

SUMMARY
Growth of new dendritic spines contributes to experience-dependent circuit plasticity in the cerebral cortex. Yet the signaling mechanisms leading to new spine outgrowth remain poorly defined. Increasing evidence supports that the proteasome is an important mediator of activity-dependent neuronal signaling. We therefore tested the role of the proteasome in activity-dependent spinogenesis. Using pharmacological manipulations, glutamate uncaging, and two-photon imaging of GFP-transfected hippocampal pyramidal neurons, we demonstrate that acute inhibition of the proteasome blocks activity-induced spine outgrowth. Remarkably, mutation of serine 120 to alanine of the Rpt6 protea-somal subunit in individual neurons was sufficient to block activity-induced spine outgrowth. Signaling through NMDA receptors and CaMKII, but not PKA, is required to facilitate spine outgrowth. Moreover, abrogating CaMKII binding to the NMDA receptor abolished activity-induced spinogenesis. Our data support a model in which neural activity facilitates spine outgrowth via an NMDA receptor- and CaM-KII-dependent increase in local proteasomal degradation.
INTRODUCTION
Rapid and stable modification of neural circuits is thought to underlie learning and memory. The signaling pathways that mediate this circuit plasticity are thought to drive both functional and structural changes in existing synapses, as well as the addition of new synapses. In the mammalian cerebral cortex, the addition of new synapses during experience-dependent plasticity has been associated with the addition of dendritic spines (Comery et al., 1995; Knott et al., 2002; Trachtenberg et al., 2002). Moreover, the appearance of new persistent spines has been associated with novel sensory experience and learning new tasks (Hofer et al., 2009; Holtmaat et al., 2006; Roberts et al., 2010; Xu et al., 2009; Yang et al., 2009). While new dendritic spines tend to be short lived (Trachtenberg et al., 2002), those that stabilize are capable of rapid functional maturation (Zito et al., 2009). These data support that the formation and stabilization of new dendritic spines is a key structural component underlying synaptic plasticity.
Although the detailed signaling mechanisms that initiate the outgrowth of new dendritic spines during experience-dependent plasticity remain poorly defined, there is strong evidence that increased neural activity can enhance new spine growth (Engert and Bonhoeffer, 1999; Kwon and Sabatini, 2011; Maletic-Savatic et al., 1999; Papa and Segal, 1996). Multiple studies demonstrate that activity-induced spine outgrowth is dependent on NMDA receptor signaling (Engert and Bonhoeffer, 1999; Kwon and Sabatini, 2011; Maletic-Savatic et al., 1999). What further signaling mechanisms act downstream of activity to initiate new spine growth? Over the past decade, evidence has been rapidly accumulating that the proteasome is an important mediator of activity-induced neuronal signaling (Bingol and Sheng, 2011; Tai and Schuman, 2008). Neural activity regulates proteasomal activity (Bingol and Schuman, 2006; Djakovic et al., 2009), resulting in alterations in the abundance of synaptic proteins (Ehlers, 2003). Moreover, activity-dependent changes in the rate of proteasome-mediated degradation can occur rapidly and can be localized to a small region of the stimulated dendrite (Bingol and Schuman, 2006; Djakovic et al., 2009). These data suggest that the proteasome is a key downstream mediator of localized activity-dependent neuronal signaling and therefore may play a role in activity-dependent spinogenesis.
In this study, we used pharmacological and genetic manipulations in combination with time-lapse two-photon microscopy and two-photon glutamate uncaging to investigate the role of the proteasome in new spine growth on dendrites of hippo-campal pyramidal neurons. We show that acute inhibition of the proteasome rapidly reduces the rate of spine outgrowth. Synaptic activity, NMDA receptors, and CaMKII, but not PKA, are upstream regulators of proteasome-mediated spine outgrowth, which is dependent upon interaction between CaMKII and the GluN2B subunit of the NMDA receptor. The S120 residue of the Rpt6 proteasomal subunit is critical for proteasome-dependent spine outgrowth in individual neurons, indicating that the proteasome acts postsynaptically and in a cell-autonomous manner to regulate spine outgrowth. Our data support a model in which synaptic activity promotes spine outgrowth via an NMDA receptor- and CaMKII-mediated regulation of local proteasomal degradation.
RESULTS
Proteasome Inhibition Acutely Reduces New Spine Outgrowth
To determine whether the proteasome plays a role in regulating the growth of new dendritic spines, we used pharmacological manipulations and time-lapse two-photon microscopy to measure the effect of acute inhibition of the proteasome on the rate of spine outgrowth. Hippocampal pyramidal neurons in or-ganotypic slice cultures were transfected with enhanced green fluorescent protein (EGFP) and imaged using a two-photon microscope. Dendrites of EGFP-expressing CA1 neurons were imaged at 15 or 20 min intervals before and after treatment with drugs or vehicle (Figure 1A).
Figure 1. Acute Inhibition of the Protea-some Rapidly Decreases New Spine Growth.
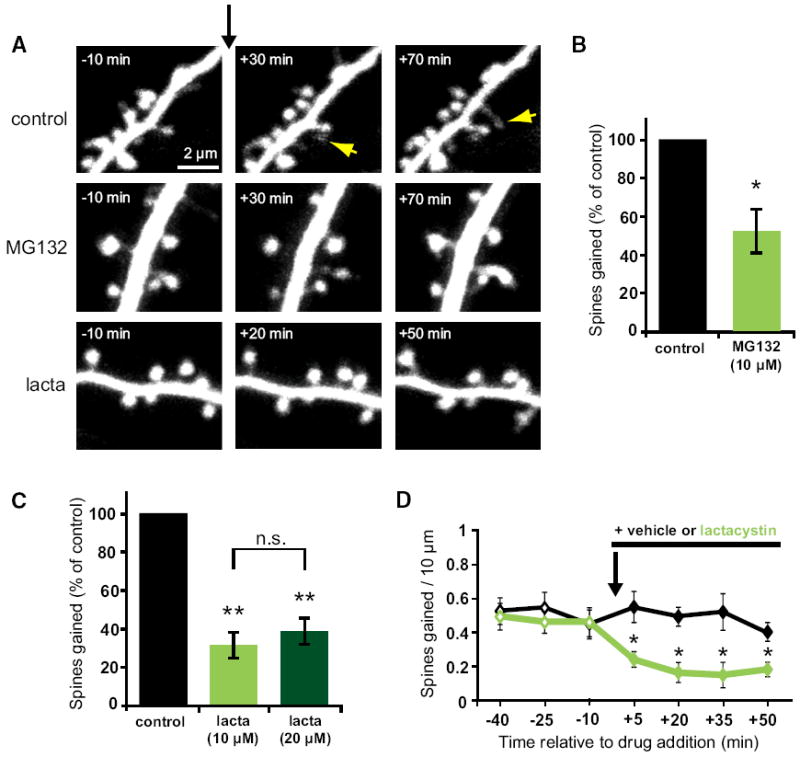
(A) Images of dendrites from EGFP-expressing neurons at 7–12 DIV before and after the addition of vehicle, MG132, or lactacystin at t = 0 (black arrow). Yellow arrows indicate new spines. (B) MG132 (10 μM) decreased the rate of spine outgrowth (green bar; 34 spines, 5 cells) as compared to vehicle control (black bar; 68 spines, 6 cells; p < 0.05). (C) Lactacystin (10 μM) decreased the rate of spine outgrowth (light green bar; 33 spines, 6 cells) as compared to vehicle control (black bar; 131 spines, 8 cells; p < 0.01). Doubling the concentration of lactacystin (20 μM) inhibited spine outgrowth to a comparable extent (dark green bar; 25 spines, 6 cells; p < 0.001). (D) New spine growth was significantly reduced within 5 min after addition of lactacystin (10 μM) and remained significantly reduced for all subsequent time points (p < 0.05). Error bars represent SEM.
Treatment with the proteasome inhibitor MG132 (10 μM) reduced the rate of spine outgrowth to half (52% ± 12%) that of vehicle-treated control cells (100% ± 13%; p < 0.05; Figure 1B). Because MG132 inhibits the activity of calpains as well as the proteasome, we confirmed our findings with the proteasome-specific inhibitor lactacystin. Treatment with lactacystin (10 μM) reduced the rate of spine outgrowth by 68% (32% ± 7%) as compared to vehicle control (100% ± 13%; p < 0.001; Figure 1C). Similar reductions in spine out-growth were observed for both apical and basal dendrites (see Figure S1A available online). The reduction in spine outgrowth due to lactacystin was not significantly different than that due to MG132 (p = 0.2), suggesting that reduced spine outgrowth in the presence of MG132 is specifically due to inhibition of the proteasome. To ensure that the effect of proteasome inhibition was saturated, we doubled the concentration of lactacystin in the bath. Treatment with 20 μM lactacystin resulted in a 62% reduction in new spine growth (38% ± 7%), which was not significantly different than that observed in the presence of 10 μM lactacystin (p = 0.6; Figure 1C), suggesting that spine outgrowth observed in the presence of lactacystin is proteasome independent.
A variety of cellular processes, including the endocytosis of transmembrane proteins, are dependent on proteolysis-independent ubiquitination (Acconcia et al., 2009; Hicke, 2001). It is conceivable that a drop in free ubiquitin levels caused by proteasome inhibition (Schubert et al., 2000) could interfere with new spine growth via a secondary effect on endocytosis. We think that this is unlikely for two reasons. First, the reduction in new spine growth in response to proteasomal inhibition was very rapid; we observed a significant reduction in spine outgrowth within 5 min of drug application (p < 0.05; Figure 1D). Second, a reduction in endocytosis might be expected to cause an increase in spine volume or density, as spine volume and stability are tightly linked to glutamate receptor content (Hsieh et al., 2006; Matsuzaki et al., 2004). Within the time course of our experiments, we saw no change in spine volume or spine density in response to MG132 treatment (data not shown). The lack of change in spine density might appear inconsistent with the significantly decreased rate of spine addition in response to MG132; however, because most new spines are transient, reduced new spine outgrowth is expected to be accompanied by reduced spine loss, which we observed (Figure S1B). Our data suggest that the reduction in new spine growth in response to proteasome inhibitors is due to acute inhibition of proteasomal activity.
Synaptic Activity Promotes New Spine Growth in a Proteasome-Dependent Manner
Because synaptic activity can enhance both spine outgrowth (Engert and Bonhoeffer, 1999; Kwon and Sabatini, 2011) and the activity of the proteasome (Bingol and Schuman, 2006; Djakovic et al., 2009), we next examined whether the protea-some plays a role in regulating activity-induced spine outgrowth (Figure 2). Treatment with bicuculline (30 μM), which strongly enhanced synaptic activity (Figure S2), resulted in a 69% increase in spine outgrowth (169% ± 16%) relative to vehicle-treated controls (100% ± 13%; p < 0.05; Figures 2A and 2B). The activity-induced increase in spine outgrowth was blocked by concurrent application of MG132 (10 μM), which instead caused a 34% decrease in spine outgrowth (66% ± 9%; p < 0.05; Figure 2B), an effect that was indistinguishable from treatment with MG132 alone (p = 0.4). Thus, we conclude that proteasomal degradation is necessary for activity-induced spine outgrowth.
Figure 2. Synaptic Activity Promotes New Spine Growth in a Proteasome- and NMDA Receptor-Dependent Manner.
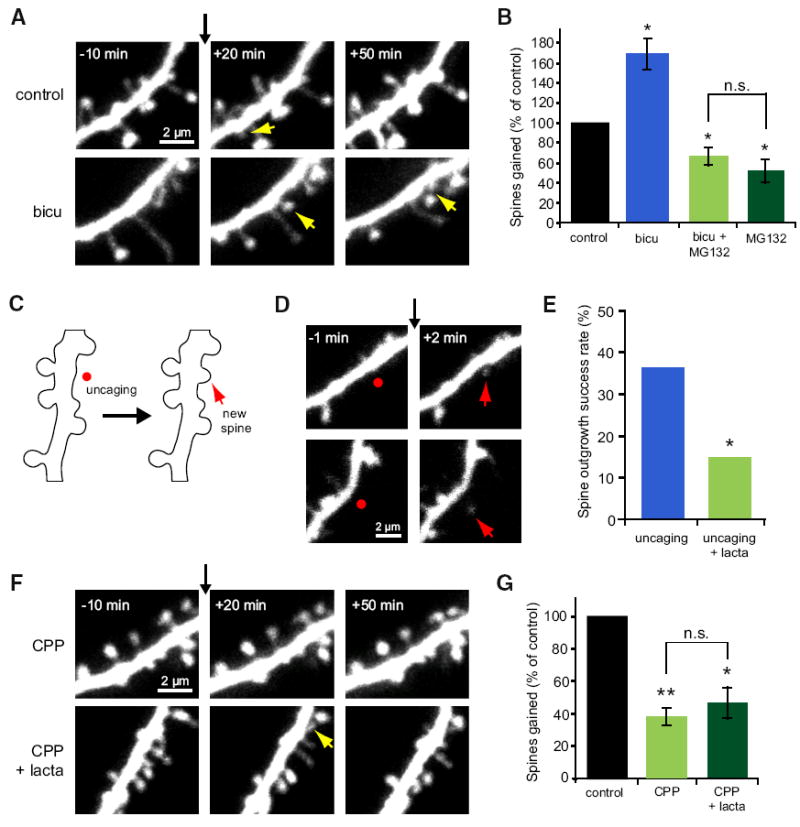
(A) Images of dendrites from EGFP-expressing neurons at 5–10 DIV before and after the addition of vehicle or bicuculline (30 μM) at t = 0 (black arrow). Yellow arrows indicate new spines. (B) Elevated neural activity in response to bicuculline increased the rate of new spine addition (blue bar; 116 spines, 6 cells) as compared to vehicle control (black bar; 68 spines, 6 cells; p < 0.01). Inhibiting the proteasome with MG132 blocked the bicu-culline-induced increase in spine outgrowth and decreased baseline new spine growth (light green bar; 46 spines, 7 cells; p < 0.05) to a level similar to that in the presence of MG132 alone (dark green bar; data from Figure 1; p = 0.4). (C) Schematic of uncaging-induced spine outgrowth. A new spine (red arrow) was induced by focal photolysis of MNI-caged glutamate (red dot) adjacent to a section of dendrite devoid of spines. (D) Images of dendrites from EGFP-expressing neurons at 7–8 DIV before and after focal photolysis of MNI-glutamate (red dots) at t = 0 (black arrow). Red arrows indicate new spines. (E) Uncaging-induced spine outgrowth (blue bar; 16 successes out of 44 trials on 15 cells) was significantly reduced in the presence of lactacystin (10 μM; green bar; 6 successes out of 40 trials on 12 cells; p < 0.05). (F) Images of dendrites from EGFP-expressing neurons at 7–11 DIV before and after the addition of CPP (30 μM) or CPP + lactacystin (10 μM) at t = 0 (black arrow). Yellow arrows indicate new spines. (G) The rate of new spine addition decreased in the presence of CPP (light green bar; 39 spines, 8 cells) as compared to vehicle control (black bar; 105 spines, 8 cells; p < 0.001). No further decrease in spine outgrowth was observed with simultaneous blockade of the proteasome with lactacystin (dark green bar; 45 spines, 7 cells; p = 0.4). Error bars represent SEM.
Because bicuculline alters global neural activity levels in our slice cultures, we chose also to use a more localized dendritic stimulus to examine the role of the proteasome in activity-dependent spine outgrowth. A recent study using focal photolysis of caged glutamate demonstrated that direct glutamatergic stimulation of the dendrite can result in rapid spine outgrowth (Kwon and Sabatini, 2011). Using two-photon uncaging of MNI-glutamate and pharmacological inhibition, we examined whether proteasomal degradation is necessary for glutamate uncaging-induced spine outgrowth (Figures 2C and 2D). We found that uncaging-induced spine outgrowth was significantly reduced in the presence of lactacystin (15% success rate) as compared to control (36% success rate; p < 0.05; Figure 2E). This local activity-dependent spine outgrowth occurred with high spatial specificity such that most new spines (>87%) grew within 1 μm of the uncaging location. These results further demonstrate that proteasomal degradation acts to facilitate activity-dependent spine outgrowth in a spatially precise manner.
Signaling through the NMDA Receptor Is Required for Proteasome-Dependent Dendritic Spine Outgrowth
Stimulation of proteasomal degradation by neural activity has been shown to occur via the action of NMDA receptors (Bingol and Schuman, 2006; Djakovic et al., 2009), which also have been shown to play a role in activity-induced outgrowth of new spines (Engert and Bonhoeffer, 1999; Kwon and Sabatini, 2011). Indeed, NMDA treatment increased the degradation of a fluorescent proteasome substrate in dendrites of CA1 pyramidal neurons in our cultured hippocampal slices (Figure S3). We therefore investigated the role of the NMDA receptor in pro-teasome-dependent new spine growth (Figures 2F and 2G).
Inhibition of NMDA receptors with CPP (30 μM) resulted in a 62% reduction in spine outgrowth (38% ± 5%) as compared to vehicle control (100% ± 9%; p < 0.001; Figure 2G), suggesting that baseline spine outgrowth is partially activity dependent. The reduction in spine outgrowth observed after CPP treatment was not different than that after lactacystin treatment (p = 0.3), suggesting that the NMDA receptor and the proteasome act in the same pathway to induce new spine growth. To test this idea, we simultaneously applied lactacystin and CPP. Simultaneous treatment with both lactacystin and CPP resulted in a 53% reduction in new spine growth (47% ± 9%) as compared to vehicle control (100% ± 8%; p < 0.05; Figure 2G), a decrease that was not different than that observed in response to CPP (p = 0.7) or lactacystin (p = 0.3; Figure 1C) alone. Our data demonstrate that the NMDA receptor and the proteasome act in the same pathway to promote new spine growth in response to neural activity.
Mutation of Serine 120 to Alanine in the Rpt6 Proteasomal Subunit Blocks Activity-Induced New Spine Growth
Which signaling mechanisms act downstream of the NMDA receptor and the proteasome to translate neural activity into enhanced spine growth? Recent work has identified serine 120 of the Rpt6 proteasomal subunit as an important target site for CaMKII phosphorylation (Bingol et al., 2010; Djakovic et al., 2012). To test whether the S120 residue of Rpt6 is necessary for activity-dependent spine outgrowth, we used time-lapse imaging to monitor new spine growth on dendrites of neurons transfected with EGFP alone or with both EGFP and hemagglutinin (HA)-tagged Rpt6-WT or Rpt6-S120A (Figure 3A). Expression levels were equivalent for both of the HA-tagged Rpt6 constructs, as verified through retrospective immunostaining (data not shown).
Figure 3. Mutation of Serine 120 to Alanine of the Rpt6 Proteasomal Subunit Blocks Activity-Induced New Spine Growth.

(A) Images of dendrites from hippocampal neurons transfected with EGFP, EGFP + Rpt6-WT, or EGFP + Rpt6-S120A at 9–10 DIV before and after the addition of vehicle or bicuculline at t = 0 (black arrow). Yellow arrows indicate new spines. (B) Transfection with Rpt6-WT (gray bar; 121 spines, 7 cells; p < 0.05) did not interfere with the bicuculline-induced increase in spine outgrowth (blue bar; 149 spines, 8 cells; p < 0.05) relative to vehicle-treated controls (black bar; 73 spines, 7 cells). In contrast, Rpt6-S120A transfection reduced spine outgrowth in bicuculline-treated cells (green bar; 56 spines, 7 cells; p < 0.001) relative to vehicle-treated controls. (C) Transfection with Rpt6-S120A decreased the rate of spine outgrowth (gray bar; 32 spines, 6 cells) as compared to cells transfected with EGFP alone (black bar; 74 spines, 6 cells; p < 0.01). Treatment of Rpt6-S120A transfected neurons with lactacystin (light green bar; 33 spines, 6 cells, p = 0.48) or with CPP (dark green bar; 44 spines, 7 cells, p = 0.24) did not further reduce rates of spine outgrowth over that observed for untreated S120A transfected cells. Error bars represent SEM.
As expected, we found that spine outgrowth increased in the presence of bicuculline for neurons transfected with EGFP alone (166% ± 13%, p < 0.05) or with EGFP and Rpt6-WT (161% ± 19%, p < 0.05) as compared to vehicle control (100% ± 11%; Figure 3B). Remarkably, neurons transfected with EGFP and Rpt6-S120A did not show a bicuculline-dependent increase in new spine growth and instead showed a 44% decrease in spine outgrowth (56% ± 8%; p < 0.05; Figure 3B), a level indistinguishable from the 55% decrease in outgrowth observed in vehicle-treated neurons expressing Rpt6-S120A (45% ± 5%; p = 0.4; Figure 3C). Treatment of Rpt6-S120A transfected cells with lactacystin did not further depress spine outgrowth (50% ± 5%; Figure 3C) compared to vehicle-treated Rpt6-S120A cells (p = 0.5), suggesting that the Rpt6-S120 residue is necessary for all proteasome-mediated spine outgrowth. Furthermore, treatment of Rpt6-S120A transfected cells with 30 μM CPP did not result in a further decrease in spine outgrowth (54% ± 6%; Figure 3C) compared to untreated Rpt6-S120A controls (p = 0.2), suggesting that the S120A residue of Rpt6 is a key regulatory site for proteasome- and NMDA receptor-dependent facilitation of new spine growth.
Inhibition of CaMKII, but Not PKA, Decreases the Rate of New Spine Growth
Recent work has demonstrated that both PKA (Zhang et al., 2007) and CaMKIIα (Djakovic et al., 2009) are capable of phos-phorylating the Rpt6 subunit of the proteasome at serine 120. In addition, CaMKIIα has been shown to mediate activity-dependent recruitment of proteasomes to dendritic spines (Bingol et al., 2010), and phosphorylation of Rpt6 by CaMKIIα increases the rate of proteasomal degradation (Djakovic et al., 2012). Finally, both CaMKII (Jourdain et al., 2003) and PKA (Kwon and Sabatini, 2011) have been implicated in activity-dependent spine outgrowth. We therefore chose to investigate the roles of CaMKII and PKA in activity- and proteasome-dependent new spine growth (Figures 4A and 4B).
Figure 4. CaMKII Activity and Its Interaction with the NMDA Receptor Subunit GluN2B Are Necessary for Activity-Dependent Spine Outgrowth.

(A) Images of dendrites from EGFP-expressing hippocampal neurons at 7–10 DIV before and after the addition of myristoylated PKI 14–22 (20 μM), Rp-cAMPS (5 μM), or KN-93 (30 μM) at t = 0 (black arrow). Yellow arrows indicate new spines. (B) Inhibition of PKA with PKI 14–22 (dark gray bar; 58 spines, 7 cells; p = 0.9) or Rp-cAMPS (light gray bar; 66 spines, 7 cells; p = 0.4) did not alter rates of spine outgrowth relative to vehicle-treated controls (black bar; 54 spines, 7 cells). In contrast, inhibition of CaMKs with KN-93 decreased spine outgrowth (light green bar; 40 spines, 7 cells; p < 0.001) but did not further decrease spine outgrowth in cells transfected with Rpt6-S120A (dark green bar; 34 spines, 7 cells; p = 0.6). (C) Images of dendrites from EGFP-expressing WT or GluN2B L1298A/R1300Q knockin (GluN2B KI) mouse neurons at 8–11 DIV, before and after treatment with vehicle, bicuculline (30 μM), or lactacystin (10 μM) at t = 0 (black arrow). Yellow arrows indicate new spines. (D) In neurons from WT mice, treatment with bicuculline increased spine outgrowth (solid blue bar; 76 spines, 5 cells; p = 0.01) relative to vehicle-treated controls (solid black bar; 40 spines, 4 cells), while lactacystin decreased spine outgrowth (solid green bar; 16 spines, 5 cells) relative to controls (solid black bar; 54 spines, 5 cells). In contrast, in neurons from GluN2B KI mice, neither bicuculline (open blue bar; 41 spines, 5 cells; p = 0.6) nor lactacystin (open green bar; 42 spines, 5 cells; p = 0.8) altered spine outgrowth relative to vehicle-treated controls (open black bar; 45 spines, 5 cells). Error bars represent SEM.
Inhibition of CaMKII with KN-93 (30 μM), which inhibits Ca2+-and CaM-dependent kinases, resulted in a 65% reduction in new spine growth (35% ± 6%) as compared to vehicle control (100% ± 11%; p < 0.001; Figure 4B). In contrast, the rate of new spine outgrowth was not significantly different from vehicle controls (100% ± 20%) when PKA was inhibited with either myristoylated PKI 14–22 amide (5 μM; 104% ± 11%; p = 0.9; Figure 4B) or Rp-cAMPS (20 μM; 120% ± 15%; p = 0.4; Figure 4B). In order to examine whether CaMKII inhibition might reduce spine outgrowth through blocking phosphorylation of Rpt6-S120, we treated Rpt6-S120A-transfected neurons with KN-93. Spine outgrowth in Rpt6-S120A-transfected neurons treated with KN-93 (42% ± 8%; Figure 4B) was not significantly different from KN-93-treated neurons (p = 0.8) or untreated Rpt6-S120A neurons (p = 0.7; Figure 3C), suggesting that CaMKII and the proteasome act in the same pathway to regulate spine outgrowth. Moreover, the reduction in new spine growth observed after KN-93 treatment was not significantly different than that after lactacystin (p = 0.5; Figure 1C) or CPP (p = 0.5; Figure 2G) treatment. Thus, our data suggest that CaMKII acts downstream of NMDA receptors to enhance local pro-teasomal activity via phosphorylation of the Rpt6 proteasomal subunit at serine 120.
Interaction of CaMKII with GluN2B Is Necessary for Activity- and Proteasome- Dependent Spine Outgrowth
Because interrupting CaMKII binding to the NMDA receptor subunit GluN2B has been shown to decrease spine density (Gambrill and Barria, 2011), we examined whether this interaction is important for activity- and proteasome-dependent spine growth using GluN2B-L1298A/R1300Q knockin (GluN2B KI) mice (Halt et al., 2012). Both the L1298A and R1300Q mutations reduce GluN2B interaction with CaMKII by over 85% in vitro (Strack et al., 2000), and these two mutations abrogate the activity-dependent increase in NMDA receptor-CaMKII interaction in vivo (Halt et al., 2012).
In order to determine whether interaction of CaMKII with GluN2B is necessary for activity- and proteasome-dependent spine outgrowth, we transfected hippocampal slice cultures from WT and GluN2B KI mice with EGFP and examined the consequences of treatment with bicuculline (30 μM) or lactacys-tin (10 μM) on rates of spine outgrowth (Figures 4C and 4D). As expected, we found that treatment of WT mouse neurons with bicuculline resulted in a 50% increase in spine outgrowth (150% ± 14%) relative to vehicle-treated WT control neurons (100% ± 10%; p < 0.05; Figure 4D). Remarkably, treatment with bicuculline did not alter outgrowth in GluN2B KI neurons (93% ± 6%) relative to vehicle-treated GluN2B KI controls (100% ± 11%; p = 0.6; Figure 4D). Conversely, treatment with lactacystin reduced spine outgrowth in WT neurons by 69% (31% ± 6%) relative to vehicle-treated WT controls (100% ± 6%; p < 0.001), while GluN2B KI neurons were unaffected by lactacystin treatment (92% ± 12%) as compared to vehicle-treated GluN2B KI controls (100% ± 21%; p = 0.8; Figure 4D). Thus, we conclude that the interaction between CaMKII and GluN2B is necessary for activity- and proteasome-dependent spinogenesis.
Surprisingly, we found that baseline spine outgrowth on GluN2B KI control neurons was not different than that on WT control neurons (Table S1; p = 0.7). We predict that compensatory mechanisms are involved, whereby GluN2B KI mice experience an increase in activity- and proteasome-independent spine outgrowth. To further confirm this possibility, we tested the effect of blocking NMDA receptors with CPP on spine outgrowth in WT and GluN2B KI mice. As expected, we found that treatment with CPP reduced spine outgrowth on neurons from WT mice by 42% (58% ± 9%) relative to vehicle-treated WT control neurons (100% ± 10%, p < 0.05) but had no effect on neurons from GluN2B KI mice (96% ± 12%) relative to vehicle-treated GluN2B KI controls (100 ± 24, p = 0.9; Figure S4). These data support that spine outgrowth on GluN2B KI neurons is both activity and proteasome independent.
DISCUSSION
Pharmacological inhibition of the proteasome can lead to presynaptic, postsynaptic, and potentially circuit-wide effects (Bingol and Sheng, 2011; Tai and Schuman, 2008). Which site or sites of action are relevant for activity-induced new spine growth? We observed that expression of Rpt6-S120A in individual neurons inhibited activity-induced spine outgrowth. Because this genetic manipulation was carried out in sparsely transfected neurons, and thus any nearby presynaptic neurons were untransfected, our results demonstrate that postsynaptic proteasomal function is necessary to facilitate new spine growth. In addition, because global pharmacological inhibition was not more effective at reducing spine outgrowth than overexpression of Rpt6-S120A in individual postsynaptic cells, our data also suggest that independent presynaptic and circuit-wide effects do not contribute significantly to the observed reduction in new spine growth. Finally, uncaging-induced spine outgrowth, which is independent of presynaptic activity, was also significantly reduced by blocking the proteasome, emphasizing the role of localized postsynaptic signaling. Our results strongly support a postsynaptic site of action for the proteasome in activity-induced new spine outgrowth.
How might synaptic activity and the proteasome act together to facilitate new spine growth? One possibility is that synaptic activity enhances proteasome function to cause the emergence of new spines. Alternatively, the synaptic stimulus could be the primary cause of spine outgrowth, and normal steady-state levels of proteasomal degradation are required for activity-induced new spine growth. We think that the latter possibility is unlikely because expression of Rpt6-S120A for 4 days does not produce any noticeable effects on cell health compared to untransfected neurons, suggesting that general proteasomal function is not significantly disrupted. In addition, the Rpt6-S120A mutation does not interfere with normal steady-state levels of proteasome-mediated protein degradation in heterologous cells (Djakovic et al., 2012). Instead, because the Rpt6-S120A mutation blocks CaMKII-mediated enhancement of pro-teasomal degradation (Djakovic et al., 2012), our data suggest that locally enhanced proteasomal degradation, probably through CaMKII phosphorylation of Rpt6 at S120, is required for activity-induced new spine growth.
How might neural activity translate to enhanced local protea-somal degradation? Changes in neuronal activity have been shown to alter both proteasome activity (Bingol and Schuman, 2006; Djakovic et al., 2009) and localization (Bingol and Schuman, 2006; Bingol et al., 2010). Although the relative contributions of changes in unit proteasome activity versus changes in proteasome localization to facilitating new spine outgrowth are not yet clear, our findings that spine outgrowth is reduced in mice with impaired NMDA receptor-CaMKII interaction support the possibility that relocation of the proteasome in response to activity may play a role in supporting new spine growth. In these GluN2B KI mice, activity-dependent association of CaMKII with the NMDA receptor is abrogated without altering CaMKII-T286 autophosphorylation, indicating that stimulus-induced activation of CaMKII is normal (Halt et al., 2012). Thus, activity-dependent new spine growth could depend upon the local concentration of active proteasomes rather than stimulation of global proteaso-mal activity.
What target proteins are degraded by the proteasome in order to stimulate new spine growth? Due to the rapid reduction in spinogenesis observed with proteasome inhibition, we expect that the immediate protein targets normally act to inhibit spine outgrowth and are rapidly turned over in an activity- and protea-some-dependent manner. Although a large number of proteins have been shown to regulate spine morphology and dynamics, the most promising candidates for regulating new spine outgrowth are those associated with regulation of the spine actin cytoskeleton (Tashiro and Yuste, 2004; Tolias et al., 2011). One promising candidate could be Ephexin5, a RhoA guanine nucleotide exchange factor shown to negatively regulate excitatory synapse formation (Margolis et al., 2010). Another prime candidate could be Rap2, a member of the Ras family of GTPases. Rap2 overexpression causes a reduction in dendritic spine density (Ryu et al., 2008), suggesting that it may be a negative regulator of spinogenesis. Both of these candidates are plausible targets of activity-induced upregulation of proteasomal activity; in each case, enhanced degradation would be expected to lead to increased spine density.
Malfunction of the proteasome (Tai and Schuman, 2008) and alterations in dendritic spine morphologies and densities (Bhatt et al., 2009) have independently been associated with neurological disorders resulting in mental retardation. Our data support a direct role for the proteasome in the activity-induced spinogenesis thought to be critical for normal brain function and for learning and memory. Disruption of proteasome-mediated degradation would be expected to interrupt activity-induced spine outgrowth during experience-dependent circuit modifications, suggesting one plausible mechanism by which proteasomal dysfunction may lead to neurological dysfunction.
EXPERIMENTAL PROCEDURES
Hippocampal Slice Culture and Transfection
Hippocampal slices were prepared from postnatal day (P) 5–P7 Sprague-Dawley rats or wild-type or GluN2B KI mice (Halt et al., 2012), as described (Stoppini et al., 1991). Genes were delivered 2–5 days (EGFP alone) or 3–4 days (EGFP and Rpt6-WT or Rpt6-S120A) prior to imaging using biolistic gene transfer (180 PSI), as described (Woods and Zito, 2008). We coated 1.6 μmm gold beads with 10 mg of EGFP (Clontech) or with 10 μmg EGFP and 25 μmg HA-tagged Rpt6-WT or Rpt6-S120A (Djakovic et al., 2012).
Time-Lapse Imaging and Image Analysis
EGFP-transfected pyramidal neurons (5–12 days in vitro [DIV]) were imaged at 29°C in ACSF using a custom two-photon microscope (Woods et al., 2011; Supplemental Experimental Procedures). All images shown are maximum projections of three-dimensional (3D) stacks. Spine volumes, densities, and turnover rates were analyzed in 3D using custom software. Because spine addition rates can vary between cultures and experimental days, spine addition rates are reported as a percent of matched controls (calculated using all spines gained in the three posttreatment time points). Absolute spine addition and loss rates for all experiments are documented in Table S1.
Pharmacology
We prepared 1,000× stocks by dissolving MG132 (A.G. Scientific) and KN-93 (Tocris) in DMSO and bicuculline (Tocris), lactacystin (EMD Biochemicals), myristoylated PKI 14–22 amide (Tocris), Rp-cAMPS (Tocris), and CPP (Sigma) in water. Vehicle controls were matched in identity and volume to that in which the inhibitor was dissolved. When two drugs were applied, the vehicle consisted of the sum of the vehicles for both drugs.
Two-Photon Glutamate Uncaging-Induced Spine Outgrowth
Slices were imaged at 30°C in magnesium-free ACSF containing 5 μM MNI-caged-glutamate. Image stacks were acquired immediately before and after the uncaging stimulus, which consisted of 50 pulses (720 nm, ~12 mW at the sample) of 4 ms duration delivered at 5 Hz by parking the beam at a point ~0.5 μmm from the edge of a secondary or tertiary apical dendrite. No more than four uncaging trials were performed on the same neuron. The success rate of de novo spine outgrowth was determined by two blind evaluators. Comparison of success rate across conditions was made by Fisher’s exact test.
Statistics
Error bars represent standard error of the mean and significance was set at p = 0.05 (two-tailed t test, unless otherwise noted). All statistics were calculated across cells. *p < 0.05 and **p < 0.001.
Supplementary Material
Acknowledgments
We thank Judy Callis, Aldrin Gomes, and Jim Trimmer for advice and reagents; Lauren Boudewyn, Julie Heiner, and Sarah Mikula for help with experiments and analysis; and Elva Diaz, Jim Trimmer, and Georgia Woods for critical reading of the manuscript. This work was supported by a Burroughs Wellcome Career Award in the Biomedical Sciences (K.Z.), an NSF CAREER Award (0845285 K.Z. and H.V.R.), and the NIH (T32GM007377 A.M.H.; MARC-GM083894 H.V.R.; NS062736 K.Z., A.M.H., and H.V.R.; NS054732 G.N.P.; AG017502 J.W.H. and I.S.S.). H.V.R. was a participant in the BUSP Program (supported by NIH-IMSD GM056765, HHMI 52005892).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes four figures, one table, and Supplemental Experimental Procedures and can be found with this article online at doi:10.1016/j.neuron.2012.04.031.
References
- Acconcia F, Sigismund S, Polo S. Ubiquitin in trafficking: the network at work. Exp Cell Res. 2009;315:1610–1618. doi: 10.1016/j.yexcr.2008.10.014. [DOI] [PubMed] [Google Scholar]
- Bhatt DH, Zhang S, Gan WB. Dendritic spine dynamics. Annu Rev Physiol. 2009;71:261–282. doi: 10.1146/annurev.physiol.010908.163140. [DOI] [PubMed] [Google Scholar]
- Bingol B, Schuman EM. Activity-dependent dynamics and sequestration of proteasomes in dendritic spines. Nature. 2006;441:1144–1148. doi: 10.1038/nature04769. [DOI] [PubMed] [Google Scholar]
- Bingol B, Sheng M. Deconstruction for reconstruction: the role of proteolysis in neural plasticity and disease. Neuron. 2011;69:22–32. doi: 10.1016/j.neuron.2010.11.006. [DOI] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, Sheng M. Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell. 2010;140:567–578. doi: 10.1016/j.cell.2010.01.024. [DOI] [PubMed] [Google Scholar]
- Comery TA, Shah R, Greenough WT. Differential rearing alters spine density on medium-sized spiny neurons in the rat corpus striatum: evidence for association of morphological plasticity with early response gene expression. Neurobiol. Learn Mem. 1995;63:217–219. doi: 10.1006/nlme.1995.1025. [DOI] [PubMed] [Google Scholar]
- Djakovic SN, Schwarz LA, Barylko B, DeMartino GN, Patrick GN. Regulation of the proteasome by neuronal activity and calcium/ calmodulin-dependent protein kinase II. J Biol Chem. 2009;284:26655–26665. doi: 10.1074/jbc.M109.021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic SN, Marquez-Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA, Patrick GN. Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci. 2012;32:5126–5131. doi: 10.1523/JNEUROSCI.4427-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD. Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci. 2003;6:231–242. doi: 10.1038/nn1013. [DOI] [PubMed] [Google Scholar]
- Engert F, Bonhoeffer T. Dendritic spine changes associated with hippocampal long-term synaptic plasticity. Nature. 1999;399:66–70. doi: 10.1038/19978. [DOI] [PubMed] [Google Scholar]
- Gambrill AC, Barria A. NMDA receptor subunit composition controls synaptogenesis and synapse stabilization. Proc Natl Acad Sci USA. 2011;108:5855–5860. doi: 10.1073/pnas.1012676108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halt AR, Dallapiazza RF, Zhou Y, Stein IS, Qian H, Juntti S, Wojcik S, Brose N, Silva AJ, Hell JW. CaMKII binding to GluN2B is critical during memory consolidation. EMBO J. 2012;31:1203–1216. doi: 10.1038/emboj.2011.482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- Hofer SB, Mrsic-Flogel TD, Bonhoeffer T, Hübener M. Experience leaves a lasting structural trace in cortical circuits. Nature. 2009;457:313–317. doi: 10.1038/nature07487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtmaat A, Wilbrecht L, Knott GW, Welker E, Svoboda K. Experience-dependent and cell-type-specific spine growth in the neocortex. Nature. 2006;441:979–983. doi: 10.1038/nature04783. [DOI] [PubMed] [Google Scholar]
- Hsieh H, Boehm J, Sato C, Iwatsubo T, Tomita T, Sisodia S, Malinow R. AMPAR removal underlies Abeta-induced synaptic depression and dendritic spine loss. Neuron. 2006;52:831–843. doi: 10.1016/j.neuron.2006.10.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jourdain P, Fukunaga K, Muller D. Calcium/calmodulin-dependent protein kinase II contributes to activity-dependent filopodia growth and spine formation. J Neurosci. 2003;23:10645–10649. doi: 10.1523/JNEUROSCI.23-33-10645.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott GW, Quairiaux C, Genoud C, Welker E. Formation of dendritic spines with GABAergic synapses induced by whisker stimulation in adult mice. Neuron. 2002;34:265–273. doi: 10.1016/s0896-6273(02)00663-3. [DOI] [PubMed] [Google Scholar]
- Kwon HB, Sabatini BL. Glutamate induces de novo growth of functional spines in developing cortex. Nature. 2011;474:100–104. doi: 10.1038/nature09986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maletic-Savatic M, Malinow R, Svoboda K. Rapid dendritic morphogenesis in CA1 hippocampal dendrites induced by synaptic activity. Science. 1999;283:1923–1927. doi: 10.1126/science.283.5409.1923. [DOI] [PubMed] [Google Scholar]
- Margolis SS, Salogiannis J, Lipton DM, Mandel-Brehm C, Wills ZP, Mardinly AR, Hu L, Greer PL, Bikoff JB, Ho HY, et al. EphB-mediated degradation of the RhoA GEF Ephexin5 relieves a developmental brake on excitatory synapse formation. Cell. 2010;143:442–455. doi: 10.1016/j.cell.2010.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzaki M, Honkura N, Ellis-Davies GC, Kasai H. Structural basis of long-term potentiation in single dendritic spines. Nature. 2004;429:761–766. doi: 10.1038/nature02617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa M, Segal M. Morphological plasticity in dendritic spines of cultured hippocampal neurons. Neuroscience. 1996;71:1005–1011. doi: 10.1016/0306-4522(95)00490-4. [DOI] [PubMed] [Google Scholar]
- Roberts TF, Tschida KA, Klein ME, Mooney R. Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature. 2010;463:948–952. doi: 10.1038/nature08759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal-regulated kinase signaling, enhanced long-term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178–8188. doi: 10.1523/JNEUROSCI.1944-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Ott DE, Chertova EN, Welker R, Tessmer U, Princiotta MF, Bennink JR, Krausslich HG, Yewdell JW. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc Natl Acad Sci USA. 2000;97:13057–13062. doi: 10.1073/pnas.97.24.13057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L, Buchs PA, Muller D. A simple method for organotypic cultures of nervous tissue. J Neurosci Methods. 1991;37:173–182. doi: 10.1016/0165-0270(91)90128-m. [DOI] [PubMed] [Google Scholar]
- Strack S, McNeill RB, Colbran RJ. Mechanism and regulation of calcium/calmodulin-dependent protein kinase II targeting to the NR2B subunit of the N-methyl-D-aspartate receptor. J Biol Chem. 2000;275:23798–23806. doi: 10.1074/jbc.M001471200. [DOI] [PubMed] [Google Scholar]
- Tai HC, Schuman EM. Ubiquitin, the proteasome and protein degradation in neuronal function and dysfunction. Nat Rev Neurosci. 2008;9:826–838. doi: 10.1038/nrn2499. [DOI] [PubMed] [Google Scholar]
- Tashiro A, Yuste R. Regulation of dendritic spine motility and stability by Rac1 and Rho kinase: evidence for two forms of spine motility. Mol Cell Neurosci. 2004;26:429–440. doi: 10.1016/j.mcn.2004.04.001. [DOI] [PubMed] [Google Scholar]
- Tolias KF, Duman JG, Um K. Control of synapse development and plasticity by Rho GTPase regulatory proteins. Prog Neurobiol. 2011;94:133–148. doi: 10.1016/j.pneurobio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trachtenberg JT, Chen BE, Knott GW, Feng G, Sanes JR, Welker E, Svoboda K. Long-term in vivo imaging of experience-dependent synaptic plasticity in adult cortex. Nature. 2002;420:788–794. doi: 10.1038/nature01273. [DOI] [PubMed] [Google Scholar]
- Woods G, Zito K. Preparation of gene gun bullets and biolistic transfection of neurons in slice culture. J Vis Exp. 2008;12:e675. doi: 10.3791/675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woods GF, Oh WC, Boudewyn LC, Mikula SK, Zito K. Loss of PSD-95 enrichment is not a prerequisite for spine retraction. J Neurosci. 2011;31:12129–12138. doi: 10.1523/JNEUROSCI.6662-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Yu X, Perlik AJ, Tobin WF, Zweig JA, Tennant K, Jones T, Zuo Y. Rapid formation and selective stabilization of synapses for enduring motor memories. Nature. 2009;462:915–919. doi: 10.1038/nature08389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang G, Pan F, Gan WB. Stably maintained dendritic spines are associated with lifelong memories. Nature. 2009;462:920–924. doi: 10.1038/nature08577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Hu Y, Huang P, Toleman CA, Paterson AJ, Kudlow JE. Proteasome function is regulated by cyclic AMP-dependent protein kinase through phosphorylation of Rpt6. J Biol Chem. 2007;282:22460–22471. doi: 10.1074/jbc.M702439200. [DOI] [PubMed] [Google Scholar]
- Zito K, Scheuss V, Knott G, Hill T, Svoboda K. Rapid functional maturation of nascent dendritic spines. Neuron. 2009;61:247–258. doi: 10.1016/j.neuron.2008.10.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.