038/embor.2012.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rao CV, Yang YM, Swamy MV, Liu T, Fang Y, Mahmood R, et al. Colonic tumorigenesis in BubR1+/−ApcMin/+ compound mutant mice is linked to premature separation of sister chromatids and enhanced genomic instability. Proc Natl Acad Sci U S A. 2005;102:4365–4370. doi: 10.1073/pnas.0407822102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang HC, Shi J, Orth JD, Mitchison TJ. Evidence that mitotic exit is a better cancer therapeutic target than spindle assembly. Cancer Cell. 2009;16:347–358. doi: 10.1016/j.ccr.2009.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Matsuura S, Ito E, Tauchi H, Komatsu K, Ikeuchi T, Kajii T. Chromosomal instability syndrome of total premature chromatid separation with mosaic variegated aneuploidy is defective in mitotic-spindle checkpoint. Am J Hum Genet. 2000;67:483–486. doi: 10.1086/303022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suijkerbuijk SJ, van Osch MH, Bos FL, Hanks S, Rahman N, Kops GJ. Molecular causes for BUBR1 dysfunction in the human cancer predisposition syndrome mosaic variegated aneuploidy. Cancer Res. 2010;70:4891–4900. doi: 10.1158/0008-5472.CAN-09-4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cortés F, Mateos S, Pastor N, Domínguez I. Toward a comprehensive model for induced endoreduplication. Life Sci. 2004;76:121–135. doi: 10.1016/j.lfs.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Storchova Z, Pellman D. From polyploridy to aneuploidy genome instability and Cancer. Nat Rev Mol Cell Biol. 2004;5:45–54. doi: 10.1038/nrm1276. [DOI] [PubMed] [Google Scholar]
- 38.Lee HO, Davidson JM, Duronio RJ. Endoreplication: polyloidy with purpose. Genes Dev. 2009;23:2461–2477. doi: 10.1101/gad.1829209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niculescu AB, 3rd, Chen X, Smeets M, Hengst L, Prives C, Reed SI. Effects of p21(Cip1/Waf1) at both the G1/S and the G2/M cell cycle transitions: pRb is a critical determinant in blocking DNA replication and in preventing endoreduplication. Mol Cell Biol. 1998;18:629–643. doi: 10.1128/mcb.18.1.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bellanger S, de Gramont, Sobczak-Thépot J. Cyclin B2 suppresses mitotic failure and DNA re-replication in human somatic cells knocked down for both cyclins B1 and B2. Oncogene. 2007;26:7175–7184. doi: 10.1038/sj.onc.1210539. [DOI] [PubMed] [Google Scholar]
- 41.Nair JS, Ho AL, Tse AN, Coward J, Cheema H, Ambrosini G, et al. Aurora B kinase regulates the postmitotic endoreduplication checkpoint via phosphorylation of the retinoblastoma protein at serine 780. Mol Biol Cell. 2009;20:2218–2228. doi: 10.1091/mbc.E08-08-0885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim JA, Lee J, Margolis RL, Fotedar R. SP600125 suppresses Cdk1 and induces endoreplication directly from G2 phase, independent of JNK inhibition. Oncogene. 2010;29:1702–1716. doi: 10.1038/onc.2009.464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tominaga Y, Wang A, Wang RH, Wang X, Cao L, Deng CX. Genistein inhibits Brca1 mutant tumor growth through activation of DNA damage checkpoints, cell cycle arrest, and mitotic catastrophe. Cell Death Differ. 2007;14:472–479. doi: 10.1038/sj.cdd.4402037. [DOI] [PubMed] [Google Scholar]
- 44.Wonesy DR, Follettie MT. Loss of the forkhead transcription factor FoxM1 causes centrosome amplification and mitotic catastrophe. Cancer Res. 2005;65:5181–5189. doi: 10.1158/0008-5472.CAN-04-4059. [DOI] [PubMed] [Google Scholar]