

Abstract
Purpose
Glaucoma affects Americans in all economic strata. Our fragmented insurance landscape provides robust coverage for eyedrop medications for some while others pay high retail costs. We set out to determine whether a recession would have a greater impact on intraocular pressure (IOP) control in individuals without vs. those with pharmacy benefits.
Methods
Change in IOP and number of medications prescribed in glaucoma patients with and without pharmacy benefits was recorded on two consecutive visits before and during a recession.
Results
Seventy-seven patients with (mean±SD age, 66±17 years; 61% female, 52% Caucasian; 69% Medicare) and 51 patients without (mean±SD age, 63±14 years; 61% female, 53% African American; 78% Charity Care) pharmacy benefits were included. Thirty-seven (48%) subjects with pharmacy benefits had ≥1 health insurance. Mean±SD IOP, number of glaucoma medications, and number of patients on ≥2 glaucoma medications before and after recession did not change significantly for either group.
Conclusions
Our results did not show any difference in IOP control between those with and without pharmacy benefits. Other complex issues such as adherence may mask the impact of pharmacy benefit on IOP control.
Introduction
Drug delivery technology evolved in part out of the need to address the deficiencies of conventional means of administering drugs. With those, it was difficult to maintain drug levels within the narrow concentration window required to avoid toxicity due to overdose, or ineffective treatment from under-dosing. Furthermore, drugs had to be administered frequently or continuously in order to achieve a long-lasting effect, potentially resulting in patient discomfort or inconvenience, or requiring tethering to external devices. With controlled release systems, a single dose can sustain drug levels within the desired therapeutic range for long periods.1, 2 Examples of passive release systems that are readily translatable to the clinic include contact lenses that release antifungal agents for treating mycotic eye infections3, injectable liposomes that release cocktails of synergistic anesthetics such as bupivacaine and saxitoxin4, drug eluting sutures5, and in-situ crosslinkable hydrogels for controlled protein release.6 Nanoparticles can also be functionalized to bind to tissue nonspecifically upon irradiation with light7, or specifically to a membrane-bound protein (e.g. angiotensin II type I in the infarcted heart8), enabling passive release combined with spatial control.
Recently, there has been increasing interest in developing methods where drug release from the injected or implanted delivery system can be controlled by an operator, perhaps via a remote device. Ideally, such systems could determine the time, duration, dosage, and even location of drug release, and could allow remote, non-invasive, repeatable, and reliable switching of therapeutic agent flux.2 These devices are beneficial to patients across a wide spectrum of ages, since they enable drug release profiles tailored to the specific therapy. For example, insulin9 or osteoporosis therapeutics10 are most effective when delivered to the patient in short bursts. Devices that release locally- or systemically-acting pain medications would allow the patient – or a health care worker or parent – to titrate the dose to effect, providing pain relief when needed and to the extent needed. Such treatments could be useful in diseases that flare up intermittently. One can also envision depot formulations that would release drugs at seizure foci when triggered by the patient, health-care worker, and or an implanted sensing system (i.e. a closed-loop system).
Controlled release systems in general can be attractive in children, in that they can potentially provide prolonged duration of effect from a single administration. Triggerable drug delivery systems would have the same advantage, and would permit fluctuations in dosing and timing but, in pediatrics, would run into the problem that many children are either unable to manage their own drug regimen, or might not be trusted to do so. Therefore, the devices might have to be controlled by a parent or health professional in a manner analogous the manner in which nurse-controlled analgesia pumps are employed. Assuming a readily usable metric with which to follow progression of a disease state (e.g. glucose, pH, other analyte) closed-loop systems might be particularly useful contexts in which to use triggered drug delivery devices in pediatrics.
Progress in the development of remotely-triggered drug delivery systems has been enabled by cross-disciplinary studies in nanotechnology, materials science and chemistry, and these systems have been designed to react to a wide range of stimuli. For example, iron oxide nanoparticles inductively heat in the presence of an oscillating magnetic field11, and the heat generated can be used to trigger drug release from a temperature-sensitive moiety. Gold nanoparticles (e.g. nanospheres, nanorods, nanoshells, nanocages) can also be heated by inductive plasmon coupling to visible or near-infrared light, and in some cases to radio frequency and microwaves. Their absorbance spectrum is critically dependent on geometry, which can be tuned at the time of synthesis12, and multiplexed drug delivery systems that are addressed by distinct wavelengths of light have been demonstrated13. Ultrasound represents another modality for controlling drug delivery, and has been used to trigger release from microparticles, vesicles, and liposomes.14–17 Advances in microfabrication and chemical synthesis have enabled systems that respond to electric fields and visible and/or ultraviolet (UV) light.
The physics, chemistry and materials science behind triggered release systems has been reviewed- along with the considerations relating to materials synthesis, device fabrication, and physical triggering mechanism.2 Many new triggered release systems release their payload all at once, in a pulsatile fashion, or do not exhibit reproducible dosing. Ultimately, triggered release systems must exhibit a number of characteristics before they can be translated to the clinic: (1) The “on”-state drug release rate must be tunable such that the dose released is within a therapeutically relevant range. (2) The ratio between “on” and “off” states should (at least optimally) be as large as possible. “Off” state leakage must be sufficiently low so as to avoid side effects and to maintain longterm viability of the device in vivo. (3) Successive “on” states must be reproducible, and ideally the duration and magnitude of the trigger determines the duration and rate of drug release. Release kinetics must not be susceptible to degradation or biofouling of the device over time. (4) Systems must exhibit an acceptable inflammatory response, and side effects relating to the triggering mechanism itself should be considered. Moreover, the biodistribution of released nanoparticles and toxicity of any degradation products must be carefully assessed.
A number of recently-reported technologies addresses each of these points by integrating new materials within carefully engineered devices. Many have been validated in vivo, and some are now undergoing clinical trials.
Magnetically-activated membranes
The structure, synthesis, and biocompatibility of magnetic oxide nanoparticles have been well-studied, and they have been widely used in the biosciences for cell labeling, bioseparation, drug delivery and hyperthermia. Functionalized magnetic nanoparticles have also been used clinically, as a contrast agent for MRI imaging18. Iron oxide nanocrystals that exhibit single magnetic domains release heat when exposed to alternating magnetic fields. They have been used to trigger drug release by incorporation into a variety of systems: macroscale polymers (e.g. poly(n-isopropyl acrylamide) (polyNIPAm)19 or chitosan20), microspheres21, 22, liposomes23, micro- or nanocapsules24–26, and nanospheres.27–29 Since alternating magnetic fields and iron oxide nanoparticles have both been used clinically, these new triggered systems hold potential for clinical translation.30–32
Engineered stimulus-sensitive membranes can be used to create reservoir systems that can contain a large quantity of drug to be released repeatedly, with controlled dosing, over extended periods. Although applicable for a range of drugs and applications, such devices could be particularly useful for local anesthesia, with a capsule implanted near a nerve (e.g. the sciatic nerve) to achieve local blockade. Such devices could be particularly useful for delivering drug cocktails that exhibit synergistic effects, such as amino-amide local anesthetics and site 1 sodium channel blockers such as tetrodotoxin (TTX).4
Nanocomposite membranes consisting of temperature-sensitive polyNIPAm-based nanogels and magnetic iron oxide nanoparticles embedded in an ethylcellulose film were recently demonstrated.33 An alternating magnetic field heated the nanoparticles, triggering collapse of the nanogels and increasing permeability of the membrane, or turning it “on” (Figure 1A). This process was reversible, and numerous “on” and “off” cycles were demonstrated (Figure 1B). These membranes were used to deliver a wide range of model compounds, ranging from small molecules (sodium fluorescein) to macromolecules (40 kDa dextran), and are therefore expected to be effective in a delivering a wide variety of drug types.
Fig. 1.
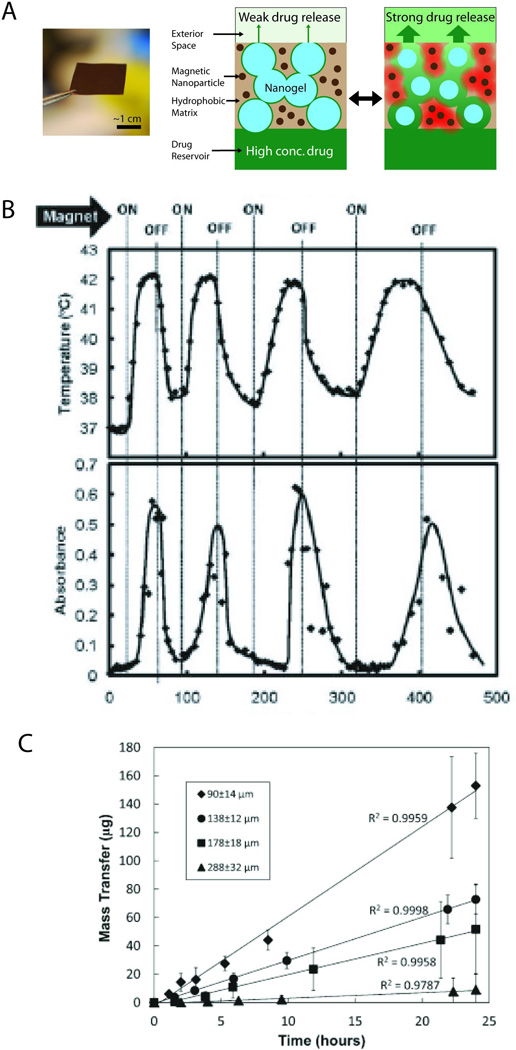
(A) (Left) Photograph of membrane containing ferromagnetic nanoparticles and temperature-sensitive nanogels, distributed throughout an ethyclellulose matrix. (Right) Schematic of a cross section of the membrane, showing nanogel particles (blue), iron oxide nanoparticles (dark brown), and ethylcellulose matrix (light brown). Upon application of an oscillating magnetic field, the magnetic nanoparticles release heat (red) and reversibly shrink the nanogel, enabling release of a drug (green) from a reservoir contained by the membrane. (B) Temperature profile and differential flux of sodium fluorescein out of a membrane-capped capsule as a function of time over four successive on/off cycles of the external magnetic field. (C) Mass transfer of sodium fluorescein as a function of “on” triggering time for membranes with different thicknesses. Adapted with permission from refs. 33 and 34. Copyright 2009 and 2011 American Chemical Society.
One advantage of the membrane capsule system is that the release rate across a membrane can be tuned by adjusting the nanogel concentration within the membrane, or the thickness of the membrane itself. Membranes with thicknesses ranging from 90 to 288 µm all exhibited zero-order release kinetics, with release rates spanning nearly two orders of magnitude (Figure 1C).34 Fine control over release kinetics has clear clinical advantages, particularly in the case of systemically toxic drugs where an effective dose of drug is desirable only at the site of triggered release.
The biocompatibility of these membranes was tested, both in vitro and in vivo. The membranes and their components were non-cytotoxic to a range of relevant cell types, including fibroblasts, macrophages, myoblasts and PC12 neuron model cells, which were chosen to represent the environment near the sciatic nerve. Membranes were intact and remained functional after up to 45 days after subcutaneous implantation in rats, suggesting that biofouling will not seriously affect sustained drug release. Hematoxylin-eosin staining indicated a generally benign tissue reaction.33 Further studies are needed to assess the therapeutic efficacy, and possible side effects, of triggered drug release from capsules.
UV-Triggered Polymers
Light ranging from UV to infrared has been used to trigger a wide variety of materials. Light as a triggering mechanisms allows spatial and temporal selectivity and at controlled doses does not harm skin or tissue. Light-triggered biomaterials are particularly useful for clinical use, since laser and LED light sources are generally cheap, portable, and relatively safe.2, 35 Maximum permissible exposures to laser irradiation have been established by the American National Standards Institute.36
Compared to longer wavelengths, light in the UV region suffers a number of drawbacks. It is strongly absorbed by skin and tissue and therefore cannot be used for deep-tissue triggering. Moreover, at substantially high powers it can cause photochemical injury by free radical generation, leading to eye injury, premature skin aging, skin cancer, and/or skin burning.35 Nevertheless, tissues such as skin, the ear, or the back of the eye are excellent candidates for treatment, or regions that are accessible by a fiber optic cable, so long as the irradiation power is safe. Numerous chemical changes, such as covalent bond cleavage and isomerization, can generally only be achieved with light in the UV or visible range.2, 35
UV-sensitive caging groups can be used to create light triggerable nanoparticles that will target any tissue that is illuminated. Nanoparticles were covalently functionalized with the peptide sequence YIGSR, which adheres to the β1 integrins present on all cell surfaces. This peptide was rendered biologically inert by linkage to to a caging group (4,5-dimethoxy-2-nitrobenzyl (DMNB)). Illumination with UV light released the caging group from the peptide, which allowed the particles to bind cells (Fig. 2).37 Particle attachment was demonstrated to both human umbilical vein endothelial cells (HUVECs) and mesenchymal stem cells (MSCs), selectively to cells irradiated by UV light. This form of triggering differs from the others we have alluded to; it is the release of a drug delivery system that is being triggered. In so doing, this system also achieves spatiotemporally controlled targeting of a sustained release system even in the absence of a specific ligand.
Fig. 2.
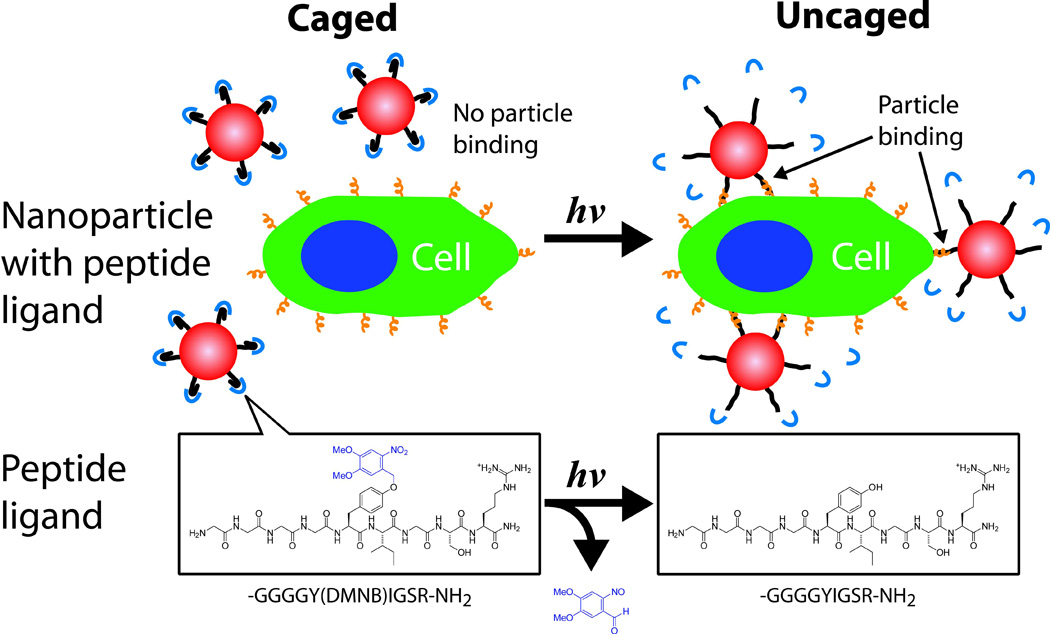
Phototargeted nanoparticles. A nonspecific ligand on the surface of the nanoparticles is inactivated by caging. Upon illumination, the caging group is released, the ligand is activated, and the nanoparticles can bind to the illuminated tissue. The lower portion of the figure shows the chemistry of the targeting moiety as it relates to the events schematized above. The GGGGYIGSR-NH2 peptide is caged on tyrosine with 4,5-dimethoxy-2-nitrobenzyl (DMNB) (Blue). Adapted with permission from ref. 37. Copyright 2009 American Chemical Society.
Nanoparticles that release an encapsulated drug upon irradiation provide an alternative and perhaps complementary route to triggered drug delivery. Toward this end, photoswitchable nanoparticles have been demonstrated that are based on spiropyan (SP), which undergoes a reversible ring-opening reaction to form merocyanine (MC) under UV irradiation (Fig. 3A).38 Such nanoparticles reversibly collapsed under UV irradiation, as the spiropyran photoisomerized into more hydrophobic merocyanine (Figs. 3B,C). To evaluate their usefulness for triggered drug release, the nanoparticles were used to encapsulate a variety of model compounds, including rhodamine, coumarin, calcein, cyanine, paclitaxel, docetaxel, doxorubicin and proparacaine. Figure 3D shows release profiles in PBS for rhodamine-loaded particles, where particle collapse was triggered with 30-second UV pulses. Release kinetics were increased approximately by a factor of four after activation.38 Therapeutic effect can be enhanced and toxicity reduced by surface modification with ligands that enable intracellular penetration and/or targeting of specific tissues. Particles modified with a cell penetration peptide (Cpp) were readily incorporated into HeLa cells, and Cpp-modified particles loaded with doxorubicin were substantially more cytotoxic than those not conjugated to Cpp.
Fig. 3.
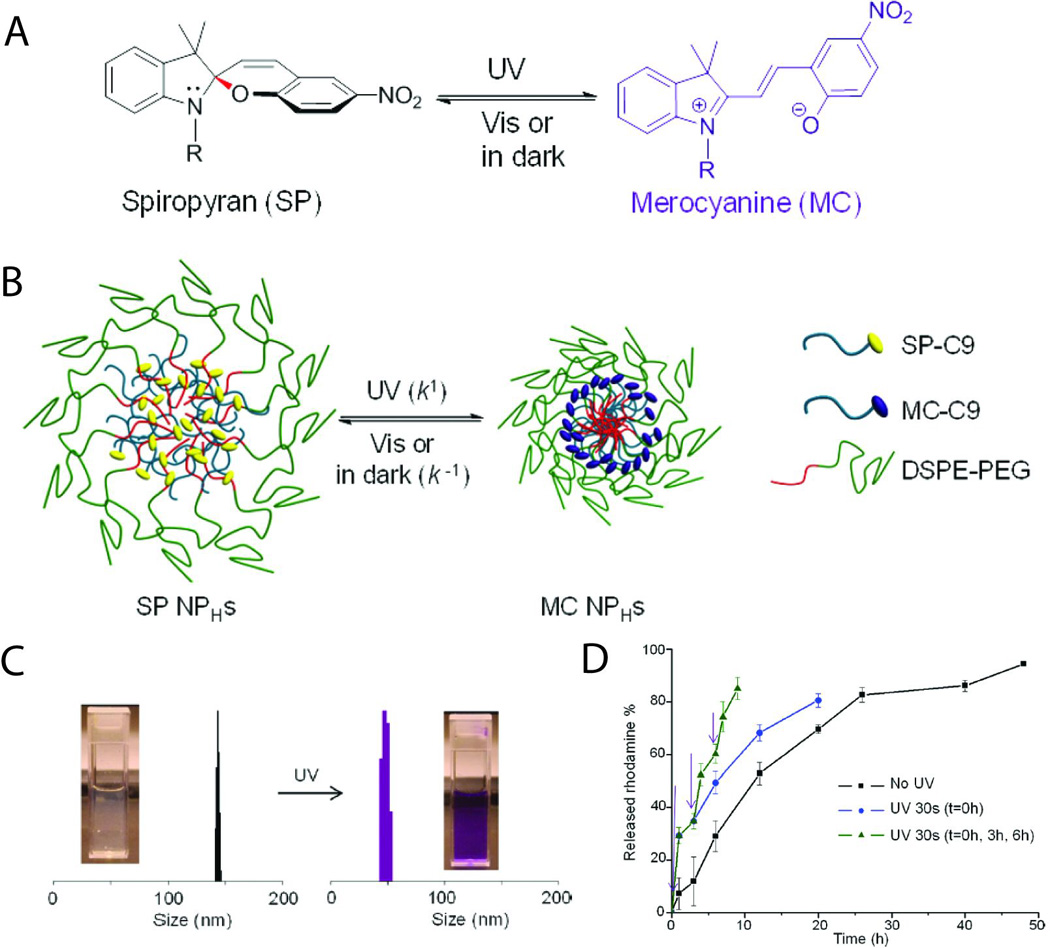
(A) Structure and photoisomerization reaction between spiropyran (SP) and merocyanine (MC). (B) Scheme of photoswitching spiropyran-based nanoparticles (SP NPHs). (C) Dynamic light scattering measurement of size changes of spiropyran-based nanoparticles upon alternating UV (30 s) and visible light (3 min) illumination. Inset: the solution of spiropyran-based nanoparticles before and after UV irradiation. (D) Release profiles in PBS for rhodamine B loaded in spiropyran-based nanoparticles under different conditions: without irradation; with UV irradiation for 30 s at 0 h; with repetitive UV irradiation at 0, 3, and 6 h. The times of irradiation are indicated by purple arrows. Data are means ± SD, N = 6. Adapted with permission from ref. 38. Copyright 2012 American Chemical Society.
Thermosensitive liposomes
Liposomes have been studied extensively over the past 30 years as carriers of a vast array of pharmaceuticals. Liposomes enable controlled, passive release of an encapsulated drug, can entrap both hydrophobic and hydrophilic molecules, protect encapsulated drugs from harmful external conditions, and can be designed to deliver pharmaceuticals into cells or sometimes into specific intracellular compartments. Liposome chemistry has been extensively studied and reviewed elsewhere.39 Numerous liposomal formulations have been approved for clinical use for treating cancer, meningitis, or fungal infections, among other conditions.40
Liposomes can be made containing temperature-sensitive moieties which cause them to deform or lyse when heated beyond a critical temperature. Such liposomes have been used in triggered release systems. Lyso-thermosensitive liposomes have been conjugated with gold or iron oxide nanoparticles and are respectively triggered with light or magnetic fields, which inductively heat the nanoparticles 2. Rupture can also be triggered by an external energy source such as radio frequency or focused ultrasound, both of which cause local heating in tissues.14, 41
A formulation of lyso-thermosensitive liposomal doxorubicin (LTLD), ThermoDox, has been developed by Celsion Corp. (Lawrenceville, NJ) and is now undergoing Phase III clinical trials (ClinicalTrials.gov ID: NCT00617981). It is used to treat hepatocellular carcinoma (HCC), the fourth leading cause of cancer death worldwide. The formulation is used in conjunction with radio frequency ablation (RFA), which by itself kills cells by hyperthermia. RFA raises the temperature of LTLD above 39.5°C, causing the liposome to burst and release the encapsulated doxorubicin. LTLD is administered systemically, but concentrates in the liver and spleen because of its size. Moreover, LTLD is much more likely to extravasate into tumors rather than healthy tissue since tumors have much higher microvascular permeability than normal blood vessels. This effect is further enhanced by RFA-induced hyperthermia, which has been shown to increase the pore size in tumor blood vessels. Pre-clinical studies have shown that doxorubicin released from LTLD exhibits 15-fold higher drug concentration in tumors compared to nonliposomal doxorubicin administered at the same dose. Phase I clinical were consistent with the hypothesis that LTLD significantly increases RFA efficiency.42, 43
Triggered release from liposomes has also been achieved with ultrasound, which causes either localized heating or mechanical disruption.40, 41 Triggered release from thermosensitive liposomes can be improved by proximity to air-filled microbubbles, which oscillate and cavitate in the presence of low-frequency ultrasound, and increase the difference between baseline and peak release rates of drugs encapsulated by the liposomes.14 In-situ cross-linking hydrogel matrices have been developed that incorporate both liposomes and microbubbles. At physiological temperature the hydrogel network formed around the liposomes, maintaining their proximity to the cosuspended microbubbles.44 Such hydrogels containing dye-loaded liposomes disintegrated and released the dye repeatedly after ultrasound pulses (Fig. 4A,B). These materials performed well in vivo, exhibiting little inflammatory response and demonstrating a low baseline leak rate and triggerability for up to 14 days (Fig. 4C).
Fig. 4.
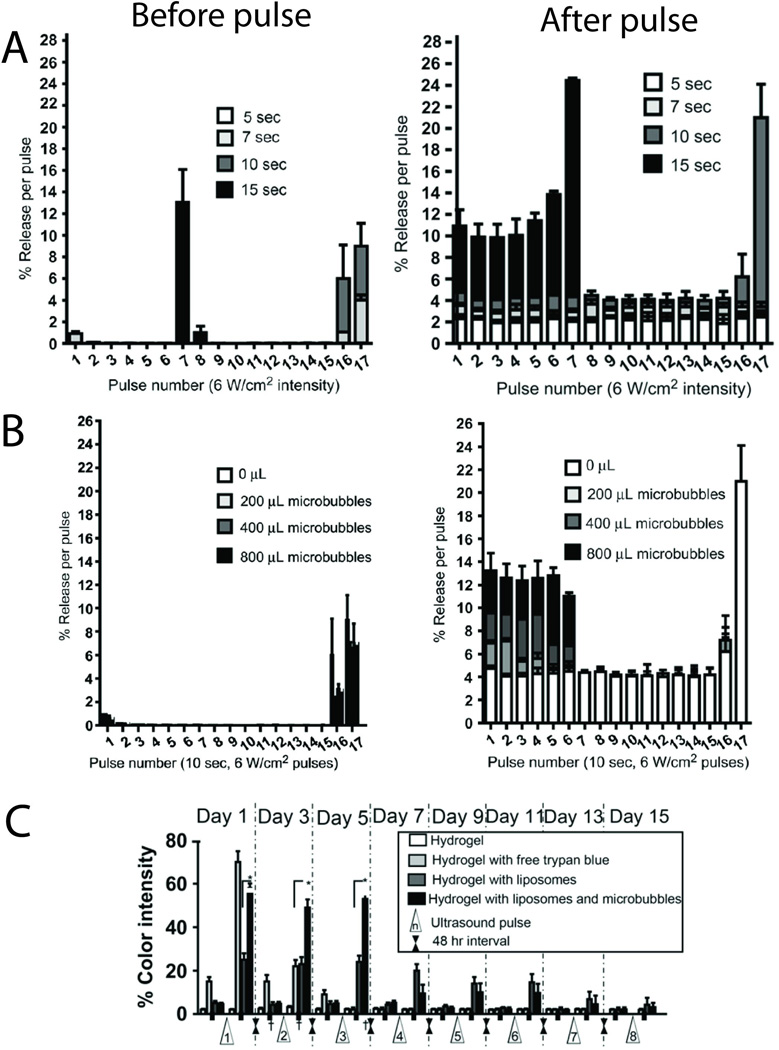
(A) The effect of low-frequency ultrasound pulse intensity duration on in vitro release of trypan blue from 200 µl of negative liposomes in hydrogels at 37°C. (B) Effect of microbubble concentration (0, 200, 400 and 800 µl added to the hydrogel) using a 10-s pulse at 6 W/cm2. For both parameters, dye release before (left panel) and after (right panel) each pulse is shown. Data are means ± SD, N = 4. (C) Ultrasound-triggered dye release profile of trypan blue from liposomes suspended with microbubbles in hydrogels, injected subcutaneously into mice. Data are means ± SD, N = 8; * p<0.001. Adapted with permission from ref. 44. Copyright 2010 Elsevier, Inc.
Controlled release microchips
A wirelessly controlled microchip-based delivery system developed by MicroCHIPS, Inc. (Waltham, MA) recently underwent testing in humans (Fig. 5A).10 The microchip itself consisted of an array of reservoirs etched into a silicon substrate, where each reservoir held tens of nanoliters of solution. The reservoirs were sealed by gold electrodes, which were electrochemically dissolved by an applied electric field, releasing the drug contained within. Because of the small size of the reservoirs, thousands of them could in principle be fabricated into a single implantable chip, with the gold cap on each individually addressed via microscale leads (Fig. 5B and C). The devices used in clinical studies contained 20 reservoirs and were packaged along with a battery and control and communication electronics, enabling computer-controlled, wirelessly-triggered drug release.45
Fig. 5.
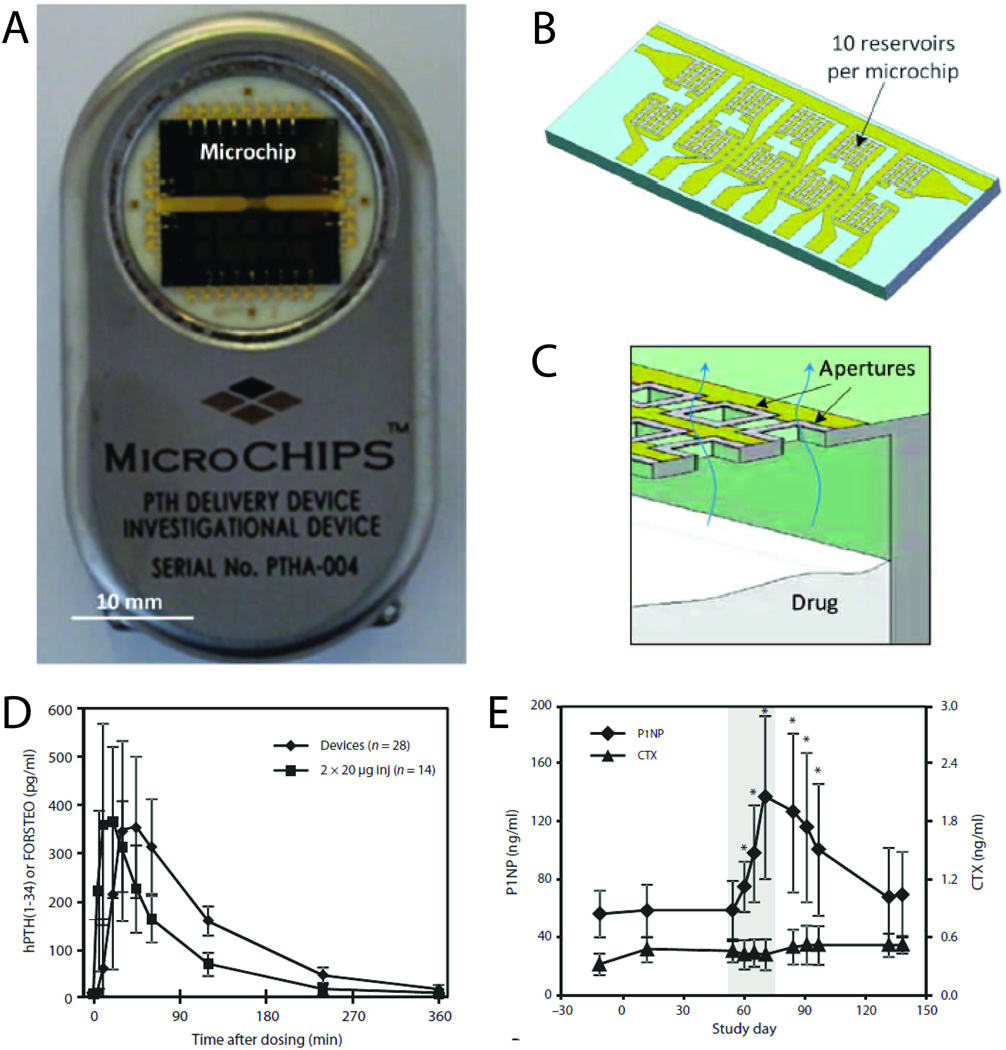
Wirelessly-triggered drug delivery device developed by MicroCHIPS, Inc. (A and B) Implantable drug delivery device (54 mm × 31 mm × 11 mm, l × w × h) (A) containing two microchips with 10 reservoirs each (B). (C) Cross-section schematic of a microchip assembly showing drug release from a single reservoir. (D) Mean plasma concentration of hPTH versus time after release of 40-µg dose from the implanted microchip device (N = 7 patients × four doses) and injection of 2×20-µg doses of FORSTEO (N = 7 patients × two doses). Data are means ± SD. (E) P1NP and CTX bone marker concentrations in serum before, during and after implant-mediated drug dosing. The shaded area encloses the 2 weeks during which individual 40-µg doses of hPTH were released from the implant once daily. Data are means ± SD (N = 7). *P < 0.05 compared to day – 12, pairwise t test. Adapted with permission from ref. 10. Copyright 2012 Science Publishing Group.
The device was used to deliver human parathyroid hormone fragment (hPTH), the only approved treatment for anabolic osteoporosis. The drug requires daily injections, making patient compliance an obstacle to effective treatment. Chips were implanted into eight osteoporotic women for 4 months, and programmed to release doses once daily for up to 20 days. Pharmacokinetics were assessed by measuring hPTH in blood plasma. The concentration of hPTH peaked within two hours of triggered release, and the release profile was consistent throughout the duration of the trial. On average, time to peak concentration and terminal half life were slightly longer than observed after injections of FORTEO (Eli Lilly and Company), the commercially-available formulation that contains a similar dose of hPTH as the active component (Fig. 5D). Drug administered by the two methods may form differently-sized boluses, affecting the rate of absorption into the bloodstream. However, there was no evidence that these differences affected skeletal response to hPTH. Patients treated with the implantable devices exhibited increases in serum type I collagen propeptide (P1NP), consistent with an anabolic increase in bone formation and confirming the therapeutic effectiveness of the device. In contrast, Type I collagenolysis fragment (CTX), a marker for bone resorption, remained constant throughout the trial (Fig. 5E).10
Conclusions and Prospects
Remotely triggered release devices are realized as either implants or injectable materials, and may be triggered by light, magnetic fields, ultrasound, or electric fields. Remote triggering can enable drug release with repeated, reproducible, on-demand dosing, potentially reducing systemic toxicity and increasing efficacy. Where desirable, these methods can also provide highly localized drug release. These advances have been brought about by the convergence of numerous disciplines including chemistry, physics, materials science, electrical engineering, and chemical engineering.1, 2 This convergence is likely to play an important part in the current trend to develop systems that can provide more than one therapeutic modality.
Among the vast array of triggered release systems that have been reported in the literature2, relatively few have been translated into clinical devices. In the case of preclinical systems, the importance of biocompatibility cannot be overstated. These studies should include not only an assessment of the biomaterial itself, both in vitro and in vivo, but also an assessment of local tissue reaction to the drug released.46 The latter point is especially important in the case of sustained or programmed release systems, where pharmacokinetics may differ substantially from those of injected doses. In particular local drug levels may be much higher than are generally achieved by systemic delivery, and for much longer durations. Appropriate studies could range from local cytotoxicity studies to toxicogenomic analyses.46, 47 Finally, the device must be assessed in terms of biofouling, degradation, and biodistribution of any potential degradation products. The solid-state devices produced by MicroCHIPS, for example, were extremely stable, but those composed of soft materials (e.g. polymer formulations) may behave differently. Ultimately, each device and application should be assessed on a case-by-case basis.
Future drug delivery devices are likely to incorporate novel materials that enable enhanced functionality. Methods already exist that combine magnetically guided targeting and triggered drug release for local, on-demand applications, or combine hyperthermia and chemical release for more effective cancer treatment. There is a similar trend toward closed-loop systems that both diagnose and treat (i.e., theranostics). Nanoelectronic devices have been recently demonstrated that can detect small molecules, pH, individual viruses, or low concentrations of protein down to the femtomolar level,48–50 or that can interface with cultured cells49, 51 or tissue52, 53 at the subcellular level. Other systems can harness energy from light54 or mechanical motion55 and might serve as local power sources for nanoelectronic sensing or release mechanisms. Nanoelectronic-enabled devices might complement traditional passive and active drug release technologies1, widening the repertoire of delivery modalities. Recently, such devices have been incorporated into engineered tissues, in which they could sense electrical activity and chemical changes.56 Such cues could in turn be used to trigger drug release within tissue constructs.45 Given the chronic shortage of transplantable organs, these are potentially important developments, particularly in pediatrics. We note that by providing pulsatile or programmed variable drug release patterns, devices of the type described here have the potential to enable regimens that take into account the potential effects of circadian and other rhythms57 on drug efficacy, interaction, and safety.
Acknowledgements
This research was funded by the National Institutes of Health (NIH, grant GM073626 to D.S.K.) and the Sanofi Biomedical Innovation Awards Program (to D.S.K.). B.P.T. acknowledges an NIH Ruth L. Kirschstein National Research Service Award (grant F32GM096546).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest
The authors have indicated that they have no conflicts of interest with regard to the content of the article.
References
- 1.Timko BP, Whitehead K, Gao WW, et al. Advances in Drug Delivery. Annual Review of Materials Research. 2011;Vol 41:1–20. [Google Scholar]
- 2.Timko BP, Dvir T, Kohane DS. Remotely triggerable drug delivery systems. Adv Mater. 2010;44:4925–4943. doi: 10.1002/adma.201002072. [DOI] [PubMed] [Google Scholar]
- 3.Ciolino JB, Hudson SP, Mobbs AN, et al. A prototype antifungal contact lens. Invest Ophthalmol Vis Sci. 2011;9:6286–6291. doi: 10.1167/iovs.10-6935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Epstein-Barash H, Shichor I, Kwon AH, et al. Prolonged duration local anesthesia with minimal toxicity. Proc Natl Acad Sci U S A. 2009;17:7125–7130. doi: 10.1073/pnas.0900598106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weldon CB, Tsui JH, Shankarappa SA, et al. Electrospun drug-eluting sutures for local anesthesia. J Control Release. 2012 doi: 10.1016/j.jconrel.2012.05.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Epstein-Barash H, Stefanescu CF, Kohane DS. An in situ cross-linking hybrid hydrogel for controlled release of proteins. Acta Biomater. 5:1703–1709. doi: 10.1016/j.actbio.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dvir T, Banghart MR, Timko BP, et al. Photo-targeted nanoparticles. Nano Lett. 1:250–254. doi: 10.1021/nl903411s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dvir T, Bauer M, Schroeder A, et al. Nanoparticles targeting the infarcted heart. Nano Lett. 10:4411–4414. doi: 10.1021/nl2025882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kost J. Pulsed and self-regulated drug delivery. Boca Raton: CRC Press; 1990. [Google Scholar]
- 10.Farra R, Sheppard NF, Jr, McCabe L, et al. First-in-human testing of a wirelessly controlled drug delivery microchip. Sci Transl Med. 2012;122:122ra21. doi: 10.1126/scitranslmed.3003276. [DOI] [PubMed] [Google Scholar]
- 11.Jeong U, Teng XW, Wang Y, et al. Superparamagnetic colloids: Controlled synthesis and niche applications. Advanced Materials. 2007;1:33–60. [Google Scholar]
- 12.Huang XH, Neretina S, El-Sayed MA. Gold Nanorods: From Synthesis and Properties to Biological and Biomedical Applications. Advanced Materials. 2009;48:4880–4910. doi: 10.1002/adma.200802789. [DOI] [PubMed] [Google Scholar]
- 13.Wijaya A, Schaffer SB, Pallares IG, et al. Selective release of multiple DNA oligonucleotides from gold nanorods. ACS Nano. 2009;1:80–86. doi: 10.1021/nn800702n. [DOI] [PubMed] [Google Scholar]
- 14.Ferrara KW. Driving delivery vehicles with ultrasound. Adv Drug Deliv Rev. 2008;10:1097–1102. doi: 10.1016/j.addr.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Frenkel V. Ultrasound mediated delivery of drugs and genes to solid tumors. Adv Drug Deliv Rev. 2008;10:1193–1208. doi: 10.1016/j.addr.2008.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mitragotri S. Healing sound: the use of ultrasound in drug delivery and other therapeutic applications. Nat Rev Drug Discov. 2005;3:255–260. doi: 10.1038/nrd1662. [DOI] [PubMed] [Google Scholar]
- 17.Schroeder A, Kost J, Barenholz Y. Ultrasound, liposomes, and drug delivery: principles for using ultrasound to control the release of drugs from liposomes. Chem Phys Lipids. 2009;1-2:1–16. doi: 10.1016/j.chemphyslip.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 18.Na HB, Song IC, Hyeon T. Inorganic Nanoparticles for MRI Contrast Agents. Advanced Materials. 2009;21:2133–2148. [Google Scholar]
- 19.Satarkar NS, Hilt JZ. Magnetic hydrogel nanocomposites for remote controlled pulsatile drug release. Journal of Controlled Release. 2008;3:246–251. doi: 10.1016/j.jconrel.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 20.Hu SH, Liu TY, Liu DM, et al. Controlled pulsatile drug release from a ferrogel by a high-frequency magnetic field. Macromolecules. 2007;19:6786–6788. [Google Scholar]
- 21.Muller-Schulte D, Schmitz-Rode T. Thermosensitive magnetic polymer particles as contactless controllable drug carriers. Journal of Magnetism and Magnetic Materials. 2006;1:267–271. [Google Scholar]
- 22.Zhang J, Misra RDK. Magnetic drug-targeting carrier encapsulated with thermosensitive smart polymer: Core-shell nanoparticle carrier and drug release response. Acta Biomaterialia. 2007;6:838–850. doi: 10.1016/j.actbio.2007.05.011. [DOI] [PubMed] [Google Scholar]
- 23.Tai LA, Tsai PJ, Wang YC, et al. Thermosensitive liposomes entrapping iron oxide nanoparticles for controllable drug release. Nanotechnology. 2009;13 doi: 10.1088/0957-4484/20/13/135101. [DOI] [PubMed] [Google Scholar]
- 24.Hu SH, Tsai CH, Liao CF, et al. Controlled Rupture of Magnetic Polyelectrolyte Microcapsules for Drug Delivery. Langmuir. 2008;20:11811–11818. doi: 10.1021/la801138e. [DOI] [PubMed] [Google Scholar]
- 25.Katagiri K, Nakamura M, Koumoto K. Magnetoresponsive Smart Capsules Formed with Polyelectrolytes, Lipid Bilayers and Magnetic Nanoparticles. Acs Applied Materials & Interfaces. 2010;3:768–773. doi: 10.1021/am900784a. [DOI] [PubMed] [Google Scholar]
- 26.Liu TY, Liu KH, Liu DM, et al. Temperature-Sensitive Nanocapsules for Controlled Drug Release Caused by Magnetically Triggered Structural Disruption. Advanced Functional Materials. 2009;4:616–623. [Google Scholar]
- 27.Hu SH, Chen SY, Liu DM, et al. Core/single-crystal-shell nanospheres for controlled drug release via a magnetically triggered rupturing mechanism. Advanced Materials. 2008;14:2690. doi: 10.1002/adma.200800193. [DOI] [PubMed] [Google Scholar]
- 28.Thomas CR, Ferris DP, Lee JH, et al. Noninvasive Remote-Controlled Release of Drug Molecules in Vitro Using Magnetic Actuation of Mechanized Nanoparticles. Journal of the American Chemical Society. 2010;31:10623–10625. doi: 10.1021/ja1022267. [DOI] [PubMed] [Google Scholar]
- 29.Hayashi K, Ono K, Suzuki H, et al. High-Frequency, Magnetic-Field-Responsive Drug Release from Magnetic Nanoparticle/Organic Hybrid Based on Hyperthermic Effect. Acs Applied Materials & Interfaces. 2010;7:1903–1911. doi: 10.1021/am100237p. [DOI] [PubMed] [Google Scholar]
- 30.Zhang L, Gu FX, Chan JM, et al. Nanoparticles in medicine: therapeutic applications and developments. Clin Pharmacol Ther. 2008;5:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 31.Stark DD, Weissleder R, Elizondo G, et al. Superparamagnetic Iron-Oxide - Clinical-Application as a Contrast Agent for Mr Imaging of the Liver. Radiology. 1988;2:297–301. doi: 10.1148/radiology.168.2.3393649. [DOI] [PubMed] [Google Scholar]
- 32.Lin MM, Kim DK, El Haj AJ, et al. Development of Superparamagnetic Iron Oxide Nanoparticles (SPIONS) for Translation to Clinical Applications. Ieee Transactions on Nanobioscience. 2008;4:298–305. doi: 10.1109/TNB.2008.2011864. [DOI] [PubMed] [Google Scholar]
- 33.Hoare T, Santamaria J, Goya GF, et al. A Magnetically Triggered Composite Membrane for On-Demand Drug Delivery. Nano Letters. 2009;10:3651–3657. doi: 10.1021/nl9018935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hoare T, Timko BP, Santamaria J, et al. Magnetically Triggered Nanocomposite Membranes: A Versatile Platform for Triggered Drug Release. Nano Letters. 2011;3:1395–1400. doi: 10.1021/nl200494t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tong R, Kohane DS. Shedding light on nanoparticles. Rev Nanomed Nanobiotechnol. 2012 doi: 10.1002/wnan.1188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.American National Standards I, Laser Institute of A. American National Standard for the safe use of lasers. Orlando, FL: The Institute; 1993. [Google Scholar]
- 37.Dvir T, Banghart MR, Timko BP, et al. Photo-targeted nanoparticles. Nano Lett. 2009;1:250–254. doi: 10.1021/nl903411s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong R, Hemmati HD, Langer R, et al. Photoswitchable nanoparticles for triggered tissue penetration and drug delivery. J Am Chem Soc. 2012;21:8848–8855. doi: 10.1021/ja211888a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jesorka A, Orwar O. Liposomes: technologies and analytical applications. Annu Rev Anal Chem (Palo Alto Calif) 2008:801–832. doi: 10.1146/annurev.anchem.1.031207.112747. [DOI] [PubMed] [Google Scholar]
- 40.Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. Nat Rev Drug Discov. 2005;2:145–160. doi: 10.1038/nrd1632. [DOI] [PubMed] [Google Scholar]
- 41.Schroeder A, Kost J, Barenholz Y. Ultrasound, liposomes, and drug delivery: principles for using ultrasound to control the release of drugs from liposomes. Chemistry and Physics of Lipids. 2009;1–2:1–16. doi: 10.1016/j.chemphyslip.2009.08.003. [DOI] [PubMed] [Google Scholar]
- 42.Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: an adjuvant to increase the cure rate of radiofrequency ablation in liver cancer. Future Oncol. 2011;8:937–945. doi: 10.2217/fon.11.73. [DOI] [PubMed] [Google Scholar]
- 43.Poon RT, Borys N. Lyso-thermosensitive liposomal doxorubicin: a novel approach to enhance efficacy of thermal ablation of liver cancer. Expert Opin Pharmacother. 2009;2:333–343. doi: 10.1517/14656560802677874. [DOI] [PubMed] [Google Scholar]
- 44.Epstein-Barash H, Orbey G, Polat BE, et al. A microcomposite hydrogel for repeated on-demand ultrasound-triggered drug delivery. Biomaterials. 2010;19:5208–5217. doi: 10.1016/j.biomaterials.2010.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Santini JT, Cima MJ, Langer R. A controlled-release microchip. Nature. 1999;6717:335–338. doi: 10.1038/16898. [DOI] [PubMed] [Google Scholar]
- 46.Kohane DS, Langer R. Biocompatibility and drug delivery systems. Chemical Science. 2010;4:441–446. [Google Scholar]
- 47.Shichor I, Shomron N, Lawlor MW, et al. Toxicogenomic analysis of a sustained release local anesthetic delivery system. Biomaterials. 2012;13:3586–3593. doi: 10.1016/j.biomaterials.2012.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Stern E, Klemic JF, Routenberg DA, et al. Label-free immunodetection with CMOS-compatible semiconducting nanowires. Nature. 2007;7127:519–522. doi: 10.1038/nature05498. [DOI] [PubMed] [Google Scholar]
- 49.Patolsky F, Timko BP, Yu G, et al. Detection, stimulation, and inhibition of neuronal signals with high-density nanowire transistor arrays. Science. 2006;5790:1100–1104. doi: 10.1126/science.1128640. [DOI] [PubMed] [Google Scholar]
- 50.Timko BP, Cohen-Karni T, Qing Q, et al. Design and Implementation of Functional Nanoelectronic Interfaces With Biomolecules, Cells, and Tissue Using Nanowire Device Arrays. Ieee Transactions on Nanotechnology. 2010;3:269–280. doi: 10.1109/TNANO.2009.2031807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tian B, Cohen-Karni T, Qing Q, et al. Three-dimensional, flexible nanoscale field-effect transistors as localized bioprobes. Science. 2010;5993:830–834. doi: 10.1126/science.1192033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Timko BP, Cohen-Karni T, Yu G, et al. Electrical recording from hearts with flexible nanowire device arrays. Nano Lett. 2009;2:914–918. doi: 10.1021/nl900096z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Qing Q, Pal SK, Tian B, et al. Nanowire transistor arrays for mapping neural circuits in acute brain slices. Proc Natl Acad Sci U S A. 2010;5:1882–1887. doi: 10.1073/pnas.0914737107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tian B, Kempa TJ, Lieber CM. Single nanowire photovoltaics. Chemical Society Reviews. 2009;1:16–24. doi: 10.1039/b718703n. [DOI] [PubMed] [Google Scholar]
- 55.Wang ZL. Piezoelectric nanostructures: From growth phenomena to electric nanogenerators. Mrs Bulletin. 2007;2:109–116. [Google Scholar]
- 56.Tian B, Liu J, Dvir T, et al. Macroporous nanowire nanoelectronic scaffolds for synthetic tissues. Nat Mater. 2012 doi: 10.1038/nmat3404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levi F, Schibler U. Circadian rhythms: mechanisms and therapeutic implications. Annu Rev Pharmacol Toxicol. 2007;47:593–628. doi: 10.1146/annurev.pharmtox.47.120505.105208. [DOI] [PubMed] [Google Scholar]