

Abstract
Rationale
Cardiac resynchronization therapy (CRT) is an effective clinical treatment for heart failure patients with conduction delay, impaired contraction and energetics. Our recent studies have revealed that mitochondrial post-translational modifications (PTM) may contribute to its benefits, motivating the present study of the oxidative regulation of mitochondrial ATP synthase.
Objectives
Here, we tested whether CRT alteration of ATP synthase function is linked to cysteine (Cys) oxidative PTM (Ox-PTM) of specific ATP synthase subunits.
Methods and Results
Canine left ventricular myocardium was collected under conditions to preserve Ox-PTM from control, dyssynchronous heart failure (DHF) or hearts that have undergone CRT. In-gel ATPase activity showed that CRT increased ATPase activity by ~2 fold (p<0.05) over DHF, approaching control levels and this effect was recapitulated with a reducing agent. ATP synthase function and three Ox-PTM: disulfide bond, S-glutathionylation and S-nitrosation were assessed. ATP synthase from DHF hearts contained two novel disulfide bonds, between ATP synthase alpha subunits themselves and between alpha and gamma subunits, both of which were decreased in CRT hearts (4.38±0.13 and 4.23±0.36-fold, respectively, p<0.01). S-glutathionylation of ATP synthase alpha subunit occurred in DHF hearts and was reversed by CRT (1.56±0.16-fold, p<0.04). In contrast, S-nitrosation of ATP synthase alpha subunit in DHF hearts was lower than in CRT hearts (1.53±0.19-fold, p<0.05). All modifications occurred at ATP synthase alpha subunit Cys294 and Cys to Ser mutation indicated that this residue is critical for ATP synthase function.
Conclusions
A selective Cys in ATP synthase alpha subunit is targeted by multiple Ox-PTM suggesting that this Cys residue may act as a redox sensor modulating ATP synthase function.
Keywords: cardiac resynchronization therapy, ATP synthase, Cys oxidative modification
Introduction
Cardiac resynchronization therapy (CRT) is a clinically effective treatment for dyssynchronous heart failure (DHF); however, the underlying molecular and cellular mechanisms remain poorly understood.1 An early clinical finding indicated that CRT improves chamber energetic efficiency2 and our recent work has pinpointed that CRT directly impacts ATP production by mitochondria, profoundly affecting the mitochondrial subproteome.3 Specifically, DHF and CRT alter the mitochondrial subproteome by modifying proteins involved in cellular redox control and oxidative phosphorylation (OxPhos) pathways, as manifested by changes in both protein quantity and post-translational modifications (PTM).3
The mitochondrial electron transport chain is a primary source of reactive oxygen and nitrogen species (ROS/RNS), which can mediate oxidative-PTM (Ox-PTM), particularly targeting important mitochondrial components including the respiratory chain and matrix enzymes, as well as membrane phospholipids.4,5 Emerging evidence links Ox-PTM to dysfunction in ATP synthesis in the failing heart, highlighting potential therapeutic targets.6–8 On the other hand, Ox-PTM in the heart may serve as redox switches, sensing cellular redox state to regulate protein function.9–11 This is not unprecedented, for example, in plants, chloroplast F1FO-ATPase is subject to redox regulation, whereby ATP hydrolytic activity is regulated by the formation and reduction of a disulfide bond located in the gamma subunit.12 Changes in redox state do not affect the rate of ATP binding to the catalytic site(s) or the torque for rotation, but long pauses caused by ADP inhibition are more frequent in the oxidized state.12 As well, introducing the redox modulated region of the plant ATPase into the yeast gamma-subunit causes a defect in oxidative phosphorylation.13
In this study, we report several ROS/RNS-related PTM that occur on ATP synthase in failing dyssynchronous hearts (DHF) and demonstrate that CRT can ameliorate these Ox-PTM, supportive of a protected phenotype and improved ATP production.
Methods
Animal model and sample preparation
Adult mongrel dogs underwent either a DHF or a CRT protocol, as previously described.3 Heart tissues were harvested under conditions to preserve Ox-PTM.14 Mitochondria were isolated by a differential centrifugation protocol as previously described3 and adapted to preserve Ox-PTM. Rat heart tissues were from Pel-Freez Biologicals (Rogers, AR) and mitochondria were isolated in the absence of N-ethylmaleimide (NEM). ATP synthase was isolated using ATP synthase immunocapture kit (Mitosciences) according to the manufacture’s protocol. See Online Supplement.
Cell culture
HEK 293 cells used for exogenous expression of Cys mutants were cultured at 37°C in a 5% CO2 incubator in DMEM media (Mediatech) supplement with 10% FBS (HyClone).
Genetic manipulation
The human ATP5A1 and ATP5C1 ORF and full length cDNA were obtained from Open Biosystems. Cys to Ser mutants were created by QuickChange site-directed mutagenesis kit (Stratagene) with primers list in Online Table I. siRNAs for ATP5A1 and ATP5C1 gene were obtained from Applied Biosystems. Expression plasmids and siRNAs were introduced into HEK 293 cells by transient co-transfection with Lipofectamine™ RNAiMAX Reagent (Invitrogen) according to the manufacture’s protocol. See Online Supplement.
Induction of Ox-PTM in isolated mitochondria and ATP synthase complex with oxidants
Isolated mitochondria or ATP synthase complex were resuspended in reaction buffer containing different reagents and induction performed as described in figure legend. See Online Supplement.
Gel electrophoresis and immunoblot
Clear Native PAGE (CNP) and Blue Native PAGE (BNP) were used to resolve intact mitochondrial protein complexes.15,16 2D BNP/SDS-PAGE was performed to analyze complex subunit composition.16 1D SDS-PAGE was performed as described.3 For immunoblotting, proteins were transferred to PVDF (polyvinylidene fluoride) membranes prior to blotting. Densitometry was performed on scanned gels and immunoblots using Progenesis Workstation 2005 software (Nonlinear Dynamics). See Online Supplement for method and antibodies.
Biotin switch assay
S-nitrosation (SNO) was detected with a modified biotin switch assay as described.17 After streptavidin pulldown, the S-nitrosated ATP synthase alpha subunit was detected with anti-Complex V α subunit antibody (Invitrogen). See Online Supplement.
ATPase activity determination
ATPase activity was determined by in-gel (CNP) ATPase assay according to a previously described method.15 See Online Supplement.
Mitochondria respiration measurement
Mitochondria were freshly isolated from LV endocardium of a normal dog in the absence of NEM as described above. Mitochondria respiration was measured as described.18 See Online Supplement.
Mass Spectrometry
Gel bands or spots were excised, digested with trypsin, and analyzed on an LTQ-Orbitrap LC-MS/MS (liquid chromatography-tandem mass spectrometry, Thermo Finnigan) with proteins identified by SEQUEST (Sage-N Research) according to our published methods.3,19 IPI data base (IPI_RAT_v3.62) was used, protein name redundancy was removed and isoforms identified positively only if a peptide(s) was observed to correspond to an isoform unique amino acid sequence. See Online Supplement.
Molecular Modeling
The three-dimensional structure of Bovine mitochondrial F1 ATP synthase from protein data bank (PDB entry # 1E79) was modeled using PyMOL (Delano Scientific LLC).
Statistics
All data are expressed as means ± SD. Comparisons between different groups were performed via 2-tailed unpaired Student t test with p<0.05 being considered significant.
Results
Reversal of Cys oxidative modification contributes to the beneficial effect of CRT on ATP synthase activity
To test whether Cys oxidative modification correlates with mitochondrial dysfunction, we measured mitochondrial ATPase activity from adult mongrel dogs subjected to either DHF, CRT, or no tachypacing (Sham) using CNP and subsequent in-gel ATPase activity assay (a method more sensitive than BNP15). Figure 1A shows ATP synthase activity in mitochondria from LV endocardium obtained from DHF, CRT and Sham and isolated under Cys modification preserving conditions, unlike previous work on this model, where the focus was on other non-oxidative PTM.3 Under these conditions, ATPase activity was significantly lower in DHF compared to control (Figure 1A, bottom panel) (26.13 ± 7.71 vs. 47.04 ± 16.9, p<0.05). CRT significantly increased activity to control levels (53.99 ± 13.53 vs 47.04 ± 16.93, p=0.24). Importantly, incubation of mitochondria isolated from DHF hearts with 1 mmol/L DTT restored ATPase activity to control levels (Figure 1A), suggesting that reversal of Cys oxidative modifications contributed to the beneficial effects of CRT on ATP synthase activity. Interestingly, the ATPase activity from Sham mitochondria was also DTT sensitive, suggesting that oxidative modifications are present under baseline conditions. However, it is important to note that the degree of DTT sensitivity was much greater in DHF hearts and was significantly reduced in CRT hearts.
Figure 1. Reversal of the cysteine oxidative modification contributes to the beneficial effect of CRT on ATPase activity.
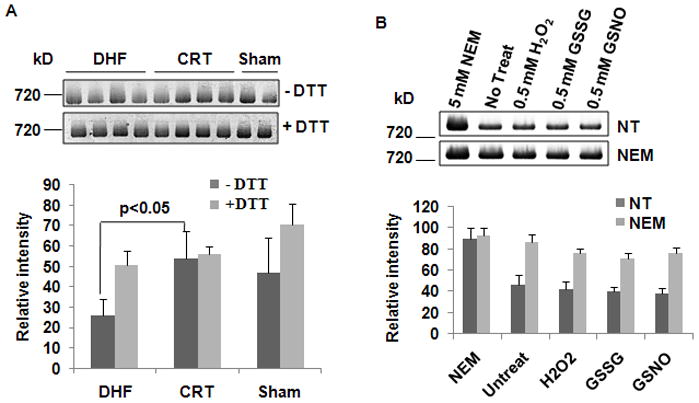
Isolated mitochondria were solubilized with 2% digitonin and clear native PAGE was run as described in Methods. A, Top: Representative images of in-gel ATPase activity assay on DHF, CRT and Sham under nonreducing (−DTT) and reducing conditions (+DTT). Bottom: Quantification of in-gel ATPase activity based on densitometry. ATPase activity was normalized for ATP synthase complex protein content (n=4 DHF, 4 CRT, 2 Sham, in 3 replicates). B, Cys Ox-PTM has profound effects on ATPase activity. Mitochondria were isolated from normal dogs in the absence of (no treatment, NT, n=4) or in the presence of NEM (NEM: 20 mmol/L, n=4) and the in-gel ATPase activity assay was carried out after different treatments. Top: Representative image of in-gel ATPase activity assay with 5 mmol/L NEM, NT, 0.5 mmol/L H2O2, 0.5 mmol/L GSSG or 0.5 mmol/L GSNO. Bottom: Quantification of in-gel ATPase activity showing that blocking free thiols significantly increase ATPase activity.
The ATPase activity of mitochondria from sham animals, obtained in the presence of alkylating or oxidizing reagents, is shown in Figure 1B. Blocking thiols on any free Cys residue with the alkylating reagent NEM during mitochondrial isolation greatly improved ATPase activity at baseline and rendered it resistant to oxidant treatment. This enhanced basal activity is probably due to reversal of modest oxidation of the exposed free thiols by oxygen in the solution during the process of mitochondrial isolation, despite our best efforts to preserve a reducing environment (see methods section for details). However, importantly, the difference between the NEM-treated and untreated groups was large (~2 fold), suggesting that the Cys oxidative modifications have a profound effect on ATPase activity. To confirm that the effect of oxidation on ATPase activity in DHF dogs occurs in vivo, independently of NEM treatment, we also measured the ATPase activity in mitochondria isolated from DHF, CRT and Sham dogs that were not treated with NEM. As shown in Online Figure I, in the absence of NEM, the DHF group still had significantly lower ATPase activity compared to controls (~30%), whereas the activity was restored to control levels by CRT. This result is similar to our previous study3 in the same model, where CRT induces a 20% increase in ATPase activity compared to DHF. Taken together, these results demonstrate that the beneficial effect of CRT with respect to ATPase activity is independent of NEM. Since NEM and DTT may have broad effects on mitochondrial proteins other than ATP synthase, we next studied the effects of NEM and DTT on oxygen consumption rate (OCR) in freshly isolated mitochondria from control dogs. As shown in Online Figure II, 20 mmol/L NEM treatment severely impaired respiration supported by either glutamate/malate or succinate. This suggests that NEM could inhibit multiple components of OxPhos (e.g., substrate transport, the Krebs cycle, or electron transport) in addition to restoring ATP synthase activity in the in-gel assay. By contrast, treatment with 1 mmol/L DTT increased respiration substantially for both substrates. On a relative basis, state 2 increased more than state 3 respiration. As a result, the net effect of DTT on OCR (glutamate/malate) was a reduction from 5 to 2.3. This finding, while consistent with the enhanced ATP synthase activity observed in the in-gel assay, indicates that the reversal of mitochondrial oxidative modifications by DTT probably affects mitochondrial respiration at more than one site.
DHF is correlated with disulfide bond formation in the ATP synthase complex
To assess the types of Cys modification present in the various experimental groups, we first assessed the presence of disulfide bonds in the ATP synthase by classical 2D-BNP/SDS-PAGE under non-reducing conditions, and compared the results with those obtained under reducing conditions from DHF, CRT and Sham hearts. Figure 2A shows that mitochondria from DHF hearts have crosslinked products of about ~120 kD and ~100 kD within the ATP synthase complex. These were reversible after DTT treatment (Figure 2A, bottom panel) and blocked by pretreatment with NEM (Figure 2C, left panel), indicating that crosslinking was a Cys-based modification, i.e., disulfide bond formation. 2D non-reducing/reducing SDS-PAGE demonstrated that the disulfide bond complex was composed of ATP synthase alpha subunit (ATPα) and ATP synthase gamma subunit (ATPγ) (Online Figure III), confirmed by western blot and verified by LC-MS/MS (Online Table II). These results suggest that the crosslinks formed in DHF dog hearts are disulfide bonds. Figure 2B shows that the quantity of the ATPα-ATPα and ATPα-ATPγ disulfide bond complexes was significantly reduced (~4 fold) after CRT treatment, compared to DHF animals. There were trace amounts of disulfide crosslinked complexes detected in Sham dogs.
Figure 2. DHF results in ATPα-ATPα and ATPα-ATPγ disulfide bond formation.
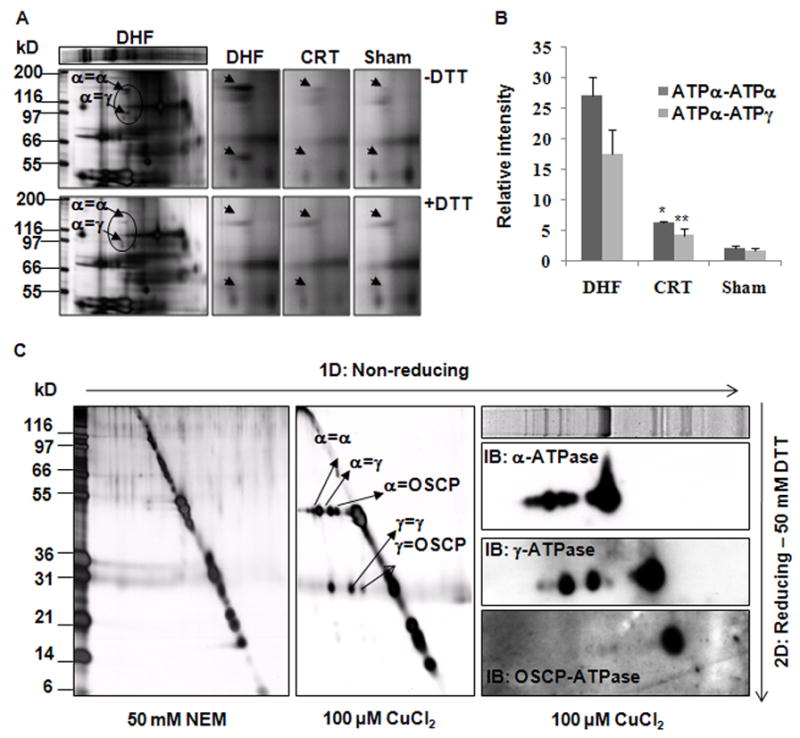
A, Representative silver stained images of 2D-BNP/SDS-PAGE show DHF increases the quantity of DTT-reversible cysteine crosslinks. The close-ups show that CRT reverses the ATPα-ATPα and ATPα-ATPγ disulfide bonds induced by DHF. B, Quantification of disulfide bonds based on densitometry. Each sample value was normalized to its ATPβ content (n=3, *, P<0.006; **, p<0.008). C, In vitro induction of disulfide bonds in isolated rat heart mitochondrial ATP synthase. 10 μg ATP synthase from rat heart mitochondria were incubated with 20 mmol/L NEM or 100 μmol/L CuCl2 at room temperature for 10 min and used for 2D-nonreducing/reducing SDS-PAGE. Left Panel: Silver stained images show extensive Cys crosslinking among ATPα, ATPγ and OSCP subunits. Right Panel: Confirmation for the components of the crosslinked products by western blot.
To validate these data, we also tested whether disulfide bonds could be induced in vitro with a generic oxidant treatment applied to isolated ATP synthase complex from rat heart mitochondria. As shown in Figure 2C middle panel, CuCl2 treatment (100 μmol/L) resulted in extensive disulfide bond formation within the ATP synthase complex. As a control, NEM was used to block free Cys, and no crosslinked product was detected (n=3) (Figure 2C, left panel). With CuCl2 treatment, besides ATPα-ATPα and ATPα-ATPγ disulfide bonds, ATPα and the ATP synthase oligomycin sensitivity conferral protein (OSCP) disulfide bonds were also observed, as well as those between ATPγ-ATPγ, and ATPγ-OSCP. As before, the identity of each subunit was confirmed by western blot (Figure 2C, right panel) and by LC-MS/MS analysis (Online Table II). Thus, it is feasible that these additional disulfide bonds could occur with extreme oxidative stress.
DHF increases S-glutathionylation of the ATP synthase complex
We next tested whether ATPα was also the target of other Cys modifications, such as protein S-glutathionylation (S-Glu) and SNO. The extent of ATPα S-Glu was determined in isolated mitochondria from DHF, CRT and Sham dogs by western blot using an anti-GSH antibody (Figure 3A). Compared to sham animals, the extent of ATPα S-Glu was significantly increased in DHF, whereas the levels of S-Glu were partially normalized in CRT (Figure 3B). For validation, isolated ATP synthase complex from rat heart mitochondria was treated with GSSG and the resulting protein S-Glu was detected by western blot. As shown in Figure 3C, the S-Glu of ATPα could be induced by GSSG in a dose-dependent manner.
Figure 3. S-Glu of ATPα occurs both in vitro and in vivo.
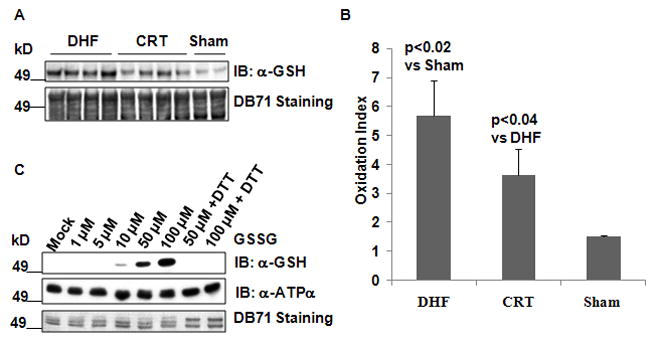
A, CRT reverses S-Glu of ATPα in DHF dogs. Top: Immunoblotting of S-Glu of ATPα with anti-GSH anitbody in isolated mitochondrial from DHF (n=4), after CRT (n=4), and in Sham (n=2) hearts. Bottom: DB71 stained PVDF membrane after protein transfer to verify equal loading based on the ~49kD area. B, Quantification of S-Glu in DHF, CRT and Sham dogs by densitometry of the western blot of panel A. C, Induction of the ATPα S-Glu in isolated rat heart mitochondrial ATP synthase. 2 μg ATP synthase were incubated with Mock or GSSG at room temperature for 30 min. Top: Immunoblotting of S-Glu with anti-GSH antibody. Middle: A parallel blot with the same samples as top panel probed with anti-ATPα. Bottom: DB71 stained PVDF membrane after protein transfer, shown are ATPα and ATPβ subunits.
CRT increases SNO of the ATP synthase complex
SNO of ATPα was investigated by a modified biotin switch assay.17 As shown in Figure 4A, S-nitrosoglutathione (GSNO) treatment resulted in a significant increase of SNO of ATPα in isolated mitochondria when conditions for preserving Cys modifications were not employed in both DHF and CRT dogs. When mitochondria were isolated under Cys modification preserving conditions, untreated mitochondria from DHF had significantly less SNO compared to CRT or Sham as shown in Figure 4B. However, the extent of SNO induced by CRT was not as pronounced as other modifications. This suggests that SNO modification can only partly account for reversal of DHF-induced crosslinking with CRT, leaving the majority of Cys free in the CRT hearts. GSNO-induced dose-dependent SNO of ATPα could also be demonstrated in the isolated rat heart mitochondrial ATP synthase complex (Figure 4C).
Figure 4. SNO of ATPα occurs both in vitro and in vivo.
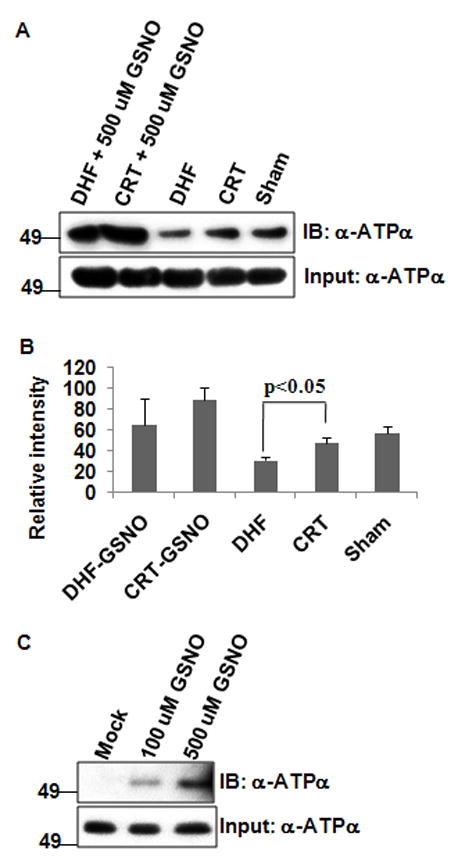
100 μg mitochondria were incubated either with or without 500 μmol/L GSNO and Biotin switch assay was performed as described in Methods. A, Top: Representative blot showed CRT leads to high level of S-nitrosated ATPα as compared to DHF (n=3 for each type). Bottom: Input for biotin switch assay: 1/20 (~5 μg total mitochondrial protein) of the solution just before streptavidin pull down were saved for western blot with anti-ATPα antibody. B, Quantification of SNO in DHF, CRT and Sham dogs by densitometry normalized to input ATPα content. C, Validation of ATPα SNO by in vitro GSNO treatment. 2 μg ATP synthase from rat heart mitochondria were incubated with mock, 100 μmol/L and 500 μmol/L GSNO at 37°C for 15 min and used for modified biotin switch assay as above. Top: Western blot shows ATPα could be S-nitrosated by GSNO in a dose-dependent manner. Bottom: Input for biotin switch assay performed as in panel A.
Cys294 of ATPA is the site for disulfide bond formation, S-Glu, and SNO
We next performed mass spectrometry (MS) to identify the specific Cys residues modified by either disulfide bond formation or S-Glu. A differential labeling and MS strategy, outlined in Figure 5A, was used to identify the Cys residues involved in disulfide bonds. ATPγ contains one Cys, Cys103, and as expected, this Cys was differentially labeled by d5NEM in the presence of 100 μmol/L CuCl2 (Online Figure IV). However, in ATPα, both Cys244 and Cys294 were labeled with d5NEM (Online Figure IV), indicating that both could be oxidized by CuCl2 treatment. To determine which of the two Cys residues of ATPα were responsible for the disulfide bond, C-terminally FLAG tagged C244S and C294S mutants were prepared and expressed in HEK 293 cells by transient transfection. As shown in Figure 5B, with CuCl2 treatment, C294S, but not C244S, prevent the formation of the ATPα-ATPα disulfide bond and the disulfide bond in C244S mutant was still reversible by DTT treatment. This result clearly demonstrates that Cys294 of ATPα is specifically involved in the disulfide bond formation.
Figure 5. Identification of ATPα C294 for disulfide bond formation.
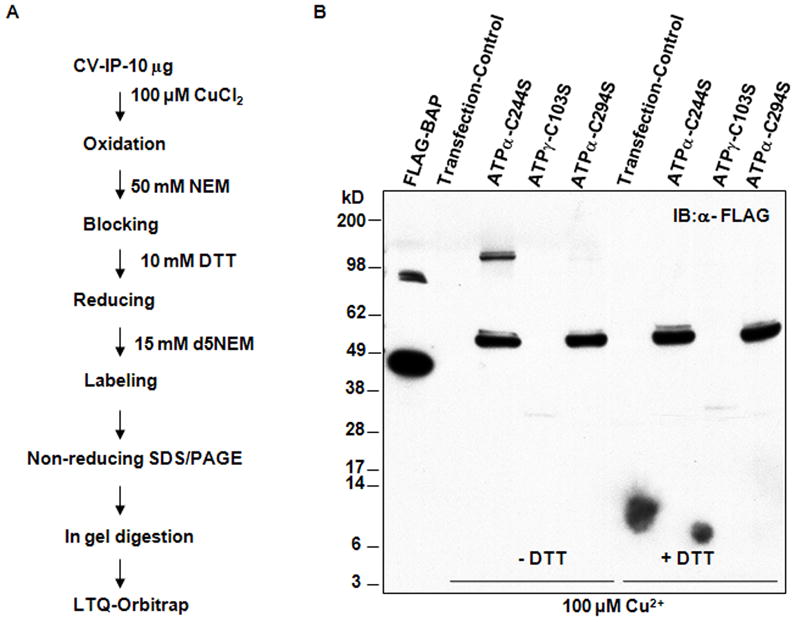
A, Outline of differential labeling and MS strategy to map the site for Cys disulfide bond formation in ATP synthase complex. B, Site directed mutagenesis for identifying the site for disulfide bond formation in HEK 293 cells. Total lysates of the expressed mutants were treated with 100 μmol/L CuCl2 and boiled without or with DTT. Representative western blot shows that only ATPα C244S mutant can form the ATPα-ATPα disulfide and this was reversed by DTT treatment (n=3 independent transfection experiments).
To determine the Cys residues that undergo S-Glu, we subjected S-glutathionylated ATPα, obtained by treating isolated rat heart mitochondrial ATP synthase with GSSG, to trypsin digestion and analyzed by LC-MS/MS. Peptides with a mass difference of 305 Da, representing one glutathione moiety were determined. Both Cys244 and Cys294 of ATPα were glutathionylated (Online Figure V). Interestingly, Cys294 has been previously shown to be S-nitrosated by GSNO treatment in isolated rat17 or mouse20 heart mitochondria. Taken together, these data show that Cys294 of ATPα is actively involved in various oxidative modifications including intermolecular disulfide bond formation, S-Glu and SNO.
Based on the x-ray crystallographic protein structure of bovine mitochondrial F1 ATP synthase (PDB 1E7921), both Cys294 of ATPα and Cys103 of ATPγ are located on the surface of the ATP synthase, which makes them accessible for oxidant attack (Online Figure VI). To gain insight into the importance of these residues on the redox regulation of ATP synthase activity, Cys244 and Cys294 of ATPα, and Cys103 of ATPγ, were mutated to Ser and the mutant subunits were expressed in HEK cells. Exogenous expression of the constructs containing only the open reading frame of the corresponding subunit was induced, while RNAi was used to knock down the expression of endogenous ATPα and ATPγ subunits. As shown in Online Figure VIIA, siRNA knock down decreased each of the endogenous subunits by 90%, with the majority of these being replaced with mutant ones. The mutants were able to form the intact F1F0-ATP synthase complex (Online Figure VIIA). Unexpectedly, however, measurements of the ATPase activity (normalized to the corresponding quantity of ATP synthase complex based on Coomassie blue staining; Online Figure VIIB) showed that the expression of the wild type subunit construct, in the presence of the siRNA, did not restore ATPase activity to levels found with the scrambled RNA transfection control. The reason for this is not clear, but since ATP synthase is a multisubunit protein complex, the function of this complex depends on the correct expression and assembly of each subunit. It is known that the 3′-untranslated region (3′-UTR) is involved in the regulation of expression of ATP synthase subunits.22 Therefore, we made additional expression constructs of the full length cDNA for mutagenesis and transfection experiments (Figure 6A). Overexpression of subunit constructs containing the 3′-UTR partially restored the ATP synthase activity when the corresponding wild type protein was present. As shown in Figure 6B, both C244S and C294S mutants have significantly lower ATP synthase activities when compared to the wild type ATPα re-expression group (~50% decrease), indicating that Cys244 and Cys294 are required for the functionality of ATP synthase complex.
Figure 6. Effects of Cys mutants (C244S, C294S of ATPα and C103S of ATPγ) on ATPase activity in HEK cells.
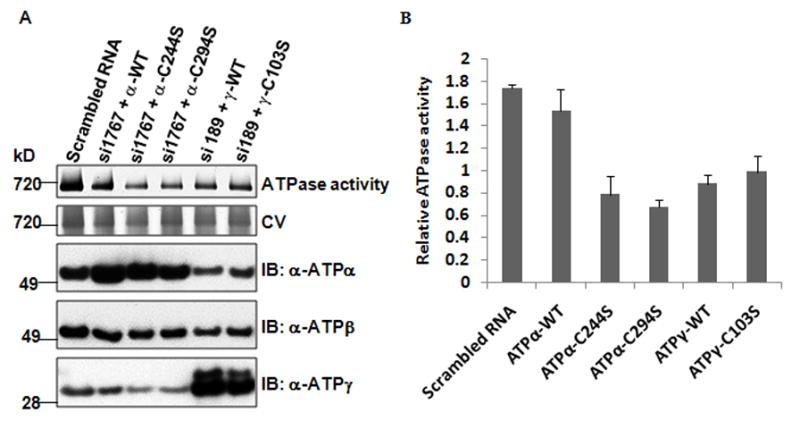
Full length cDNA of WT and C244S, C294S and C103S mutant plasmids were co-transfected into HEK cells along with the corresponding siRNA for the native subunit (si1767 for ATPα and si189 for ATPγ) to knockdown the endogenous ones. A, In-gel ATPase activity of different Cys mutants. ATP synthase complex was visualized with Coomassie blue staining after removing lead phosphate precipitate in destaining solution (20% Methanol, 10% Acetic acid). ATPα, ATPβ and ATPγ were determined by western blot in total mitochondria from the same transfection. B, Quantification of ATPase activity in different Cys mutants based on densitometry (n=3 independent transfection experiments). ATP syntase activity was normalized to ATP synthase complex protein content.
Both wild type and the C103S mutant of ATPγ also had significantly lower ATPase activities when compared to the transfection control despite our efforts to optimize the knock down and transfection protocol (online Table III), indicating that there may be unknown mechanisms of PTM(s) or chaperones missing that are required to regulate the gamma subunit assembly into ATP synthase complex. For example, the newly identified chaperon, DAPIT (Diabetes-associated Protein in Insulin-sensitive Tissue), is required for the folding of ATP synthase.23 However, the C103S mutant had slightly more activity than its wild type counterpart (~10% increase). When the mutant cell lines were treated with 100 μmol/L CuCl2 or 1 mmol/L DTT, no significant differences in ATP synthase activity were detected for the mutants in response to either treatment when compared to its corresponding wild type subunit (online Figure VIII), suggesting that the site-specific oxidations occurring in vivo are likely to be functionally important, although it currently remains elusive whether the rotation of gamma subunit is needed for ATP hydrolysis.24
Discussion
In our previous study using the CRT model3, we showed that mitochondrial ATPase activity was increased with CRT as compared to DHF, and this correlated with reduced proteolysis and modest dephosphorylation. The largest changes in CRT, however, were up-regulation of several key redox enzymes, leading to the current hypothesis that Ox-PTM might contribute to the beneficial effect of CRT. To maximize the detection of Ox-PTM, here we performed the tissue collection and all downstream operations under conditions to preserve reversible Cys oxidative modifications. In this study, we have shown for the first time that Cys oxidative modifications lead to mitochondrial ATP synthase dysfunction in DHF, and that this effect is partially reversed by CRT. A number of Cys oxidative modifications can occur on the ATP synthase, including disulfide bond formation, S-Glu, and SNO, and they are specific to either DHF or CRT. With DHF, several subunits of the ATP synthase are crosslinked via intermolecular disulfide bonds. Specifically, disulfide bonds form between F1FO ATP synthase ATPα-ATPα and ATPα-ATPγ subunits, and S-Glu of ATPα was detected. These disulfide bonds were significantly decreased in CRT animals, as was the level of S-Glu. Although, at present, we cannot quantify the extent of disulfide bonds versus S-Glu in the ATP synthase complex, it is clear that both can exist in the same sample, and both positively correlate with the loss of function of ATP synthase. In contrast, ATP synthase modification by SNO shows the opposite profile; it is reduced in DHF and then increased in CRT. In particular, we found that CRT can induce SNO of a modified cysteine after reversing the cysteine crosslinking and this might reverse the impaired function of the ATP synthase.
Location of reactive Cys
In the present study, we have found an intermolecular disulfide bond between ATPα through Cys294 and an intermolecular disulfide bond between ATPα and ATPγ through Cys294 and Cys103, respectively. In addition, Cys294 of ATPα can also be modified by S-Glu and SNO. Based on the molecular model, Cys294 is located on the nucleotide binding domain surface, surrounded by several positively charged residues (Online Figure VI). Thus, it would probably be deprotonated at physiological pH, making it a good candidate for S-Glu and SNO.
Within an individual ATP synthase complex, the Cys294 of one of the ATPα subunits is located farther than 5 Å (the distance required for a disulfide bond to occur) from its neighboring ATPα or the Cys103 of ATPγ. Hence, it is unlikely that a disulfide bond between either ATPα-ATPα or ATPα-ATPγ occurs within a single complex. It is also unlikely that disulfide bonds occur between the ATPα from different ATP synthase complexes, since it is well documented that the subunits e and g are involved in the ATP synthase dimer interfaces resulting in the F1 subunits pointing away from each other.25,26
An alternate explanation is that these disulfide bonds could be formed before the individual subunits assemble into the ATP synthase complex, or when (or if) the protein/complex is aggregated. In all cases, the ATP synthase complex assembly will be compromised. In fact, DHF dog hearts have ~20% less ATPα and ATP synthase beta subunit (ATPβ) content, and intact complex content than normal dogs, but this increases with CRT back to near control levels.3 This suggests that disulfide bond formation may represent a misfolded/aggregated form of the complex (but one with all subunits present).
The situation is different for both S-Glu and SNO, for which the physical spacing within the complex is not a factor. Rather, the main determinant would be whether the residues are accessible and if the Cys is buried or exposed, although for SNO there is no agreement on an amino acid consensus sequence.27,28 Importantly, Cys294 and Cys244 of ATPα are both found on the complex surface and there is no amino acid sequence homology within 15 amino acid residues on either side of the modifiable Cys in the human or rat sequence.
Heart failure-induced oxidative stress causes redox imbalance, with low GSH levels and high GSSG levels, which could induce protein S-Glu.29 ATPα has already been reported to be the major mitochondrial protein that becomes glutathionylated under oxidative stress, at least in rat brain or in liver mitochondria that were isolated under a discontinuous percoll gradient.30 Under these conditions, S-Glu of ATPα led to a substantial decrease of ATPase activity.30 Similarly, DHF dog hearts have a significantly higher level of S-Glu of ATPA compared to controls and this increase correlated with the loss of function of the ATP synthase complex. S-Glu of Cys294 of ATPα, which adds a bulky negatively charged group to this residue, could potentially disrupt nucleotide binding to decrease ATPase activity.
Therapeutic benefit of CRT and altered Cys Ox-PTM
Cys modifications are dependent on the redox status and antioxidant capacity of the myocyte. Our data indicate that DHF is associated with increased oxidative stress, while CRT improves antioxidant defenses, in particular, the thioredoxin/peroxiredoxin pathway.3 Enhanced ROS scavenging might also prevent NOS uncoupling and preserve physiological NO signaling mechanisms, including SNO.31 We and others have previously shown that ATPα could be S-nitrosated by in vitro GSNO treatment in a large scale study mapping SNO sites in isolated rat heart mitochondria.17,32 Also, it has been established that SNO modification of this protein occurs after ischemic preconditioning (IPC) of hearts in a mouse model, and these authors suggested that SNO of this protein serves as a cardioprotective mechanism in failing heart.32,33 In the present study, we have found that SNO of ATPα was significantly decreased in DHF dog hearts, whereas it was recovered in the CRT dog hearts. This indicates that CRT can reverse Cys crosslinks and induce SNO modification. However, the extent of crosslink reversal is more than can be accounted for by induction of SNO modification, while the overall ATPase activity is recovered in CRT dogs.
Supplementary Material
Novelty and significance.
What Is Known?
Cardiac resynchronization therapy (CRT) has become an effective clinical intervention for dyssynchronous heart failure (DHF) patients leading to improvement in heart function, clinical symptoms and survival.
Mitochondrial ATP synthase function is attenuated in HF and is restored at least in part by CRT.
CRT affects the mitochondrial subproteome by specifically altering proteins that control the cellular redox state and oxidative phosphorylation (OxPhos) pathways, as manifested by changes in both protein quantity and post-translational modifications (PTM) within the mitochondria.
What New Information Does This Article Contribute?
ATP synthase undergoes a number of cysteine-specific oxidative PTMs, disulfide bond formation, S-glutathionylation and S-nitrosation in the failing heart.
CRT reverses disulfide bond formation and S-Glu but induces S-nitrosation
A specific Cys residue (ATP synthase alpha subunit Cys294) competes for disulfide bond formation, glutathionylation and nitrosation and thus, may be a “redox switch” sensing redox status and altering ATP synthase function
CRT is a clinically effective treatment for DHF; however, the molecular mechanisms underlying the beneficial effects of CRT remain largely unknown. We show that in failing hearts ATPα undergoes specific oxidative modifications by three different Ox-PTM: intermolecular disulfide bonds, S-glutathionylation and nitrosation These modifications can occur at Cys294, suggesting that in comparison with other cysteine residues present in ATP synthase, this amino acid has a high redox sensitivity. This implies that ATPα Cys294 acts as a redox switch that senses the redox potential of the local cellular environment, thereby regulating ATPase activity. With CRT, antioxidant protective systems are enhanced, and under these conditions, the disulfide bonds are reversed and S-glutathionylation decreases, resulting in greatly improved ATPase activity. Remarkably, CRT hearts appear to “sense” the improvement in the redox environment and respond by activating NO signaling and thereby inducing S-nitrosylation of Cys294, which may be potentially protective.
Acknowledgments
Source of funding
This work was supported by National Heart, Lung, and Blood Institute PPG on Pathology of Cardiac Dyssynchrony and Resynchronization P01HL77189-01 to J.E.V.E., B. O’R., and D.A.K.
Non-standard Abbreviations and Acronyms
- 2D BNP/SDS-PAGE
two dimensional BNP/SDS-polyacrylamide gel electrophoresis
- ATPα
ATP synthase alpha subunit
- ATPβ
ATP synthase beta subunit
- ATPγ
ATP synthase gamma subunit
- BNP
blue Native polyacrylamide gel electrophoresis
- CNP
clear native polyacrylamide gel electrophoresis
- CRT
cardiac resynchronization therapy
- Cys
cysteine
- DB71
Direct Blue 71
- DHF
dyssynchronous heart failure
- DTPA
diethylenetriaminepentaacetic acid
- DTT
dithiothreitol
- HBSS
Hank’s Buffered Salt Solution
- H2O2
hydrogen peroxide
- LC-MS/MS
liquid chromatography-tandem mass spectrometry
- LV
left ventricular
- MS
mass spectrometry
- NEM
N-ethylmaleimide
- OCR
Oxygen consumption rate
- OSCP
ATP synthase oligomycin sensitivity conferral protein
- Ox-PTM
oxidative post-translational modifications
- PBS
phosphate buffered saline
- PTM
post-translational modifications
- PVDF
polyvinylidene fluoride
- ROS/RNS
reactive oxygen/nitrogen species
- Ser
serine
- S-Glu
S-glutathionylation
- SNO
S-nitrosation
- TBS
tris buffered saline
Footnotes
Disclosure
None
References
- 1.Kass DA. Pathobiology of cardiac dyssynchrony and resynchronization. Heart Rhythm. 2009;6:1660–1665. doi: 10.1016/j.hrthm.2009.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chakir K, Kass DA. Rethinking Resynch: Exploring Mechanisms of Cardiac Resynchroniztion Beyond Wall Motion Control. Drug Discov Today Dis Mech. 2010;(2):e103–e107. doi: 10.1016/j.ddmec.2010.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. doi: 10.1161/CIRCGENETICS.109.871236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paradies G, Petrosillo G, Paradies V, Ruggiero FM. Oxidative stress, mitochondrial bioenergetics, and cardiolipin in aging. Free Radic Biol Med. 15(48):1286–1295. doi: 10.1016/j.freeradbiomed.2010.02.020. [DOI] [PubMed] [Google Scholar]
- 5.Wen JJ, Garg N. Oxidative modification of mitochondrial respiratory complexes in response to the stress of Trypanosoma cruzi infection. Free Radic Biol Med. 2004;37:2072–2081. doi: 10.1016/j.freeradbiomed.2004.09.011. [DOI] [PubMed] [Google Scholar]
- 6.Giordano FJ. Oxygen, oxidative stress, hypoxia, and heart failure. J Clin Ivest. 2005;115:500–508. doi: 10.1172/JCI200524408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Je JH, Lee TH, Kim DH, Cho YH, Lee JH, Kim SC, Lee SK, Lee J, Lee MG. Mitochondrial ATP synthase is a target for TNBS-induced protein carbonylation in XS-106 dendritic cells. Proteomics. 2008;8:2384–2393. doi: 10.1002/pmic.200700962. [DOI] [PubMed] [Google Scholar]
- 8.Nojiri H, Shimizu T, Funakoshi M, Yamaguchi O, Zhou H, Kawakami S, Ohta Y, Sami M, Tachibana T, Ishikawa H, Kurosawa H, Kahn RC, Otsu K, Shirasawa T. Oxidative stress causes heart failure with impaired mitochondrial respiration. J Biol Chem. 2006;281:33789–33801. doi: 10.1074/jbc.M602118200. [DOI] [PubMed] [Google Scholar]
- 9.Foster DB, Van Eyk JE, Marbán E, O’Rourke B. Redox signaling and protein phosphorylation in mitochondria: progress and prospects. J Bioenerg Biomembr. 2009;41:159–168. doi: 10.1007/s10863-009-9217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ying J, Clavreul N, Sethuraman M, Adachi T, Cohen RA. Thiol oxidation in signaling and response to stress: detection and quantification of physiological and pathophysiological thiol modifications. Free Radic Biol Med. 2007;43:1099–1108. doi: 10.1016/j.freeradbiomed.2007.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Burwell LS, Brookes PS. Mitochondria as a target for the cardioprotective effects of nitric oxide in ischemia-reperfusion injury. Antioxid Redox Signal. 2008;10:579–599. doi: 10.1089/ars.2007.1845. [DOI] [PubMed] [Google Scholar]
- 12.Kim Y, Konno H, Sugano Y, Hisabori T. Redox regulation of rotation of the cyanobacterial F1-ATPase containing thiol regulation switch. J Biol Chem. 2011;286:9071–9078. doi: 10.1074/jbc.M110.200584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shen H, Walters DE, Mueller DM. Introduction of the chloroplast redox regulatory region in the yeast ATP synthase impairs cytochrome c oxidase. J Biol Chem. 2008;283:32937–32943. doi: 10.1074/jbc.M805310200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Skalska J, Bernstein S, Brookes P. Measurement of extracellular (exofacial) versus intracellular protein thiols. Methods Enzymol. 2010;474:149–164. doi: 10.1016/S0076-6879(10)74009-X. [DOI] [PubMed] [Google Scholar]
- 15.Wittig I, Karas M, Schägger H. High resolution clear native electrophoresis for in-gel functional assays and fluorescence studies of membrane protein complexes. Mol Cell Proteomics. 2007;6:1215–1225. doi: 10.1074/mcp.M700076-MCP200. [DOI] [PubMed] [Google Scholar]
- 16.Schagger H, von Jagow G. Blue native electrophoresis for isolation of membrane protein complexes in enzymatically active form. Anal Biochem. 1991;199:223–231. doi: 10.1016/0003-2697(91)90094-a. [DOI] [PubMed] [Google Scholar]
- 17.Murray CI, Kane LA, Uhrigshardt H, Wang SB, Van Eyk JE. Site-Mapping of in vitro S-nitrosation in cardiac citochondria: implications for cardioprotection. Mol Cell Proteomics. 2011;10:M110.004721. doi: 10.1074/mcp.M110.004721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi SW, Gerencser AA, Nicholls DG. Bioenergetic analysis of isolated cerebrocortical nerve terminals on a microgram scale: Spare respiratory capacity and stochastic mitochondrial failure. J Neurochem. 2009;109:1179–1191. doi: 10.1111/j.1471-4159.2009.06055.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gundry RL, White MY, Murray CI, Kane LA, Fu Q, Stanley BA, Van Eyk JE. Preparation of proteins and peptides for mass spectrometry analysis in a bottom-up proteomics workflow. Curr Protoc Mol Biol. 2009;Chapter10(Unit10):25. doi: 10.1002/0471142727.mb1025s88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohr MJ, Aponte AM, Sun J, Wang G, Murphy E, Gucek M, Steenbergen C. Characterization of Potential S-nitrosylation Sites in the Myocardium. Am J Physiol Heart Circ Physiol. 2011;300:H1327–H1335. doi: 10.1152/ajpheart.00997.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gibbons C, Montgomery MG, Leslie AG, Walker JE. The structure of the central stalk in bovine F(1)-ATPase at 2.4 A resolution. Nat Struct Biol. 2000;7:1055–1061. doi: 10.1038/80981. [DOI] [PubMed] [Google Scholar]
- 22.Di Liegro CM, Bellafiore M, Izquierdo JM, Rantanen A, Cuezva JM. 3′-untranslated regions of oxidative phosphorylation mRNAs function in vivo as enhancers of translation. Biochem J. 2000;352:109–115. [PMC free article] [PubMed] [Google Scholar]
- 23.Ohsakaya S, Fujikawa M, Hisabori T, Yoshida M. Knockdown of DAPIT (Diabetes-associated Protein in Insulin-sensitive Tissue) Results in Loss of ATP Synthase in Mitochondria. J Biol Chem. 2011;286:20292–20296. doi: 10.1074/jbc.M110.198523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dittrich M, Schulten K. Zooming in on ATP hydrolysis in F1. J Bioenerg Biomembr. 2005;37:441–444. doi: 10.1007/s10863-005-9487-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wittig I, Velours J, Stuart R, Schägger H. Characterization of domain interfaces in monomeric and dimeric ATP synthase. Mol Cell Proteomics. 2008;7:995–1004. doi: 10.1074/mcp.M700465-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Bornhövd C, Vogel F, Neupert W, Reichert AS. Mitochondrial membrane potential is dependent on the oligomeric state of F1F0-ATP synthase supracomplexes. J Biol Chem. 2006;281:13990–13998. doi: 10.1074/jbc.M512334200. [DOI] [PubMed] [Google Scholar]
- 27.Benoit R, Auer M. A direct way of redox sensing. RNA Biol. 2011;8:18–23. doi: 10.4161/rna.8.1.13555. [DOI] [PubMed] [Google Scholar]
- 28.Seth D, Stamler JS. The SNO-proteome: causation and classifications. Curr Opin Chem Biol. 2011;15:129–136. doi: 10.1016/j.cbpa.2010.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Donoso P, Sanchez G, Bull R, Hidalgo C. Modulation of cardiac ryanodine receptor activity by ROS and RNS. Front Biosci. 2011;16:553–567. doi: 10.2741/3705. [DOI] [PubMed] [Google Scholar]
- 30.Garcia J, Han D, Sancheti H, Yap LP, Kaplowitz N, Cadenas E. Regulation of mitochondrial glutathione redox status and protein glutathionylation by respiratory substrates. J Biol Chem. 2010;285:39646–39654. doi: 10.1074/jbc.M110.164160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Okazaki T, Otani H, Shimazu T, Yoshioka K, Fujita M, Katano T, Ito S, Iwasaka T. Reversal of inducible nitric oxide synthase uncoupling unmasks tolerance to ischemia/reperfusion injury in the diabetic rat heart. J Mol Cell Cardiol. 2011;50:534–544. doi: 10.1016/j.yjmcc.2010.12.010. [DOI] [PubMed] [Google Scholar]
- 32.Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- 33.Lin J, Steenbergen C, Murphy E, Sun J. Estrogen receptor-beta activation results in S-nitrosylation of proteins involved in cardioprotection. Circulation. 2009;21(120):245–254. doi: 10.1161/CIRCULATIONAHA.109.868729. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.