

Abstract
Piroxicam (PX), an anti-inflammatory drug, exhibits poor water solubility, dissolution and flow properties. Thus, the aim of the present study was to improve the solubility and dissolution rate of PX by freeze drying technique using dimethylformamide (DMF), chloroform and water as co-solvent system. The prepared crystals containing PX were evaluated for DMF and chloroform solvent residual by gas chromatography and solubility and in vitro dissolution. The prepared formulations were characterized by scanning electron microscopy, differential scanning calorimeter; X-ray diffraction and fourier transform infrared spectroscopy. Dissolution profile of the freeze dried crystals was compared with its recrystallized and pure samples. The samples were stored in stability chamber to investigate their physical stability. Solvent residual of DMF and chloroform was found to be within the toxic level. Freeze dried crystals exhibited decreased crystallinity and the solubility and dissolution of the PX crystals were significantly improved compared to its recrystallized and pure samples. In stability test, the release profile of the freeze dried crystals was almost unchanged as compared with the freshly prepared freeze dried crystals stored at 40°C and 75% relative humidity for 90 days. Hence, this technique can be used for formulation of PX tablets by direct compression with directly compressible tablet excipients.
Keywords: Piroxicam, Freeze drying, Compressibility, Solubility, Dissolution, Stability
INTRODUCTION
Piroxicam (PX) is a non-steroidal anti-inflammatory drug with low solubility and high permeability classified in class II of the Biopharmaceutical Drug Classification System(1). It is used as an analgesic drug and in acute or long-term treatment of osteoarthritis, rheumatoid arthritis and in a variety of other acute and chronic musculoskeletal disorders, such as dysmenorrhea(2).
After oral administration, PX is completely but slowly and gradually absorbed through the gastrointestinal tract and reaches the maximum haematic concentrations after 2-4 h(3). Since the drug is slightly soluble in biological fluids, PX dissolution rate turns out to be the absorption rate-limiting step and consequently it critically affects its analgesic effect onset. As a consequence, many strategies have been proposed with the aim of improving PX dissolution rate and to obtain formulations with fast analgesic effect onset, particularly useful in the treatment of dysmenorrhea, migraine, renal colic and postoperative pain(4–6).
Formulation and manufacture of solid oral dosage forms, and tablets in particular, have undergone rapid change and development over the last several decades. One of the most revolutionary technologies is that of direct compression. Direct compression is econo-mical, facilitates processing without the need of moisture, heat and involves small number of processing steps. In direct tabletting method, it is necessary to increase flowability and compressibility of the bulk powder in order to retain a steady supply of powder mixture to the tabletting machine and sufficient mechanical strength of the compacted tablets. In addition to increasing efficiency of the manufacturing process, it is also important to increase bioavailability of the drug by improving the solubility of the bulk drug powder. Thus, one of the major challenges to drug development today is poor solubility. As an estimation, 40% of all newly developed drugs are poorly soluble or insoluble in water(7). As a result, much research has been conducted into methods of improving drug solubility and dissolution rates to increase the oral bioavai-lability of hydrophobic drugs.
Currently, several solubilization techniques have been applied and reported to enhance the solubility of poorly water soluble drugs. Some literatures are available on enhancing the solubility and dissolution of PX like formation of microparticle by spray drying and spray chilling methods(8), novel lipid-based formu-lations enhancing the in vitro dissolution and permeability characteristics of PX(4), preparation of solid dispersions of PX in polyvinylpyrrolidone(9), enhancement of dis-solution rate of PX using liquisolid compacts(10), improved dissolution behaviour of steam-granulated PX(11), increasing the dissolution rate of PX using cyclodextrin(12), solid dispersions of PX with polyethylene glycol and polyvinyl pyrolidone K-30(13), orodis-persible PX tablets(14), solid dispersions with polyethylene glycol 4000(15), and phospho-lipid carrier and additives(16).
Lyophilization monophase solution tech-nique was developed as a suitable alternative procedure that could overcome the above men-tioned demerits of the conventional freeze drying. As previously reported, in this tech-nique, tertiary butyl alcohol (TBA), which is miscible with water in any proportion, was used as an organic co-solvent to solubilize the hydrophobic drug while the hydrophilic carrier was dissolved in water and then the mixed isotropic solution was lyophilized. TBA possesses a high vapor pressure, a high mel-ting point and has a low toxicity(17). Lyophilization monophase solution technique has been used for improvement of the physicochemical properties of an anticancer drug, flutamide(18).
The objective of the present study was to prepare freeze dried crystals of PX using freeze drying teqnique. Prepared crystals were evaluated for solvents residual. Differential scanning calorimeter (DSC), X-ray diffraction (XRD), fourier transform infrared spectro-scopy (FTIR) and scanning electron micro-scopy (SEM) analysis were performed to determine the physicochemical properties of the freeze dried crystals and its comparison with recrystallized sample and pure drug. The solubility and dissolution characteristics of the PX freeze dried crystals and their physical stability in a climate chamber at 40°C and 75% relative humidity (RH) were also determined after 90 days.
MATERIALS AND METHODS
PX was obtained as a gift sample from IPCA Lab. Mumbai, India. All chemicals and buffers used were of analytical grade.
Preparation of freeze dried crystals of piroxicam
PX (2.5 g) was dissolved in 25 ml of dimethylformamide (DMF) heated at 45°C until a clear solution was obtained. The drug solution was poured in to solvents consists of water (68 ml) and chloroform (7 ml) maintained at room temperature. Above resulted solution is a monophase solution which was then shifted to 100 ml glass bottle and then transferred to an ultra low freezer at -40°C and kept in the freezer for 24 h. The frozen drug solution was placed in a lyophilizer for 72 h using a freeze dryer (IISHIN Lab. Co. Ltd. Korea) with a conden-ser temperature of -40°C and a pressure of 7 × 10-2 mbar followed by a secondary drying at 25°C for 24 h. The resulted crystals were kept in a desiccators at room temperature until further experiment.
Recrystallization of piroxicam
PX (2.5 g) was dissolved in 25 ml DMF heated at 45°C until a clear solution was obtaimned. The drug solution was poured in to solvents consisting of water (68 ml) and chloroform (7 ml) which maintained at 20°C with occasional stirring. The crystals of PX were collected by filtration and were dried at 45°C for 12 h.
Determination of residual solvents in freeze dried crystals by gas chromatography (GC)
The GC studies were carried out on SHIMADZU model 2014 (Shimadzu Techno-logies, Japan) coupled with a split/split less injector, operated in a split-mode and FID. The computer with GC solutions software has been used to control the gas chromatograph. Rtx-5 capillary column (cross bond 5% diphenyl /95% dimethyl polysiloxane) with a length of 30 meters and an internal diameter of 0.25 mm was used throughout the study.
Differential scanning calorimetry (DSC)
The DSC study was carried out to detect possible polymorphic transition during the crystallization process. DSC measurements were performed on a DSC DuPont 9900, diffe-rential scanning calorimeter with a thermal analyzer.
Fourier transform infrared (FTIR) spectroscopy
The FTIR spectral measurements were taken at ambient temperature using a Shimadzu, Model 8033 (USA). About 2 mg of the pure drug, recrystallized and freeze dried crystals were used separately. Pure drug, freeze dried crystals and recrystallized samples were dispersed in KBr powder and the pellets were made by applying 6000 kg/cm2 pressure.
X-ray diffraction (XRD)
XRD patterns were used to detect possible polymorphic transition during the crystalli-zation process. XRD patterns were obtained at room temperature using a Philips X’ Pert MPD diffractometer, with Cu as anode material and graphite monochromator, operated at a voltage of 40 mA, 45 kV.
Scanning electron microscopy (SEM)
The SEM (Joel- LV-5600, USA, with magnification of 250x) photographs were obtained to identify and confirm spherical nature and morphological characters of the crystals.
Mechanical Properties
Tensile strength
Tensile strength of freeze dried crystals was determined by compressing 500 mg of crystals using hydraulic press at different kg/cm2 for 1 min. The compacts stored in desiccator for overnight to allow elastic recovery. The thick-ness and diameter were measured for each compact. The hardness of each compact was then measured using Pfizer hardness tester. The tensile strength (σ) of the compact (kg/cm2) was calculated using following equation:
σ = 2F/π Dt
where, F, D and t are hardness (kg/cm2), compact diameter (cm) and thickness (cm), respectively.
Heckel plot
Compressibility of granules was evaluated by the Heckel equation(1) in triplicate. Granules were lubricated with magnesium stearate and were compressed (average mass 500 mg ± 2%) at different pressures up to constant density of compacts using the 13 mm flat faced punch and die set on a hydraulic press (Spectra Lab, India). The range of different pressures applied to get constant density was 1000-5000 kg/cm2. The tablets were stored in airtight moisture-proof contain-ers for 24 h to enable elastic recovery and hardening. The compressibility behavior was studied using the Heckel equation:
ln [1/(1–D] = kP + A (1)
where, D is relative density and k and A are constants.
The slope of the straight line portion, K, is the reciprocal of the mean yield pressure, Py, of the material. From the intercept A, the relative density, DA, can be calculated using the following equation:
DA = 1 – e−A (2)
Relative density of the powder at the point when the applied pressure equals zero, D0, is used to describe the initial rearrangement phase of densification as a result of die filling.
Relative density, DB, describes the phase of rearrangement at low pressures and is the difference between DA and D0.
DB = DA – D0 (3)
Solubility studies of crystals
The solubility of PX freeze dried crystals in water was determined by taking excess quantity of freeze dried crystals and adding to screw-capped 50 ml glass vials filled with water. The vials were shaken for 24 h on mechanical shaker. The solution was filtered through Whatmann filter paper No.1 and the drug concentration was determined spectro-photometrically at 332 nm.
Dissolution studies of crystals
The dissolution of PX pure sample, freeze dried crystals and recrystallized sample was determined by using USP dissolution appara-tus XXIV-Type II (Electro Lab, Mumbai). Dissolution medium (900 ml) consisted of 7.2 phosphate buffer was used and 10 ml of dissolution medium was withdrawn at every 10 min interval for 1 h. The amount of dissolved drug was determined using UV spectrophotometric method (UV 1601 A Shimadzu, Japan) at 332 nm.
Determination of the physical stability
To determine the physical stability of freeze dried crystals was placed in a climate chamber of 40°C and 75% RH. After 90 days, the drug release percentage of PX in the freeze dried crystals was determined by dissolution study and compare with freshly prepared freeze dried crystals.
RESULTS
The GC results confirmed that there were 3.8 and 2.4 ppm residual of DMF and chloroform present in the freeze dried crystals respectively, which was much lower than the toxic level i.e. 880 and 60 ppm respectively (19,20).
The DSC thermograms showed a sharp endothermic peak for all the PX crystals. This one step melt might be due to only one crystal form (Triclinic) of the PX formed during the freeze drying process, thus indicating that PX did not undergo any crystal modification. The temperature range of the endothermic peak of all the PX crystals lies in the range of 203°C to 197°C (Fig. 1).
Fig. 1.
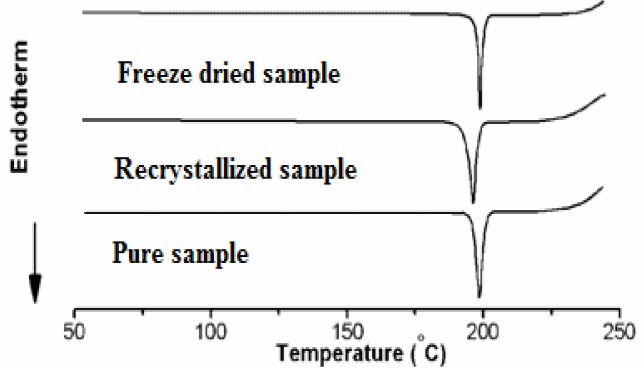
DSC thermograms of piroxicam samples
Infrared spectra of pure PX, recrystallized sample, freeze dried crystals showed charac-teristic peaks at -NH and -OH stretching which ties at 1385 cm-1, 1635 or 1625 cm-1 (N-H-CO3 stretching vibration), 1525 cm-1 (secondary -NH2 stretching), 1440 cm-1 (CH3 AND Ar-c=c stretching), 1355 cm-1 (sym. -CH3) and 1155 and 1070 cm-1 or 1050-1070 cm-1 (-SO2-N-) 770 and 740 or 740 cm-1 (Ortho-disubstituted phenyl) and showed in Fig. 2.
Fig. 2.
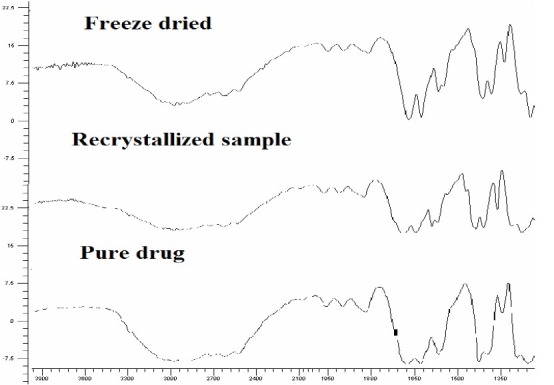
FTIR spectra of piroxicam samples
The XRD was used to analyze potential changes in the inner structure of PX crystal during the formulation of freeze dried crystals. The characteristic peak of the PX appeared in the 2θ range of 10-50°, indicating that the unprocessed PX was a crystalline material. All the samples showed similar peak positions (2θ) in XRD, formation of different polymorphs of PX was ruled out. However relative intensities of XRD peaks were modified (Fig. 3).
Fig. 3.
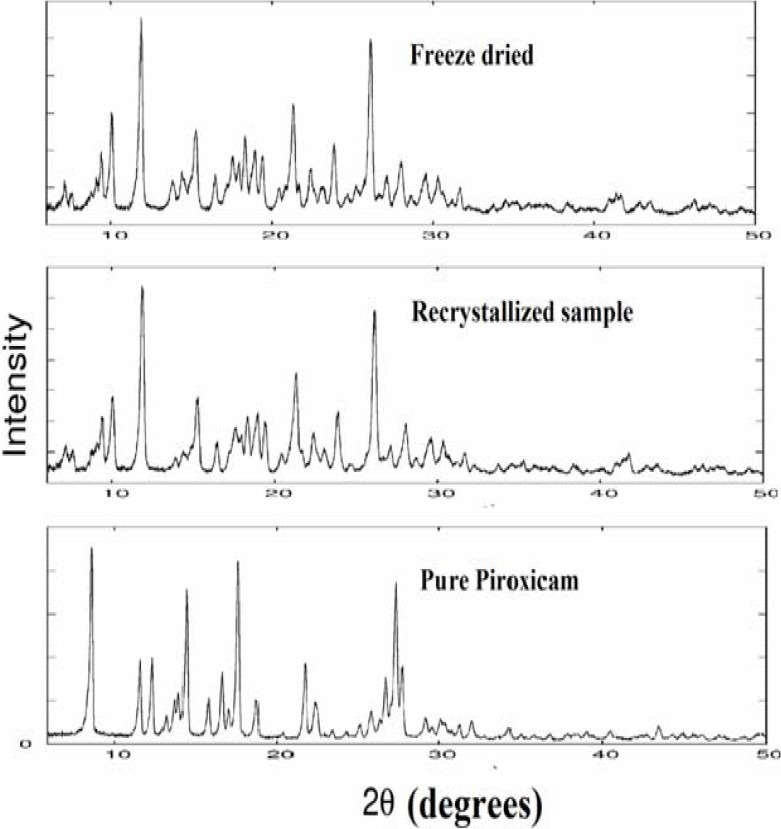
X-ray diffraction spectra of piroxicam samples
The SEM study showed that crystals of pure sample are of the smallest size (5-10 μm) and they have irregular shapes. Recrysta-llization crystals with intermediate size (7-18 μm) which had rod like shapes. The freeze dried crystals were formed by microcrystalline precipitates, so the resultant freeze dried crys tals had a smooth surface with small in size (0.70-0.85 μm) (Fig. 4).
Fig. 4.

SEM of different samples of piroxicam
The freeze dried crystals exhibited superior compressibility characteristics compared to conventional drug crystals (Fig. 5).
Fig. 5.
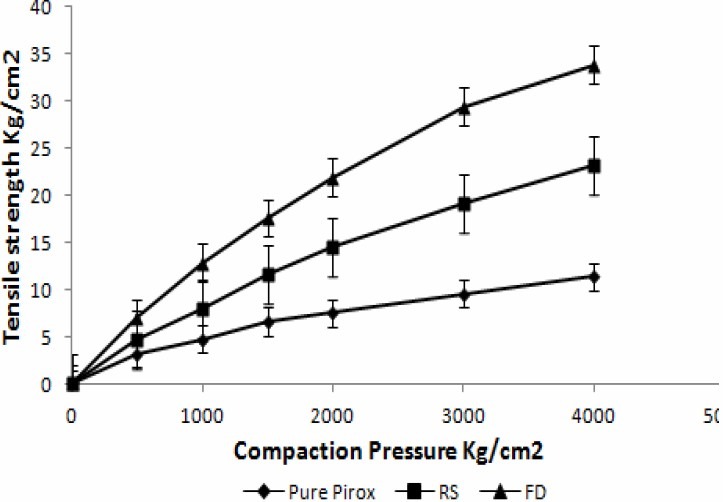
Tensile strength of pure piroxicam, recrystallized sample and freeze dried crystals
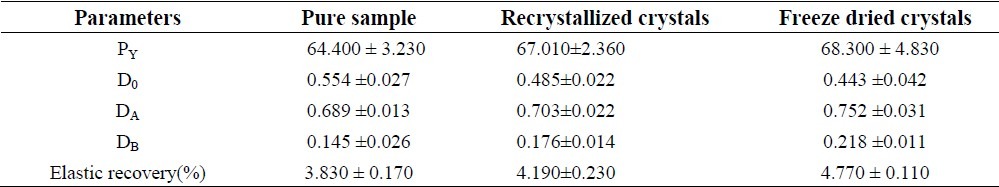
Heckle profile of pure sample and prepared crystals are shown in Fig. 6, and characteristic values of PY, DA, D0, and DB and elastic recovery are reported in Table 1 and Fig. 6.
Fig. 6.
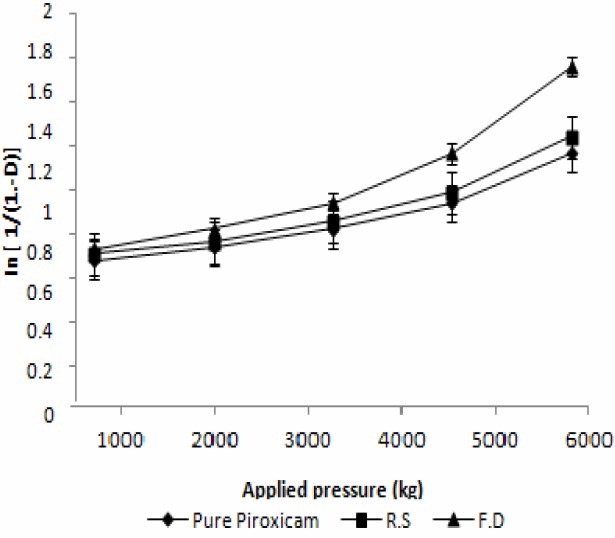
Heckel plot of Pure piroxicam, Recrystallized sample and Freeze dried crystals
Table 1.
Heckle's parameters and elastic recovery of piroxicam crystals
The freeze dried crystals showed increased solubility than the pure sample in water and increased nearly more than four fold higher (0.0378 mg/ml) than pure PX (0.0084 mg/ml). The dissolution profiles of PX (Fig. 7) exhibited improved dissolution behavior for freeze dried crystals than pure sample.
Fig. 7.
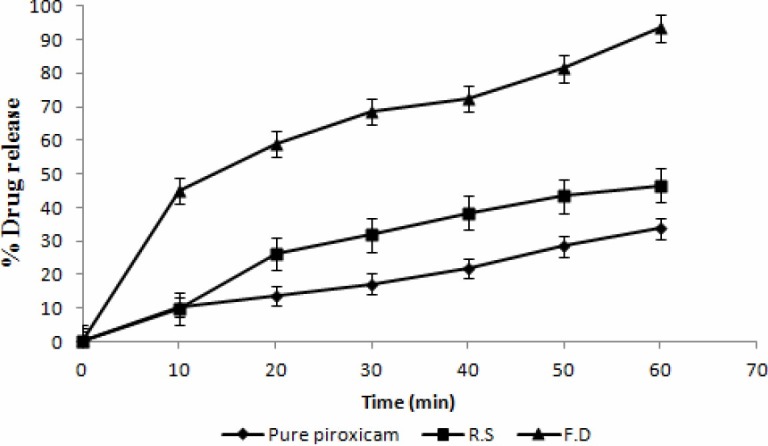
Dissolution of pure piroxicam, ecrystallized sample and freeze dried crystals
The influence of freeze dried crystals on the physical stability of PX was investigated. The drug release percentage of freeze dried crystals was almost the same i.e. (92.82%) after 90 days of storing when compared with the freshly prepared freeze dried crystals i.e. (93.42 %).
DISCUSSION
A solvent system composed of DMF, chloroform and water was used to prepare freeze dried PX crystals.. The selection of these solvent depends on the miscibility of the solvents and solubility of the drug in individual solvents. DMF is miscible in any proportion with water and chloroform(21).
Recrystallization of PX was done to find out the changes in crystal lattice, being induced by solvents which can influence the physico-chemical properties of the substance. Hence, the mechanical and dissolution properties of freeze dried crystals were compared with those of pure sample and recrystallized sample. Recrystallization of PX was carried out using same solvent composition as used for freeze drying(8).
Based upon high solubility of PX in DMF (1 g/10 ml), high viscosity of DMF and crystal morphology, DMF determined to be suitable freeze drying medium for PX. Controlling of residual DMF was needed though. DMF is a toxic organic solvent based on its concentration and has little detriment to human body. Therefore, the low level of both DMF and chloroform in the freeze dried crystals should not be harmful to both animal and human(19,20).
The low level of both DMF and chloroform in the freeze dried crystals results from their ability to form high surface area crystals and from the fact that the intermolecular forces among both DMF and chloroform molecules are not as strong as those of water. This allows both DMF and chloroform to sublime more completely and easily than water.
In the DSC curve, pure PX had a sharp endothermic peak at 203°C with enthalpy of 174.32 J/g that corresponded to the melting point of PX. Melting points show slight variation as the nature of the crystals might have been affected by the solvent. The melting endotherm for freeze dried PX was 197.09°C with decreased enthalpy of (155.43 J/g) indicating decreased crystallinity of PX in freeze dried crystals(1,2).
Specific changes in IR spectra are not very clear whichcould be due to variations in the resonance structure, rotation of a part of a molecule or certain bonds. Alteration could be due to minor distortion of bond angles, or even a result of the crystallization solvents.
The relative intensities of freeze dried crystals reduced two times than pure PX. This could be attributed to the markedly different crystal habits of the samples. Therefore, the relative abundance of the planes exposed to the X-ray source would have been altered, producing the variations in the relative intensities of the peak or may be due to differences in crystal sizes(5,6). The pure drug exhibits its characteristic diffraction peaks at various diffraction angles indicating the presence of crystallinity. The XRD of the drug recrystalized samples showed the peak corresponding to the crystalline drug molecules present in the RS, although their intensity was lower than pure drug due to the differences in crystal sizes. The XRD pattern of the freeze dried crystals showed that PX peak intensity was much lower than the pure drug and recrystalized samples samples of PX. This could be due to the increasing wettability of the freeze dried crystals. These results could explain the observed enhancement of PX solubility and dissolution in freeze dried crystals.
During the process of compression fresh surfaces are formed by fracturing crystals. Surface freshly prepared by fracture enhanced the plastic inter particle bonding, resulting in a lower compression force required for compressing the freeze dried crystals under plastic deformation compared to that of single crystal(8). PX crystals prepared by freeze drying show higher tensile strength compared to recrystallized sample and pure drug sample. Hence, crystals prepared by freeze drying are suitable for tabletting.The compression of prepared crystals begins at lower relative density, while the initial rearrangement phase without pressure increases for pure sample. This corresponds to different D0 values. DB is greater for prepared crystals indicating a greater brittle fracture tendency of these materials(22). Elastic recovery is relatively high for a brittle material, but it must be noted that tablets survive the decompression phase and show no sign of capping. Prepared PX crystals exhibited higher porosity compared to pure sample, hence require lower compression force for compressing under plastic deformation compared to commercial sample.
The higher solubility of PX from freeze dried crystals may be due to the reduction in particle size and increased wettability of PX in freeze dried crystals(8). The reason for this faster dissolution could be linked to the better wettability and reduction in particle size of the freeze dried crystals. The amount of drug dissolved in 60 min greatly varied for freeze dried crystals.
Stability result shows that freeze dried crystals of PX was stable after 90 days at 40°C and 75% RH.
CONCLUSION
Freeze dried crystals of PX were prepared by freeze drying technique. Freeze dried crystals exhibited decreased crystallinity and improved mechanical properties. DSC, FTIR and XRD studies showed that there is no change in the crystal structure of PX during the crystallization process i.e., polymorphism has not occurred. The dissolution of the freeze dried crystals was improved compared with recrystallized sample and pure PX sample. Hence this technique can be used for formulation of tablets of PX by direct compression with directly compressible tablet excipients.
ACKNOWLEDGMENT
The authors are thankful to IPCA Labs, Mumbai, India for the gift sample of PX, Dr. H.G. Shivakumar, Principal, J.S.S.College of Pharmacy, Mysore for providing facilities to carry out this work.
REFERENCES
- 1.Amidon GL, Lennernas H, Shah VP, Crison JR. A theoretical basic for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vitro bioavailability. Pharm Res. 1995;12:413–420. doi: 10.1023/a:1016212804288. [DOI] [PubMed] [Google Scholar]
- 2.Gennaro AR. Remington's Pharmaceutical Sciences. Easton: Mack Publishing Company; 1990. p. 1116. [Google Scholar]
- 3.Tagliati CA, Kimura E, Nothenberg MS, Santos JC, Oga S. Pharmacokinetic profile and adverse gastric effect of zinc-piroxicam in rats. Gen Pharmacol. 1999;33:67–71. doi: 10.1016/s0306-3623(98)00267-5. [DOI] [PubMed] [Google Scholar]
- 4.Prabhu S, Ortega M, Ma C. Novel lipid-based formulations enhancing the in vitro dissolution and permeability characteristics of a poorly water-soluble model drug, piroxicam. Int J Pharm. 2005;301:209–216. doi: 10.1016/j.ijpharm.2005.05.032. [DOI] [PubMed] [Google Scholar]
- 5.Karatas A, Yuksel N, Baykara T. Improved solubility and dissolution rate of piroxicam using gelucire 44/14 and labrasol. Farmaco. 2005;60:777–782. doi: 10.1016/j.farmac.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 6.Yuksel N, Karatas A, Ozkan Y, Savaser A, Ozkan SA, Baykara T. Enhanced bioavailability of piroxicam using Gelucire 44/14 and Labrasol: in vitro and in vivo evaluation. Eur J Pharm Biopharm. 2003;56:453–459. doi: 10.1016/s0939-6411(03)00142-5. [DOI] [PubMed] [Google Scholar]
- 7.Naseem A, Olliff CJ, Martini LG, Lioyd AW. Effects of plasma irradiation on the wettability and dissolution of compacts of griseofulvin. Int J Pharm. 2004;269:443–450. doi: 10.1016/j.ijpharm.2003.09.029. [DOI] [PubMed] [Google Scholar]
- 8.Dixit M, Kini AG, Kulkarni PK. Preparation and characterization of microparticles of piroxicam by spray drying and spray chilling methods. Res Pharma Sci. 2010;5:89–97. [PMC free article] [PubMed] [Google Scholar]
- 9.Tantishaiyakul V, Kaewnopparat N, Ingkata-wornwong S. Properties of solid dispersions of piroxicam in polyvinylpyrrolidone. Int J Pharm. 1999;181:143–151. doi: 10.1016/s0378-5173(99)00070-8. [DOI] [PubMed] [Google Scholar]
- 10.Javadzadeh Y, Siahi-Shadbad MR, Barzegar-Jalali M, Nokhodchi A. Enhancement of dissolution rate of piroxicam using liquisolid compacts. Farmaco. 2005;60:361–365. doi: 10.1016/j.farmac.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 11.Cavallari C, Albertini B, González-Rodríguez ML, Rodriguez L. Improved dissolution behaviour of steam-granulated piroxicam. Eur J Pharm Biopharm. 2002;54:65–73. doi: 10.1016/s0939-6411(02)00021-8. [DOI] [PubMed] [Google Scholar]
- 12.Nagabhushanam MV. Formulation studies on cyclodextrin complexes of piroxicam. Rasayan J Chem. 2010;3:314–320. [Google Scholar]
- 13.Patel DM, Shah RR, Jogani PD. Studies to enhance dissolution of piroxicam. Ind J Pharm Sci. 2003;65:264–267. [Google Scholar]
- 14.Ravi KN, Palanichamy S, Rajesh M, Godwin RT, Anusha V, Parasakthi N, et al. Formulation and evaluation of orodispersible piroxicam tablets. J Pharm Sci Res. 2010;2:615–621. [Google Scholar]
- 15.Fernández M, Margarit MV, Rodríguez IC, Cerezo A. Dissolution kinetics of piroxicam in solid dispersions with polyethylene glycol 4000. 1993;98:29–35. [Google Scholar]
- 16.Sabiruddin M, Inna M, Muhammad JH, James FB, Muhammad DH. Enhanced dissolution and oral bioavailability of piroxicam formulations: modulating effect of phospholipids. Pharmaceutics. 2010;2:339–350. doi: 10.3390/pharmaceutics2040339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Teagarden DL, Baker DS. Practical aspects of lyophilization using nonaqueous co-solvent systems. Eur J Pharm Sci. 2002;15:115–133. doi: 10.1016/s0928-0987(01)00221-4. [DOI] [PubMed] [Google Scholar]
- 18.Nazik E, Kadria E, Abdallah M, Ahmed E. Lyophilization monophase solution technique for improvement of the physicochemical properties of an anticancer drug, flutamide. Eur J Pharm Biopharm. 2010;74:397–405. doi: 10.1016/j.ejpb.2009.11.011. [DOI] [PubMed] [Google Scholar]
- 19.Gescher A. Metabolism of N,N-dimethylformamide: key to the understanding of its toxicity. Chem Res Toxicol. 1993;6:245–251. doi: 10.1021/tx00033a001. [DOI] [PubMed] [Google Scholar]
- 20.Heywood R, Sortwell RJ, Noel PRB, Street AE, Prentice DE, Roe FJC, et al. Safety evaluation of toothpaste containing chloroform. III. Long-term study in beagle dogs. J Environ Pathol Toxicol. 1979;2:885–851. [PubMed] [Google Scholar]
- 21.Dixit M, Kulkarni PK, Selvam RP, Kini AG, Shivakumar HG. Preparation and characterization of spherical agglomerates of piroxicam. Lat Am J Pharm. 2011;30:188–199. [Google Scholar]
- 22.Vander VMK, Bolhuis GK. Improving properties of materials for direct compression. Pharm Technol Europe. 1998;10:28–36. [Google Scholar]