

Abstract
Proteomic technologies are used to study the complexity of proteins, their roles and biological functions. It is based on the premise that the diversity of proteins, comprising their isoforms, and post translational modifications (PTMs) underlies biology. Based on an annotated human cardiac proteins 62 % have at least one PTM (phosphorylation currently dominating) while ~25% have more than one type of modification. The field of proteomics strives to observe and quantify this protein diversity. It represents a broad group of technologies and methods arising from analytical protein biochemistry, analytical separation, mass spectrometry and bioinformatics. Since the 1990s the application of proteomic analysis has been increasingly used in cardiovascular research. Technology development and adaptation has been at the heart of this progress. Technology undergoes a maturing becoming routine and ultimately obsolete being replaced by newer methods. Due to extensive methodological improvements, many proteomic studies today observe 1000-5000 proteins. Only five years ago this was not feasible. Even so, there are still road blocks. Nowadays, there is a focus on obtaining better characterization of protein isoforms and specific PTMs. Consequently, new techniques for identification and quantification of modified amino acid residues are required, as is the assessment of SNPs in addition to determination of the structural and functional consequences. In this series, four articles provide concrete examples of how proteomics can be incorporated into cardiovascular research and address specific biological questions. They also illustrate how novel discoveries can be made and how proteomic technology has continued to evolve.
Keywords: Proteomics, technology, protein isoform, posttranslational modification, polymorphorism
In this accompanying series, we explore how proteomics has matured and its application in cardiovascular research. Cardiovascular science investigates and analyzes the biological systems involved in prevention, treatment and development of disease. It is often aimed at discovering the fundamental principles that govern or underlie the molecular disease pathways. To achieve this aim, there has been and is a constant need of technology development. In this context, technology refers to techniques, instrumentation and approaches/strategies that are used to solve problems that are involved in research. Each technology must, therefore, have a particular utility and use. Proteomics is a field that has been driven by the development and strategic application of technology and it continues to rapidly evolve. Proteomic technologies are used to study the complexity of proteins, their biological roles including biophysical properties, structure and function. It is based on the premise that the diversity of proteins, comprising their isoforms, SNPs, and post translational modifications (PTMs), underlies biology. Protein alterations occurring due to physiological and pathological processes are reflective of changes at the gene, mRNA, miRNA and metabolite levels. Thus, the proteome comprises information from protein expression, PTMs, processing and turnover, localization and time. PTMs alter the chemical nature of an amino acid residue; which can induce changes in protein structure, activity, binding partners or subcellular localization. PTMs can add or remove a functional group (including a peptide) to a specific amino acid residue within a protein. PTMs include acetylation, amidation, methylation, sumolyation, ubiquitination, prenylation, myristoylatoin, S-glutathiolation, sulfation, N- and O-linked glycosylation, glycation, S-nitrosylation (SNO, S-nitrosation) proteolysis and phosphorylation among others. The field of proteomics strives to observe and quantify this protein diversity. It represents a broad group of technologies and methods arising from analytical protein biochemistry, analytical separation, mass spectrometry and bioinformatics. Many technologies are synergistic and complement each other providing different data for each proteome. Proteomic technology development can be linked to specific biological and clinical questions (Figure 1). This is a reiterative situation as the application of different technologies can provide additional insights and generate new hypotheses, which is often assisted by additional methodological refinement and optimization or even creation of new technology..
Figure 1. Schema representing the relations that are involved in discovery.

Biological or clinical questions drive the development of new technologies which, when applied, will generate new hypothesis. All technology undergoes an evolution from development to finally becoming obsolete and being replaced by new or refined methods.
i) The process of technology development
Technology development is a multiple step process (Figure 1). These steps are the i) development phase, where the method or technology is born from trial and error experimentation; ii) prototype production, where the consistency and variability is determined; iii) early mass production, where the method requires minimal changes, but is tested on a number of different situations or conditions allowing for fine tuning; iv) regular production with standardization, where problems only rarely are experienced; v) established method, where the wide scientific community are allowed robust use of the technology; and vi) obsolete, where the technology is no longer required or has been replaced with new methods that provides ”better” data (e.g. qualitative vs quantitative). Often the application of technology requires the establishment of a technical pipeline representing a number of distinct processes that are linked in order to solve a problem. Of equal value is the adaptation of new technology to more complex samples or experimental conditions. Most often methods are initially developed using simple standards such as purified peptides or proteins. The technology then must be vetted in samples of increasing complexity. Moving the technology to a cell subproteome, intact cell or tissue can be extremely time-consuming and require considerable additional experimentation at the prototype or early mass production stages. This can also be true when changing among different subproteomes, cells and tissue types (e.g. HeLa cells vs cardiac myocytes; cardiac muscle vs. liver; mouse vs. human). Proteomic analysis of cardiac myocytes and tissue are particularly challenging compared to many other tissues, and the reasons for this are multi-factorial. Chief among them is the fact that the cardiac muscle has a large dynamic range due to dominance of the myofilament and mitochondrial subproteomes, and this generally requires extensive fractionation or targeted enrichment to observe low abundant proteins [e.g. 1-10]. This unique aspect of cardiac muscle is due to its intrinsic physiology – the sustained, intense energetic requirements for continuous contractile activity, and the need for a minute to minute adaptation of heart rate, blood pressure and cardiac output. Another unique aspect of the myocyte proteome is the large number of cardiac-specific isoforms and their regulation by different PTMs. An example illustrating the biological importance of protein isoforms is that of cardiac troponin I (cTnI) which regulates muscle contraction/relaxation, but unlike the skeletal muscle isoforms is phosphorylated by protein kinase A and other kinases [e.g 11-14]. As well, it has been shown to undergo other types of PTM (O-GlcNAcylation and proteolysis) with different pathological conditions [e.g. 15-19]. In many cases, a small degree of change to one of these PTMs can result in large physiological alterations (e.g. 15% substitution of the truncated cTnI form results in ~50% reduced maximal force [20]). Since subtle PTM changes can have large functional consequences, it is necessary to develop and use techniques that can assess multiple PTMs, identify and quantify each modified residue and be very accurate (a necessity when quantifying low abundant PTMs). In addition to the problems outlined above, the final issue for cardiac muscle proteomic studies is that there are no dividing cell culture systems for ventricular cardiomyocytes. Approaches dependent upon complete saturation metabolic labeling (like Stable isotope labeling by amino acids in cell culture also known as SILAC [21]) are therefore not possible. To detail cardiac protein changes, unique metabolic labeling strategies or alternative technologies will need to be extended or adapted.
ii) Technology progression in the field of proteomics
Since the mid to late 1990s the application of proteomic analysis has been increasingly used in cardiovascular research. As shown in Figure 2, there has been a continued increase in the publication of papers employing proteomic (or proteome) in Pubmed. Two-dimensional gel electrophoresis (2DE), a traditional protein separation method in proteomics which combines separation of intact proteins by pI and molecular weight, was developed in mid 1970s [22,23] continues to increase, but over the last few years has contributed less toward the total number of proteomic based studies. This is due to the increasing number of other protein and peptide separation methods which have been developed or refined over the last decade along with improvements in the sensitivity and accuracy of mass spectrometers and more comprehensive bioinformatics. 2DE is one of few methods that allow the determination of quantity and potential PTM or isoform status of proteins, (those which differ in the pI of the protein). It has continues to be improved with methods for solubilization and loading of difficult proteins (e.g. hydrophobic or basic) onto the gel, more sensitive protein stains and labeling that allow simultaneously analysis of multiple samples, and improved image alignment and quantification (reviews [24-27]). Other intact protein separation methods involving liquid phases (free flow electrophoresis), capillary electrophoresis and liquid chromatography (LC), have similar benefits as 2DE, and interestingly, often observe a different set of proteins (reviews [28-31]). This is due to the solubilization or separation parameters. By using more than one method in a multi-dimensional arrangement (off-line or in-line with mass spectrometry (MS)) an investigator can increase proteome coverage (number of proteins observed) and often protein coverage (number of peptides observed for each protein). As examples from our group we have compared a number of different protein and peptide separation methods [1,9,32,33] and in some cases there is only < 30% overlap in the proteins observed between methods. Today, many studies observe and quantify 1000-5000 proteins, while 5 years ago this was not feasible even with extensive separation methods. To date, the type of data that proteomics can help achieving can be broken into 1) protein identification (which allow ones to create a database in order to determine the protein composition of a specific protein complex, organelle or cell); 2) protein quantification (either determining the relative ratio or absolute concentration); 3) identification of protein isoforms; 4) determining if a protein has a PTM, the type of PTM and identifying the specific modified amino acid residues and finally, 5) quantification of each modified residue. Certainly, the level of technical difficulty increases as one move up to more specific and quantifiable data.
Figure 2. The number of papers published per year using proteomics in the area of cardiac and heart research has been steadily increasing.
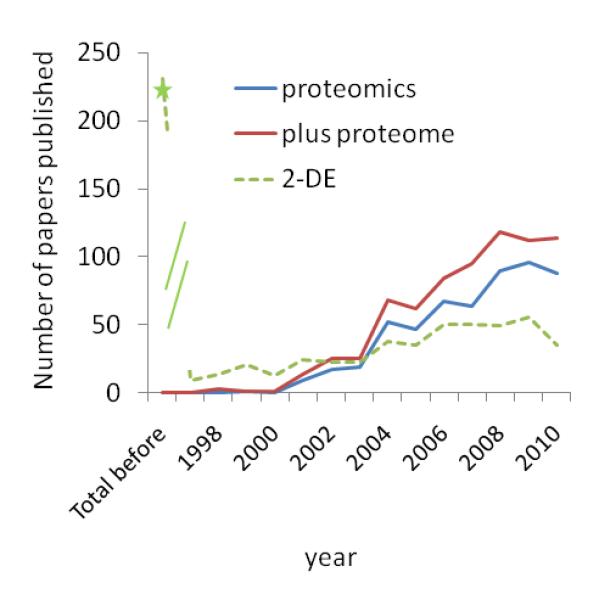
Pubmed was searched using i) “proteomics” alone or with “proteome” and ii) either “heart or cardiac” as subjects. Papers that were not in English or were cited as reviews were excluded from the total count. There were no papers prior to 1998. Note, however, that when a search was carried out where “proteomics/proteome” was replaced with 2DE papers earlier than 1998 were identified.
iii) Protein identification and Quantification
Protein identification is at the heart of most proteomics. This can be accomplished using MS or protein-specific capture/detecting regents like antibodies. Antibody detection and quantification of intact proteins has also been improving with the large scale production of antibodies against human proteins by both industry and academic. Of note is the human protein atlas (http://www.proteinatlas.org/ [34,35]) which currently has more than 13,150 antibodies which recognize gene products from 10,100 human genes. The use of antibody arrays (reversed or direct) provide the ability to screen for ever increasing numbers of proteins while the opposite, antigen arrays can allow detection of auto-antibodies (review [36,37]). This work has stimulated the produce new generation affinity (capture and sensor(detecting)) reagents including peptoids and aptimers (e.g. [38,39]).
MS is also a commonly used method that directly analyzes proteins or the respective peptides obtained via either enzymatic or chemical digestion. It is, however, the resulting peptides that provide amino acid sequence data. Although it is often possible to deduce sequence information directly from the spectra, currently the most common data analyses pipelines include comparing the experimental spectra to those derived from a theoretical digestion (i.e. in silico) of a selected protein database by way of a particular “search engine” algorithm (e.g. Mascott, XTandem, Sequest, OSSMA). To ensure rigorous standards of data analysis and reporting, a number of important issues should be considered. First, the mass accuracy of the data affects the reliability of the identifications; therefore the type of MS instrumentation used needs to be taken into consideration. Second, there are many different data search engine algorithms available, and each algorithm is likely to produce a different result from the same data. Third, many search engines will make some attempt to assign sequence information to poor quality spectra, which can result in incorrect identifications. Fourth, it is critical to select a protein database appropriate for the sample. Databases can vary with respect to accuracy and completeness as well as contain protein name redundancy whereby the same amino acid sequence may be represented by multiple accession numbers. Fifth, data clarity can be compromised if a spectrum can be assigned to multiple amino acid sequences, due to ambiguity, or if a peptide sequence can be assigned to multiple proteins, due to homology. Consequently, analysis of MS data and assignment of peptide/protein identification can be difficult.
Protein quantification is an important aspect of a proteomic analysis. There are essentially two types of quantification that is currently used in proteomics - relative ratio and absolute quantities. For the former, 2DE is most often used for proteins while quantitation at the peptide level is achieved using metabolic (SILAC, on proliferating cells) or chemical labeling (most often labeling of the digested peptides include isobaric tag for relative and absolute quantification (also called iTRAQ [40] and tandem mass tags (also called TMT) [41] or label free (based on spectral counts) [42]. Comparison is made between two or more samples allowing the relative quantitative relation between them to be determined. On the other hand, absolute quantification means that the precise value of each peptide or protein in a sample is determined independent of other samples. Previously, to obtain absolute quantitation of a protein, we were forced to rely on ELISA or quantitative western blots, both of which require high-quality antibodies. When high quality antibodies are available, these methods are very valuable. But this is rare (although becoming less rare due to the large scale efforts in reagent production as discussed previously) in research where candidate proteins arise from proteomic studies, or when specific protein isoforms (or specific modified amino acids) need to be investigated. With the recent adoption of multiple reaction monitoring (MRM), it is possible to obtain absolute quantification (relative ratios can also be determined) of target proteins without the need for antibodies. This MS method is a long-established technology in the field of metabolite quantification, but has only in the last few years been optimized for the measurement of larger molecules, including peptides [e.g 43-46]. In essence, a proteotypic peptide (peptides with unique amino acid sequences to a particular protein isoform) that results from an enzymatic digestion of the sample (e.g. trypsin) is specifically selected within the MS instrument. MRM enhances: i) protein studies across different species; ii) analysis when sample size is limited; iii) the study of proteins which have no (or poor quality) antibodies; iv) simultaneous quantification of multiple proteins (note upwards of 40 proteins in a single analysis [47]; v) quantification of protein isoforms; and vi) quantification of site-specific PTMs within one or more proteins [review comparing ELISA and MRM see [48]).
iv) Targeting the analysis of PTMs
The area of PTM proteomics is challenging. Many PTMs are transient or labile with respect to MS analysis and are often present in low abundance, resulting in incomplete amino acid sequence coverage and a failure to fully characterize the PTMs. Biological complexity further impedes analysis; a single protein can have a number of amino acid residues that are modified, comprising one or different types of PTMs. Furthermore, each modified residue may have a different effect on function. Quantitative data on each modified amino acid residue are needed if we are to understand the dynamic changes of the proteome, the crosstalk and selectivity of different PTMs, and their impacts on signaling cascades. Many useful methods for PTM analysis have the following characteristics: i) utilize enrichment of low abundance modified proteins (or peptides), ii) simultaneously identify and quantify a protein, iii) identify the actual modified amino acid residue, iv) quantify the extent of a PTM under different conditions and v) quantify the relative differences of a particular modified residue in a protein with multiple modified residues. Most PTMs do not have such methods developed yet. Even so, progress has been made – either through the use of multiple methods each allowing one to observe more of the PTMs proteome or via a continuous refinement, often in incremental steps. Phosphorylation is an example where multiple synergistic approaches have been developed [reviews 49-51]. It is the (by far) most common PTM described today (Figure 3). This PTM is of particular interest to the broader scientific community. The analysis of phospho-proteins can be done by multiple approaches – each with their own advantages and disadvantages. Often 2DE is used, as a change in pI of a protein in the first dimension can be indicative of phosphorylation (or dephosphorylation) due to the addition (or loss) of a charged phosphate group. However, a shift in pI can be due to a number of PTMs and hence a secondary set of experiments is required to prove that the protein is phosphorylated. The treatment of the sample prior to 2DE with a phosphatase and monitoring the reverse shift in pI can be one unambiguous approach [e.g. 52,53]. However, a particular phosphatase (even those with broad specificity like alkaline phosphatases) does not dephosphorylate all proteins. Thus, if a protein does not show a shift after treatment this does not mean that the protein is not phosphorylated. ProQ diamond is a protein stain that displays enhanced binding to negatively charged proteins, like those which are phosphorylated [54]. However, false positives can occur thus secondary proof is required. The new phos-tag one dimensional gels can retard phosphorylated proteins which can provide insight in to extent of modification (most often based on western blots)[55]. None of these protein based methods provide information about the actual phosphorylated amino acid residues. That requires additional MS analysis. To determine phospho-sites there are also multiple techniques: direct analysis based on neutral loss and the phospho-peptide enrichment methods (immobilized metal affinity chromatography and TiO2 affinity chromatography, among other). Importantly, each method only sees a subset of phospho-peptides. This means that it is difficult to unambiguously detect all phosphorylation sites in either an isolated protein or a complex protein mixture. Over the last few years, there has been considerable work defining the phosphoproteome within some of the subproteomes of cardiac muscle. This includes extensive analysis of the myofilament [56], 20S proteasome [57] and the mitochondria using both direct targeting of phospho-peptides [58] or proteins [e.g.59-61]. However, none of these methods provide quantitative data without additional steps.
Figure 3. The number of annotated human cardiac proteins with one or more different types of PTMs.
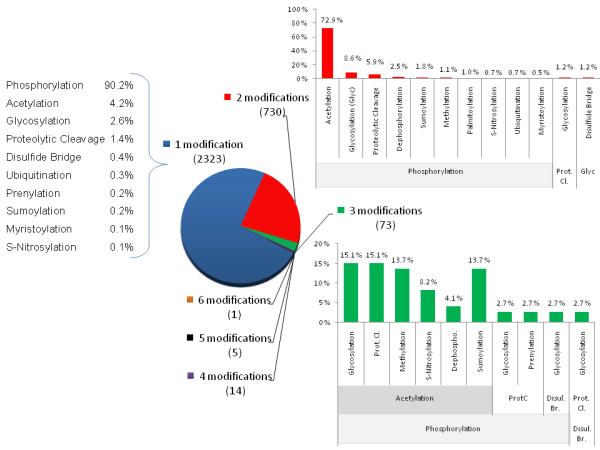
The human protein reference data base (currently contains 30,047 protein entries and 93710 PTM sites) was searched for proteins that are known to be expressed in the heart. For this subgroup of proteins, all the different PTMs annotated were determined (phosphorylation (combined with dephosphorylation); acetylation, glycosylation (N- and O-linked, Glyc), proteolytic cleavage (Prot.Cl.), disulfide bridge (Disul. Br), ubiquitination, prenylation, sumoylation, myristoylation, and SNO). The number of different PTMs were determined and represented as a pie chart. The percentage of each PTM for those proteins which have only one, two or three types of modifications are shown.
As mentioned above, in some cases, there is a continuous improvement of technology aimed at one particular problem. An example is the PTM, O-GlcNAcylation (O-GlcNAc), which was discovered over twenty-five years ago [63]. Today it is clear that O-GlcNAc rivals protein phosphorylation in terms of abundance, dynamic cycling, and distribution on nuclear, mitochondrial and cytoplasmic proteins [64-70]. Within the heart this PTM has roles in aging, protection and disease (e.g. [71-76]; reviews 77-80]). However, this understanding was slow to be gained as GlcNAcylation is difficult to detect and quantify by standard methods and required continued technology development [81-84}. The current MS-based method can simultaneously quantify site occupancy for both O-GlcNAc and phosphate on hundreds of proteins (recent reviews [85,86]). The interplay between these different types of PTM and the potential to influence the action of each other is becoming a more common theme not only for O-GlcNAc vs phosphorylation but also with other PTMs. For example, interplay between phosphorylation and SNO is also possible [e.g. 87]. SNO modification has been found for a number of phosphatases in a number of systems, although not yet in the heart. To follow up this and other PTM’s interplay in the future, technologies targeting less common PTMs will be essential. It is clear that this is an area of future growth.
v) Proteome complexity
It is generally accepted that the proteome is complex and that some of this complexity arises from PTMs. To grabble with this we have asked two questions “what is the extent of PTMs reported in the human heart” and “how many proteins have more than one type of PTM, a potential indication of signaling system cross talk. We turned to the well annotated Human Protein Reference Database (HPRD, www.hprd.org) [88] that was established by Dr. Akhilesh Pandey (Johns Hopkins University). HPRD involves a large team of biologists, bioinformaticists and software engineers who oversee and manually curate the information based on the literature with frequent reannotation. Our search was limited to only those human proteins which have been shown to be present in the heart. In total there was 5079 human cardiac proteins annotated. 62% of these have at least one documented PTM. For a breakdown regarding the number of proteins with a specific PTM and also heart proteins with specific cellular component/localization see the upcoming review in this series. To investigate the potential for cross talk between different types of signaling pathways that result in PTM, we determined the number of annotated human cardiac proteins with one or more different types of PTMs (Figure 3). Of these, a total of 3146 proteins, 75% have one PTM type. Phosphorylation dominates, accounting for over 90% of all proteins with a single modification type. Not surprisingly this is also true if the protein has more than one type of modification. It is somewhat surprising that acetylation is the next most prominent PTM in cardiac proteins followed by glycosylation (combing both N and O-linked). There are a number of caveats to this information. The first, this database is only for human proteins (note: PTMs observed only in other species are not included). Second, the database is continuously being updated, but only reflects what is present to date. Third, the stage of technology development for the different PTMs and that the information in the HPRD reflects only those proteins for which site information (the actual modified amino acid) is known. There has been continuous development of technology tools for the identification of phosphorylated amino acids residues in part driven by the wide interest in this particular PTM as a regulator of biological processes (as described above in section targeting the analysis of PTMs). This has resulted in several studies identifying large number of phospho-sites in human cells or tissues. For examples, recently 2225 phospho-sites were identified in 1023 proteins isolated from human liver [89] while Mann and colleagues reported more than 20,000 phosphorylation sites in 6027 proteins from HeLa cells [90]. As tools are developed for other PTMs to allow easier determination of the actual modified amino acid residues it will be very interesting to see how these numbers change.
vi) The future expanding proteomic coverage and understanding
Although often inferred in protein identification it is often difficult to distinguish specific protein isoforms, mutant or SNP and polymorphisms. This is most likely due to incomplete detection by the MS (a result of inherent sampling biased, ionizablity of individual peptides and instrument sensitivity thus reducing the coverage of a protein) and that our bioinformatics tools and protein databases have yet to be sufficiently encompassing to allow the specific peptides to be identified. This is needed. In view of the great amount of data being generated from genomic studies (such as genome wide association studies, GWAS) about SNPs and their link to human diseases, proteomic methods must move towards linking with this knowledge. Thus, more sensitive MS instruments, more robust methods to increase coverage (e.g. enhanced protein and peptide separation and use of multiple enzymes) and expanded databases will help. One can envision means to specifically target disease linked SNPs or mutations which would allow for quantification of the different forms. One possibility is MRM, because this is a peptide tagged method. This level of proteome characterization will occur, but the method still needs to develop into the “regular production stage” of development. Already the triple quadruple MS instruments are in clinical laboratories where it is currently used for absolute quantification of metabolites and drugs. Thus, one can see a program development for protein/peptide based quantification of biomarkers in that setting [91]. Yet, what is needed is to also move MRM into regular science research laboratories as a means to replace quantitative western blots and ELISA technology. The driving force will be the ability to greatly expand the number of analytes to quantify and to provide more accurate quantification with a larger dynamic range. To do this will require robust MS instruments that are user friendly and a large number of consumables or kits comprising standard labeled peptides. The purchase of a MRM peptide kit for a particular protein, isoform, mutant, SNP or PTM would be equivalent to the purchase of an antibody.
Equally important to the field of proteomics has been the simultaneous development of methods for large scale protein characterization. These include initiatives in protein structure (e.g. Protein Structure Initiative (PSI) e.g., review [92,93];) and currently it is feasible to determine the 3-dimensional structure of >100 proteins per year. There has also been a push to define protein interacting partners also referred to as the “interactome”. The interactome can target the identification of binding partners to a single protein but also can include the analysis of “complete” signaling cascades (e.g TGF beta [94]), proteins which bind common small signaling molecules (e.g. cAMP, [95]) or specific classes of proteins (e.g. kinase/phosphatases [96]). These studies on protein complexes have increased our understanding of basic protein biochemical properties as well as pushed the field towards broad network analysis (e.g. [97]). Along these same lines, the biophysical protein characterization (e.g. Structural Genomics Consortium (SGC); review [98]) and in vivo cell localization (review [99, 100]) are now being carried out in higher throughput mode although often in model systems.
vii) Conclusions
Proteomics is becoming integrated and incorporated into cardiovascular research and as such will impact new discoveries. In this series, four papers are presented which provide concrete examples of how proteomics can be incorporated into research and address specific biological questions. Mayr and colleagues present how proteomics has allowed a better definition of stem cell populations and their phenotype; Costello and colleagues have summarized the importance of oxidative modifications and their growing role and definition in cardiovascular disease; Agnetti et al. discuss organelles and their interplay with PTM regulation and at the end, Gerzneten and colleagues describe the pipeline required to developed circulating biomarkers with clinical relevance. These papers summarize some novel aspects within cardiovascular research where proteomics has helped to provide solutions and insights into biology which would have been difficult without these approaches. Nonetheless, considerable roadblocks remain to fully integrate proteomics into cardiovascular research and translational medicine As discussed throughout these document these include additional PTM specific mapping tools, routine quantitative methods, better and extensive databases, more sensitive and cost effective instrumentation, to name a few. Furthermore, in order to exploit the proteomic data, we must find ways to integrate the data and the development of models to understand the complexity of the cell and organ. As well, we need better approaches to link between a proteome change and direct function and cell localization in context to health and disease. Excitingly, however, technology does not stand still and it will continue to undergo transformation; with older technologies improving and newer methods being developed. Proteomics research within the cardiovascular field is evolving rapidly and if the past predicts the future, even more insights and novel findings will occur as the broader scientific community adopts and uses proteomics methods.
Acknowledgments
Joe Coresh and Miroslava Stastna for their thoughts, Cathrine Husberg for her thoughts and work analyzing the cardiac proteome documented in the human protein reference database (HPRD), A Pandey for development of HPRD.
Sources of Funding This was funded by NHLBI - HV-10-05 (2); NHLBI-P50 HL 084946-01; and the Clinical Translational Science Award at Johns Hopkins University.
Abbreviations
- 2DE
Two-dimensional gel electrophoresis
- cTnI
cardiac troponin I
- GWAS
genome wide association study
- HPRD
human protein reference database
- ITRAQ
isobaric tag for relative and absolute quantification
- LC
liquid chromatography
- MRM
multiple reaction monitoring
- O-GlcNAc
O-GlcNAcylation
- PTM
post translational modifications
- SILAC
Stable isotope labeling by amino acids in cell culture
- SNP
single nucleotide polymorphorism
- SNO
S-nitrosylation or S-nitrosation
- TMT
Tandem mass tag
Footnotes
Disclosures: The author has no disclosures for this report.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gundry RL, Tchernyshyov I, Sheng S, Tarasova Y, Raginski K, Boheler KR, Van Eyk JE. Expanding the mouse embryonic stem cell proteome: combining three proteomic approaches. Proteomics. 2010;10:2728–32. doi: 10.1002/pmic.201000039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Warren CM, Geenen DL, Helseth DL, Jr, Xu H, Solaro RJ. Sub-proteomic fractionation, iTRAQ, and OFFGEL-LC-MS/MS approaches to cardiac proteomics. J Proteomics. 2010;73:1551–61. doi: 10.1016/j.jprot.2010.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bousette N, Kislinger T, Fong V, Isserlin R, Hewel JA, Emil A, Gramolini AO. Large-scale characterization and analysis of the murine cardiac proteome. J Proteome Res. 2009;8:1887–901. doi: 10.1021/pr800845a. [DOI] [PubMed] [Google Scholar]
- 4.Callipo L, Capriotti AL, Cavaliere C, Gubbiotti R, Samperi R, Laganà A. Evaluation of different two-dimensional chromatographic techniques for proteomic analysis of mouse cardiac tissue. J Proteomics. 2010;73:1551–61. doi: 10.1002/bmc.1487. [DOI] [PubMed] [Google Scholar]
- 5.Duan X, Young R, Straubinger RM, Page B, Cao J, Wang H, Yu H, Canty JM, Qu J. A straightforward and highly efficient precipitation/on-pellet digestion procedure coupled with a long gradient nano-LC separation and Orbitrap mass spectrometry for label-free expression profiling of the swine heart mitochondrial proteome. Proteome Res. 2009;8:2838–50. doi: 10.1021/pr900001t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Drews O, Zong C, Ping P. Exploring proteasome complexes by proteomic approaches. Proteomics. 2007;7:1047–58. doi: 10.1002/pmic.200600574. [DOI] [PubMed] [Google Scholar]
- 7.Gomes AV, Zong C, Edmondson RD, Li X, Stefani E, Zhang J, Jones RC, Thyparambil S, Wang GW, Qiao X, Bardag-Gorce F, Ping P. Mapping the murine cardiac 26S proteasome complexes. Circ Res. 2006;99:362–71. doi: 10.1161/01.RES.0000237386.98506.f7. [DOI] [PubMed] [Google Scholar]
- 8.Neverova I, Van Eyk JE. Application of reversed phase high performance liquid chromatography for subproteomic analysis of cardiac muscle. Proteomics. 2002;2:22–31. [PubMed] [Google Scholar]
- 9.McDonald T, Sheng S, Stanley B, Chen D, Ko Y, Cole RN, Pedersen P, Van Eyk JE. Expanding the subproteome of the inner mitochondria using protein separation technologies: one- and two-dimensional liquid chromatography and two-dimensional gel electrophoresis. Mol Cell Proteomics. 2006;5:2392–411. doi: 10.1074/mcp.T500036-MCP200. [DOI] [PubMed] [Google Scholar]
- 10.Taylor SW, Warnock DE, Glenn GM, Zhang B, Fahy E, Gaucher SP, Capaldi RA, Gibson BW, Ghosh SS. An alternative strategy to determine the mitochondrial proteome using sucrose gradient fractionation and 1D PAGE on highly purified human heart mitochondria. J Proteome Res. 2002;1:451–8. doi: 10.1021/pr025533g. [DOI] [PubMed] [Google Scholar]
- 11.Ouyang Y, Mamidi R, Jayasundar JJ, Chandra M, Dong WJ. Structural and kinetic effects of PAK3 phosphorylation mimic of cTnI(S151E) on the cTnC-cTnI interaction in the cardiac thin filament. J Mol Biol. 2010;400:1036–45. doi: 10.1016/j.jmb.2010.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sumandea MP, Rybin VO, Hinken AC, Wang C, Kobayashi T, Harleton E, Sievert G, Balke CW, Feinmark SJ, Solaro RJ, Steinberg SF. Tyrosine phosphorylation modifies protein kinase C delta-dependent phosphorylation of cardiac troponin I. J Biol Chem. 2008;283:22680–9. doi: 10.1074/jbc.M802396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zabrouskov V, Ge Y, Schwartz J, Walker JW. Unraveling molecular complexity of phosphorylated human cardiac troponin I by top down electron capture dissociation/electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2008;7:1838–49. doi: 10.1074/mcp.M700524-MCP200. [DOI] [PubMed] [Google Scholar]
- 14.Gallon CE. Current techniques for the study of troponin I phosphorylation in human heart. J Muscle Res Cell Motil. 2008;29:169–72. doi: 10.1007/s10974-009-9170-4. [DOI] [PubMed] [Google Scholar]
- 15.McDonough JL, Arrell DK, Van Eyk JE. Troponin I degradation and covalent complex formation accompanies myocardial ischemia/reperfusion injury. Circ Res. 1999;84:9–20. doi: 10.1161/01.res.84.1.9. [DOI] [PubMed] [Google Scholar]
- 16.Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM., Jr Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–7. doi: 10.1161/01.cir.103.16.2035. [DOI] [PubMed] [Google Scholar]
- 17.Ramirez-Correa GA, Jin W, Wang Z, Zhong X, Gao WD, Dias WB, Vecoli C, Hart GW, Murphy AM. O-linked GlcNAc modification of cardiac myofilament proteins: a novel regulator of myocardial contractile function. Circ Res. 2008;103:1354–8. doi: 10.1161/CIRCRESAHA.108.184978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng J, Schaus BJ, Fallavollita JA, Lee TC, Canty JM., Jr Preload induces troponin I degradation independently of myocardial ischemia. Circulation. 2001;103:2035–7. doi: 10.1161/01.cir.103.16.2035. [DOI] [PubMed] [Google Scholar]
- 19.Kositprapa C, Zhang B, Berger S, Canty JM, Jr, Lee TC. Calpain-mediated proteolytic cleavage of troponin I induced by hypoxia or metabolic inhibition in cultured neonatal cardiomyocytes. Mol Cell Biochem. 2000;214:47–55. doi: 10.1023/a:1007160702275. [DOI] [PubMed] [Google Scholar]
- 20.Murphy AM, Kögler H, Georgakopoulos D, McDonough JL, Kass DA, Van Eyk JE, Marbán E. Transgenic mouse model of stunned myocardium. Science. 2000;287:488–91. doi: 10.1126/science.287.5452.488. [DOI] [PubMed] [Google Scholar]
- 21.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, Mann M. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–86. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 22.MacGillivray AJ, Rickwood D. The heterogeneity of mouse-chromatin nonhistone proteins as evidenced by two-dimensional polyacrylamide-gel electrophoresis and ion-exchange chromatography. Eur J Biochem. 1974;41:181–90. doi: 10.1111/j.1432-1033.1974.tb03258.x. [DOI] [PubMed] [Google Scholar]
- 23.O’Farrell PH. High resolution two-dimensional electrophoresis of proteins. J Biol Chem. 1975;250:4007–21. [PMC free article] [PubMed] [Google Scholar]
- 24.Rabilloud T, Chevallet M, Luche S, Lelong C. Two-dimensional gel electrophoresis in proteomics: past, present and future. J Proteomics. 2010;73:2064–77. doi: 10.1016/j.jprot.2010.05.016. [DOI] [PubMed] [Google Scholar]
- 25.Chevalier F. Highlights on the capacities of “Gel-based” proteomics. Proteome Sci. 2010;8:23–26. doi: 10.1186/1477-5956-8-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rabilloud T. Variations on a theme: changes to electrophoretic separations that can make a difference. J Proteomics. 2010;73:1562–72. doi: 10.1016/j.jprot.2010.04.001. [DOI] [PubMed] [Google Scholar]
- 27.Friedman DB, Hoving S, Westermeier R, Westermeier R, Schickle H. The current state of the art in high-resolution two-dimensional electrophoresis. Methods Enzymol. 2009;463:305–28. [Google Scholar]
- 28.Neverova and Van Eyk Role of chromatographic techniques in proteomic analysis. J Chromatogr B Analyt Technol Biomed Life Sci. 2005;815:51–63. doi: 10.1016/j.jchromb.2004.11.009. [DOI] [PubMed] [Google Scholar]
- 29.Horvatovich P, Hoekman B, Govorukhina N, Bischoff R. Multidimensional chromatography coupled to mass spectrometry in analysing complex proteomics samples. J Sep Sci. 2010;33:1421–37. doi: 10.1002/jssc.201000050. [DOI] [PubMed] [Google Scholar]
- 30.Tomás R, Klepárník K, Foret F. Multidimensional liquid phase separations for mass spectrometry. J Sep Sci. 2008;31:1964–79. doi: 10.1002/jssc.200800113. [DOI] [PubMed] [Google Scholar]
- 31.Mitulovic G, Mechtler K. HPLC techniques for proteomics analysis--a short overview of latest developments. Brief Funct Genomic Proteomic. 2006;5:249–60. doi: 10.1093/bfgp/ell034. [DOI] [PubMed] [Google Scholar]
- 32.Guo Y, Ma SF, Grigoryev D, Van Eyk J, Garcia JG. 1-DE MS and 2-D LC-MS analysis of the mouse bronchoalveolar lavage proteome. Proteomics. 2005;5:4608–24. doi: 10.1002/pmic.200500052. [DOI] [PubMed] [Google Scholar]
- 33.Sheng S, Chen D, Van Eyk JE. Multidimensional liquid chromatography separation of intact proteins by chromatographic focusing and reversed phase of the human serum proteome: optimization and protein database. Mol Cell Proteomics. 2006;5:26–34. doi: 10.1074/mcp.T500019-MCP200. [DOI] [PubMed] [Google Scholar]
- 34.Berglund L, Björling E, Oksvold P, Fagerberg L, Asplund A, Al-Khalili Szigyarto C, Persson A, Ottosson J, Wernérus H, Nilsson P, Lundberg E, Sivertsson A, Navani S, Wester K, Kampf C, Hober S, Pontén F, Uhlén M. A gene-centric human protein atlas for expression profiles based on antibodies. Mol Cell Proteomics. 2008:2019–27. doi: 10.1074/mcp.R800013-MCP200. [DOI] [PubMed] [Google Scholar]
- 35.Nilsson P, Paavilainen L, Larsson K, Odling J, Sundberg M, Andersson AC, Kampf C, Persson A, Al-Khalili Szigyarto C, Ottosson J, Björling E, Hober S, Wernérus H, Wester K, Pontén F, Uhlen M. Towards a human proteome atlas: high-throughput generation of mono-specific antibodies for tissue profiling. Proteomics. 2005 Nov;5:4327–37. doi: 10.1002/pmic.200500072. [DOI] [PubMed] [Google Scholar]
- 36.Voshol H, Ehrat M, Traenkle J, Bertrand E. van Oostrum Antibody-based proteomics: analysis of signaling networks using reverse protein arrays. J.FEBS. 2009;276:6871–9. doi: 10.1111/j.1742-4658.2009.07395.x. [DOI] [PubMed] [Google Scholar]
- 37.Sharp V, Utz PJ. Technology insight: can autoantibody profiling improve clinical practice? Nat Clin Pract Rheumatol. 2007;3:96–103. doi: 10.1038/ncprheum0404. [DOI] [PubMed] [Google Scholar]
- 38.Uhlén M, Hober S. Generation and validation of affinity reagents on a proteome-wide level. J Mol Recognit. 2009;22:57–64. doi: 10.1002/jmr.891. [DOI] [PubMed] [Google Scholar]
- 39.Kodadek T. Synthetic receptors with antibody-like binding affinities. Curr Opin Chem Biol. 2010 doi: 10.1016/j.cbpa.2010.07.001. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]; Colas P. The eleven-year switch of peptide aptamers. J Biol. 2008;7:2–6. doi: 10.1186/jbiol64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ross PL, Huang YN, Marchese JN, Williamson B, Parker K, Hattan S, Khainovski N, Pillai S, Dey S, Daniels S, Purkayastha S, Juhasz P, Martin S, Bartlet-Jones M, He F, Jacobson A, Pappin DJ. Multiplexed protein quantitation in Saccharomyces cerevisiae using amine-reactive isobaric tagging reagents. Mol. Cell. Proteomics. 2004;3:1154–69. doi: 10.1074/mcp.M400129-MCP200. [DOI] [PubMed] [Google Scholar]
- 41.Thompson A, Schäfer J, Kuhn K, Kienle S, Schwarz J, Schmidt G, Neumann T, Johnstone R, Mohammed AK, Hamon C. Tandem mass tags: a novel quantification strategy for comparative analysis of complex protein mixtures by MS/MS. Anal. Chem. 2003;75:1895–904. doi: 10.1021/ac0262560. [DOI] [PubMed] [Google Scholar]
- 42.Bantscheff M, Schirle M, Sweetman G, Rick J, Kuster B. Quantitative mass spectrometry in proteomics: a critical review. Anal. Bioanalytical Chem. 2007;389:1017–31. doi: 10.1007/s00216-007-1486-6. [DOI] [PubMed] [Google Scholar]
- 43.Kitteringham NR, Jenkins RE, Lane CS, Elliott VL, Park BK. Multiple reaction monitoring for quantitative biomarker analysis in proteomics and metabolomics. J Chromatogr B Analyt Technol Biomed Life Sci. 2008;877:1229–39. doi: 10.1016/j.jchromb.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 44.Fortin T, Salvador A, Charrier JP, Lenz C, Lacoux X, Morla A, Choquet-Kastylevsky G, Lemoine J. Clinical quantitation of prostate specific antigen biomarker in the low nanogram/mL range by conventional bore liquid chromatography-tandem mass spectrometry (MRM) coupling and correlation with ELISA tests. Mol Cell Proteomics. 2009;8:1006–15. doi: 10.1074/mcp.M800238-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeSouza LV, Taylor AM, Li W, Minkoff MS, Romaschin AD, Colgan TJ, Siu KW. Multiple reaction monitoring of mTRAQ-labeled peptides enables absolute quantification of endogenous levels of a potential cancer marker in cancerous and normal endometrial tissues. J Proteome Res. 2008;7:3525–34. doi: 10.1021/pr800312m. [DOI] [PubMed] [Google Scholar]
- 46.Kuhn E, Wu J, Karl J, Liao H, Zolg W, Guild B. Quantification of C-reactive protein in the serum of patients with rheumatoid arthritis using multiple reaction monitoring mass spectrometry and 13C-labeled peptide standards. Proteomics. 2004;4:1175–86. doi: 10.1002/pmic.200300670. [DOI] [PubMed] [Google Scholar]
- 47.Anderson L, Hunter CL. Quantitative mass spectrometric multiple reaction monitoring assays for major plasma proteins. Mol Cell Proteomics. 2006;5:573–88. doi: 10.1074/mcp.M500331-MCP200. [DOI] [PubMed] [Google Scholar]
- 48.Fu Q, Schoenhoff FS, Savage WJ, Zhang P, Van Eyk JE. Multiplex assays for biomarker research and clinical application: Translational science coming of age. Proteomics Clin Appl. 2010;4:271–284. doi: 10.1002/prca.200900217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Palmisano G, Thingholm TE. Strategies for quantitation of phosphoproteomic data. Expert Rev Proteomics. 2010;7:439–56. doi: 10.1586/epr.10.19. [DOI] [PubMed] [Google Scholar]
- 50.Grimsrud PA, Swaney DL, Wenger CD, Beauchene NA, Coon JJ. Phosphoproteomics for the masses. ACS Chem Biol. 2010;5:105–19. doi: 10.1021/cb900277e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lemeer S, Heck AJ. The phosphoproteomics data explosion. Curr Opin Chem Biol. 2009;13:414–20. doi: 10.1016/j.cbpa.2009.06.022. [DOI] [PubMed] [Google Scholar]
- 52.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. doi: 10.1161/CIRCGENETICS.109.871236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Matt P, Fu Z, Carrel T, Huso DL, Dirnhofer S, Lefkovits I, Zerkowski HR, Van Eyk JE. Proteomic alterations in heat shock protein 27 and identification of phosphoproteins in ascending aortic aneurysm associated with bicuspid and tricuspid aortic valve. J Mol Cell Cardiol. 2007;43:792–801. doi: 10.1016/j.yjmcc.2007.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Martin K, Steinberg TH, Goodman T, Schulenberg B, Kilgore JA, Gee KR, Beechem JM, Patton WF. Strategies and solid-phase formats for the analysis of protein and peptide phosphorylation employing a novel fluorescent phosphorylation sensor dye. Comb Chem High Throughput Screen. 2003;6:331–9. doi: 10.2174/138620703106298581. [DOI] [PubMed] [Google Scholar]
- 55.Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics. 2006;5:749–57. doi: 10.1074/mcp.T500024-MCP200. [DOI] [PubMed] [Google Scholar]
- 56.Yuan C, Sheng Q, Tang H, Li Y, Zeng R, Solaro RJ. Quantitative comparison of sarcomeric phosphoproteomes of neonatal and adult rat hearts. Am J Physiol Heart Circ Physiol. 2008;295:H647–56. doi: 10.1152/ajpheart.00357.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu H, Zong C, Wang Y, Young GW, Deng N, Souda P, Li X, Whitelegge J, Drews O, Yang PY, Ping P. Revealing the dynamics of the 20 S proteasome phosphoproteome: a combined CID and electron transfer dissociation approach. Mol Cell Proteomics. 2008;7:2073–89. doi: 10.1074/mcp.M800064-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Deng N, Zhang J, Zong C, Wang Y, Lu H, Yang P, Wang W, Young GW, Wang Y, Korge P, Lotz C, Doran P, Liem DA, Apweiler R, Weiss JN, Duan H, Ping P. Phosphoproteome analysis reveals regulatory sites in major pathways of cardiac mitochondria. Mol Cell Proteomics. 2010 doi: 10.1074/mcp.M110.000117. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Aponte AM, Phillips D, Hopper RK, Johnson DT, Harris RA, Blinova K, Boja ES, French S, Balaban RS. Use of (32)P to study dynamics of the mitochondrial phosphoproteome. J Proteome Res. 2009;8:2679–95. doi: 10.1021/pr800913j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Feng J, Zhu M, Schaub MC, Gehrig P, Roschitzki B, Lucchinetti E, Zaugg M. Phosphoproteome analysis of isoflurane-protected heart mitochondria: phosphorylation of adenine nucleotide translocator-1 on Tyr194 regulates mitochondrial function. Cardiovasc Res. 2008;80:20–9. doi: 10.1093/cvr/cvn161. [DOI] [PubMed] [Google Scholar]
- 61.Chu G, Egnaczyk GF, Zhao W, Jo SH, Fan GC, Maggio JE, Xiao RP, Kranias EG. Phosphoproteome analysis of cardiomyocytes subjected to beta-adrenergic stimulation: identification and characterization of a cardiac heat shock protein p20. Circ Res. 2004;94:184–93. doi: 10.1161/01.RES.0000107198.90218.21. [DOI] [PubMed] [Google Scholar]
- 62.Holt GD, Hart GW. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. J Biol Chem. 1986;261:8049–57. [PubMed] [Google Scholar]
- 63.Wells L, Hart GW. O-GlcNAc turns twenty: functional implications for post-translational modification of nuclear and cytosolic proteins with a sugar. FEBS Lett. 2003;546:154–8. doi: 10.1016/s0014-5793(03)00641-0. [DOI] [PubMed] [Google Scholar]
- 64.Watson LJ, Facundo HT, Ngoh GA, Ameen M, Brainard RE, Lemma KM, Long BW, Prabhu SD, Xuan YT, Jones SP. O-linked β-N-acetylglucosamine transferase is indispensable in the failing heart. Proc Natl Acad Sci U S A. 2010;107:17797–802. doi: 10.1073/pnas.1001907107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Marsh SA, Dell’italia LJ, Chatham JC. Activation of the hexosamine biosynthesis pathway and protein O-GlcNAcylation modulate hypertrophic and cell signaling pathways in cardiomyocytes from diabetic mice. Amino Acids. 2010 doi: 10.1007/s00726-010-0699-8. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yokoe S, Asahi M, Takeda T, Otsu K, Taniguchi N, Miyoshi E, Suzuki K. Inhibition of phospholamban phosphorylation by O-GlcNAcylation: implications for diabetic cardiomyopathy. Glycobiology. 2010;20:1217–26. doi: 10.1093/glycob/cwq071. [DOI] [PubMed] [Google Scholar]
- 67.Ngoh GA, Hamid T, Prabhu SD, Jones SP. Increased O-linked beta-N-acetylglucosamine levels on proteins improves survival, reduces inflammation and organ damage 24 hours after trauma-hemorrhage in rats. Am J Physiol Heart Circ Physiol. 2009;297:H1711–9. [Google Scholar]
- 68.Wu T, Zhou H, Jin Z, Bi S, Yang X, Yi D, Liu W. Cardioprotection of salidroside from ischemia/reperfusion injury by increasing N-acetylglucosamine linkage to cellular proteins. Eur J Pharmacol. 2009;613:93–9. doi: 10.1016/j.ejphar.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 69.Zou L, Yang S, Champattanachai V, Hu S, Chaudry IH, Marchase RB, Chatham JC. Glucosamine improves cardiac function following trauma-hemorrhage by increased protein O-GlcNAcylation and attenuation of NF-{kappa}B signaling. Am J Physiol Heart Circ Physiol. 2009;296:H515–23. doi: 10.1152/ajpheart.01025.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hu Y, Suarez J, Fricovsky E, Wang H, Scott BT, Trauger SA, Han W, Hu Y, Oyeleye MO, Dillmann WH. Increased enzymatic O-GlcNAcylation of mitochondrial proteins impairs mitochondrial function in cardiac myocytes exposed to high glucose. J Biol Chem. 2009;284:547–55. doi: 10.1074/jbc.M808518200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fülöp N, Feng W, Xing D, He K, Not LG, Brocks CA, Marchase RB, Miller AP, Chatham JC. Aging leads to increased levels of protein O-linked N-acetylglucosamine in heart, aorta, brain and skeletal muscle in Brown-Norway rats. Biogerontology. 2008;9:139–51. doi: 10.1007/s10522-007-9123-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ngoh GA, Facundo HT, Zafir A, Jones SP. O-GlcNAc signaling in the cardiovascular system. Circ Res. 2010;107:171–85. doi: 10.1161/CIRCRESAHA.110.224675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chatham JC, Marchase RB. The role of protein O-linked beta-N-acetylglucosamine in mediating cardiac stress responses. Biochim Biophys Acta. 2010;1800:57–66. doi: 10.1016/j.bbagen.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Laczy B, Hill BG, Wang K, Paterson AJ, White CR, Xing D, Chen YF, Darley-Usmar V, Oparil S, Chatham JC. Protein O-GlcNAcylation: a new signaling paradigm for the cardiovascular system. Am J Physiol Heart Circ Physiol. 2009;296:H13–28. doi: 10.1152/ajpheart.01056.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Z, Gucek M, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: site-specific phosphorylation dynamics in response to globally elevated O-GlcNAc. Proc Natl Acad Sci U S A. 2008;2008:105–13793. doi: 10.1073/pnas.0806216105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Copeland RJ, Bullen JW, Hart GW. Cross-talk between GlcNAcylation and phosphorylation: roles in insulin resistance and glucose toxicity. Am J Physiol Endocrinol Metab. 2008;295:E17–28. doi: 10.1152/ajpendo.90281.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Hart GW, Housley MP, Slawson C. Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446:1017–22. doi: 10.1038/nature05815. [DOI] [PubMed] [Google Scholar]
- 78.Wang Z, Pandey A, Hart GW. Dynamic interplay between O-linked N-acetylglucosaminylation and glycogen synthase kinase-3-dependent phosphorylation. Mol Cell Proteomics. 2007;6:1365–79. doi: 10.1074/mcp.M600453-MCP200. [DOI] [PubMed] [Google Scholar]
- 79.Whelan SA, Lane MD, Hart GW. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. J Biol Chem. 2008;283:21411–7. doi: 10.1074/jbc.M800677200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song M, Kim HS, Park JM, Kim SH, Kim IH, Ryu SH, Suh PG. o-GlcNAc transferase is activated by CaMKIV-dependent phosphorylation under potassium chloride-induced depolarization in NG-108-15 cells. Cell Signal. 2008;20:94–104. doi: 10.1016/j.cellsig.2007.09.002. 2008. [DOI] [PubMed] [Google Scholar]
- 81.Rexach JE, Rogers CJ, Yu SH, Tao J, Sun YE, Hsieh-Wilson LC. Quantification of O-glycosylation stoichiometry and dynamics using resolvable mass tags. Nat Chem Biol. 2010;6:645–51. doi: 10.1038/nchembio.412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang Z, Udeshi ND, O’Malley M, Shabanowitz J, Hunt DF, Hart GW. Enrichment and site mapping of O-linked N-acetylglucosamine by a combination of chemical/enzymatic tagging, photochemical cleavage, and electron transfer dissociation mass spectrometry. Mol Cell Proteomics. 2010;9:153–60. doi: 10.1074/mcp.M900268-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zachara NE, Cole RN, Hart GW, Gao Y. Detection and analysis of proteins modified by O-linked N-acetylglucosamine. Curr Protoc Protein Sci. 2001 doi: 10.1002/0471140864.ps1208s25. Chapter 12:Unit 12.8. [DOI] [PubMed] [Google Scholar]
- 84.Wells L, Vosseller K, Cole RN, Cronshaw JM, Matunis MJ, Hart GW. Mapping sites of O-GlcNAc modification using affinity tags for serine and threonine post-translational modifications. Mol Cell Proteomics. 2002;1:791–804. doi: 10.1074/mcp.m200048-mcp200. [DOI] [PubMed] [Google Scholar]
- 85.Hu P, Shimoji S, Hart GW. Site-specific interplay between O-GlcNAcylation and phosphorylation in cellular regulation. Mol Biol Cell. 2010;21:1922–36. doi: 10.1016/j.febslet.2010.04.044. [DOI] [PubMed] [Google Scholar]
- 86.Zeidan Q, Hart GW. The intersections between O-GlcNAcylation and phosphorylation: implications for multiple signaling pathways. J Cell Sci. 2010;123:13–22. doi: 10.1242/jcs.053678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Foster DB, Van Eyk JE, Marbán E, O’Rourke B. Redox signaling and protein phosphorylation in mitochondria: progress and prospects. J Bioenerg Biomembr. 2009;41:159–68. doi: 10.1007/s10863-009-9217-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Prasad TSK, Goel R, Kandasamy K, Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R, Shafreen B, Venugopal A, Balakrishnan L, Marimuthu A, Banerjee S, Somanathan DS, Sebastian A, Rani S, Ray S, Harrys Kishore CJ, Kanth S, Ahmed M, Kashyap MK, Mohmood R, Ramachandra YL, Krishna V, Rahiman BA, Mohan S, Ranganathan P, Ramabadran S, Chaerkady R, Pandey A. Human Protein Reference Database - 2009 Update. Nucleic Acids Research. 2009;37:D767–72. doi: 10.1093/nar/gkn892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Han G, Ye M, Liu H, Song C, Sun D, Wu Y, Jiang X, Chen R, Wang C, Wang L, Zou H. Phosphoproteome analysis of human liver tissue by long-gradient nanoflow LC coupled with multiple stage MS analysis. Electrophoresis. 2010;31:1080–9. doi: 10.1002/elps.200900493. [DOI] [PubMed] [Google Scholar]
- 90.Olsen JV, Vermeulen M, Santamaria A, Kumar C, Miller ML, Jensen LJ, Gnad F, Cox J, Jensen TS, Nigg EA, Brunak S, Mann M. Quantitative phosphoproteomics reveals widespread full phosphorylation site occupancy during mitosis. Sci Signal. 2010;3:ra3. doi: 10.1126/scisignal.2000475. [DOI] [PubMed] [Google Scholar]
- 91.Banks R, Van Eyk JE, Patterson S. A Strategy to Implement Next-Generation Proteomic Analyses (NGPA) to the Clinic for Patient Benefit: Pathway to Translation. Proteomics: Clin. Appl. 2010 doi: 10.1002/prca.201000113. in press. [DOI] [PubMed] [Google Scholar]
- 92.Large-scale structural biology of the human proteome. Annu Rev Biochem. 2009;78:541–68. doi: 10.1146/annurev.biochem.78.070907.103305. [DOI] [PubMed] [Google Scholar]
- 93.Terwilliger TC, Stuart D, Yokoyama S. Lessons from structural genomics. Annu Rev Biophys. 2009;38:371–83. doi: 10.1146/annurev.biophys.050708.133740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Perera M, Tsang CS, Distel RJ, Lacy JN, Ohno-Machado L, Ricchiuti V, Samaranayake LP, Smejkal GB, Smith MG, Trachtenberg AJ, Kuo WP. TGF-beta1 interactome: metastasis and beyond. Cancer Genomics Proteomics. 2010;7:217–29. [PubMed] [Google Scholar]
- 95.Scholten A, Poh MK, van Veen TA, van Breukelen B, Vos MA, Heck AJ. cAMP interactome using a chemical proteomics approach in mammalian heart tissue validates sphingosine kinase type 1-interacting protein as a genuine and highly abundant AKAP. J Proteome Res. 2006;5:1435–47. doi: 10.1021/pr0600529. [DOI] [PubMed] [Google Scholar]
- 96.Roy J, Cyert MS. Cracking the phosphatase code: docking interactions determine substrate specificity. Sci Signal. 2009 Dec 8;2(100) doi: 10.1126/scisignal.2100re9. [DOI] [PubMed] [Google Scholar]
- 97.De Las Rivas J, de Luis A. Interactome data and databases: different types of protein interaction. Comp Funct Genomics. 2004;5(2):173–8. doi: 10.1002/cfg.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vedadi M, Arrowsmith CH, Allali-Hassani A, Senisterra G, Wasney GA. Biophysical characterization of recombinant proteins: a key to higher structural genomics success. J Struct Biol. 2010;172:107–19. doi: 10.1016/j.jsb.2010.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Fero M, Pogliano K. Automated quantitative live cell fluorescence microscopy. Cold Spring Harb Perspect Biol. 2010;2:a000455. doi: 10.1101/cshperspect.a000455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Hamilton NA, Teasdale RD. Visualizing and clustering high throughput sub-cellular localization imaging. BMC Bioinformatics. 2008;9:81. doi: 10.1186/1471-2105-9-81. [DOI] [PMC free article] [PubMed] [Google Scholar]