

Abstract
Helicases are ubiquitous proteins that unwind DNA and participate in DNA metabolism including replication, repair, transcription, and chromatin organization. The highly conserved RecQ helicase family proteins are important in these transactions and have been termed the guardians of the genome. Humans have five members of this family: WRN, BLM, RECQL4, RECQL1, and RECQL5. The first three of these members are associated with premature aging and cancer prone syndromes, but the latter two proteins have not yet been implicated in any human disease. Although WRN and BLM have been fairly well characterized, RECQL4 has only recently been intensively investigated. The sum of this work to date has shown that RECQL4 has helicase activity and localizes to telomeres and mitochondria. In addition, new protein partners are emerging, implicating RECQL4 in novel processes. Here, we describe these recent findings, which place RECQL4 at the crossroads of genomic instability and aging processes.
Keywords: RecQ helicase, cancer, aging, Rothmund-Thomson syndrome
The Human RecQ helicases
DNA helicases are important ubiquitous enzymes that participate in DNA metabolism and affect genome stability. Helicases hydrolyze ATP in order to translocate along DNA to separate the two DNA strands and dislodge proteins in their path. Bacteria and yeast express a single RecQ helicase whereas humans express five: BLM, WRN, RECQL1, RECQL4, and RECQL5. All of these proteins share the core motifs of the RecQ helicase family, but outside of this homology, the proteins vary significantly (Figure 1). Loss of function of WRN or BLM leads to the autosomal recessive diseases Werner syndrome (OMIM #277700) or Bloom syndrome (OMIM, #210900), respectively. In contrast, mutations in RECQL4 give rise to three autosomal recessive diseases: Rothmund-Thomson syndrome (RTS, OMIM #268400), RAPADILINO (RAPA, OMIM #266280), or Baller-Gerold syndrome (BGS, OMIM #218600).
Figure 1.
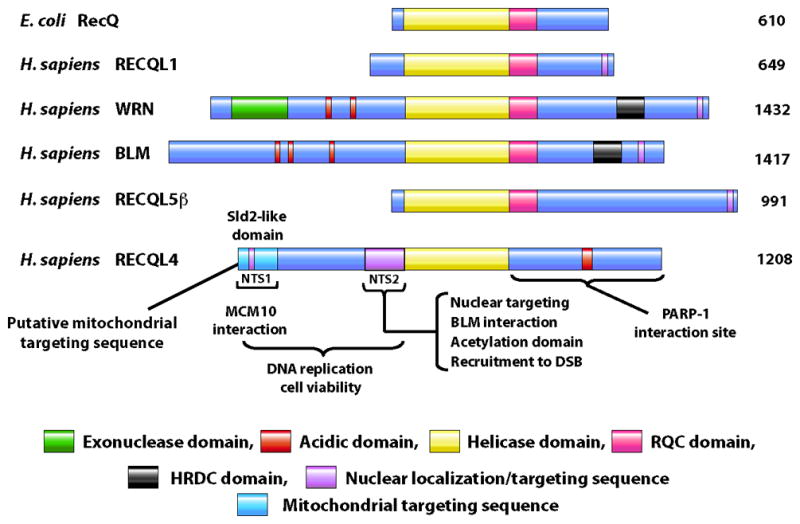
Structural organization of human RecQ helicases. All the RecQ helicase members possess a highly conserved helicase domain (yellow). The nuclear localization signal (purple) is present at the C-terminus in most of the family members, except in RECQL4 which has two nuclear targeting sequences (NTS1and NTS2) that are present at the N-terminus. The RQC (pink) and helicase and RNase C-terminal (HRDC) domain (black) are present at the C-terminus of helicase domain except for in RECQL4. The WRN protein is unique among human RecQ helicase members in having an exonuclease domain (green) at the N-terminus. The N-terminus of RECQL4 show sequence similarity with the Sld2 protein in yeast that is important for DNA replication and cell viability. A putative mitochondrial targeting sequence is present at the N-terminus of RECQL4 (light blue).
About two-thirds of patients with mutations in RECQL4 have RTS and the remaining patients have either RAPA or BGS [1,2]. Within the disorder RTS, there are two subtypes, type I and II. Type II patients have documented mutations within RECQL4, whereas type I patients have mutations in genes that have not yet been identified [3]. The common features among these diseases are short stature and skeletal abnormalities, particularly radial ray defects. RTS and RAPA patients are also at elevated risk for osteosarcomas and lymphomas, but this is not true for BGS patients [1]. Poikiloderma is another common feature of RTS and RAPA that is distinctively absent in BGS patients. RTS patients display segmental premature aging features like sparse hair/eyelashes, juvenile cataracts, and predisposition to cancer [4]. Mouse models have been constructed to mimic the loss of RECQL4, but so far none genuinely recapitulate the human phenotype (Box 1).
Box 1. Mouse Models for studying RECQL4.
Three mouse models have been developed to more closely examine the loss of function of Recql4 in mammals. The first model [58] is a mouse in which exons 5-8, which precede the helicase domain, were removed. This mouse was embryonic lethal between days 3.5–6.5. A second model was created in which exon 13 was deleted (Δ-exon 13); this is the last exon encoding the helicase domain [59]. In this study, the authors reported that the majority of the animals (95%) died within two weeks after birth. In the third mouse model, the entire helicase domain, Δ-exons 9-13, was deleted. This mouse was viable but displayed elevated rates of perinatal lethality [60]. The deletion of the entire helicase domain, Δ-exons 9-13, likely renders the protein more structurally stable than the other two deletion constructs because the helicase domain is probably an autonomously folding unit. Additionally, the neonatal and perinatal lethality associated with these models may have to do with replication failure and the burst of oxidative damage experienced at birth which these Recql4-deficient mice simply cannot handle.
In contrast to the other RecQ helicase mouse models which either show no phenotype (RECQL1 [61] and WRN [62]) or are lethal BLM [63]), the surviving Recql4 deficient mice were the first to show premature aging phenotypes. Specifically, the Δ-exon 13 Recql4 mice that survived showed skin atrophy, colorless hair, hair loss, short stature, bone dysplasia, dystrophic teeth, cataracts, and immune abnormalities which are all features associated with premature aging [59]. In the Δ-exon 9-13 Recql4 mice, distinctive skin and skeletal abnormalities were observed in addition to genomic instability, which was characterized by aneuploidy and premature centromere separation [60]. When crossed into a cancer susceptible background, APCmin, the Δ-exon 9-13 Recql4 allele led to a dramatic increase in the number and size of tumors in the intestines [60]. At this time it is not clear why the mouse model alone does not recapitulate the predisposition for osteosarcoma and lymphomas like the human RECQL4-associated diseases. Perhaps it reflects differences between mouse and man, or alternatively, there may need to be some initiating environmental insult for the loss of function of Recql4 in mice to be tumor promoting.
The gene for RECQL4 was first cloned by Kitao et al. and mapped to chromosome 8q24.3 [5], which places it on the same chromosome as WRN but on the opposite chromosomal arm. RECQL4 mRNA shows tissue- and cell cycle-specific expression patterns. Human thymus, testis, and placenta show high expression, whereas moderate expression is seen in the heart, brain, small intestines, and colon by Northern blot analysis [6]. RECQL4 mRNA expression peaks in S-phase of the cell cycle, similar to BLM expression [6]. RECQL4 mRNA is also induced in Ebstein-barr virus or simian 40 transformed cells [7]. Consistent with these observations, osteosarcomas as well as cervical and prostate cancers exhibit abnormally high levels of RECQL4 expression and this is associated with metastasis [8–12]. Additionally, RECQL4 and WRN expression are down-regulated by p53, a master transcriptional regulator of the DNA damage response which induces cell cycle arrest, inhibits DNA replication, and activates DNA repair or apoptosis [13–15]. Because p53 is mutated in numerous cancers, the regulation of both RECQL4 and WRN expression could also be perturbed, impacting the DNA damage response pathway in tumor cells.
RECQL4 is the only RecQ helicase known to be present in both the nucleus and mitochondria. In the nucleus it is predominantly localized to the nucleoplasm with a fraction of the protein co-localizing to telomeres and the nucleolus [16–18]. It dynamically associates with foci produced by DNA damaging agents including hydrogen peroxide (H2O2), ionizing radiation, topoisomerase inhibitors, and ultraviolet radiation (UV) [16,17,19–21]. RECQL4 has also been found in the cytoplasm and more recently inside the mitochondria [18,22,23]. The intracellular localization of RECQL4 appears to be cell-type specific as some cells show more cytoplasmic than nuclear localization [18]. The intracellular distribution and dynamic re-localization of RECQL4 to sites of DNA damage may contribute to the phenotypes and associated heterogeneity seen in RECQL4-associated diseases.
Structural and functional uniqueness of RECQL4
Human RECQL4 is a 1208 amino acid (aa) protein containing a highly conserved 3′ to 5′ helicase domain in the middle of the protein. RecQ helicase family members typically have the conserved helicase motifs, a RecQ conserved domain (RQC), and the helicase and RNase D conserved domain (HRDC), but curiously RECQL4 lacks the latter two (Figure 1). Both the N- and C-terminal regions of RECQL4 contain distinct sequences from the other RecQ family helicases. The N-terminal region of RECQL4 is a multifunctional domain that contains both nuclear and mitochondrial targeting sequences. The C-terminal region of RECQL4 is not well characterized but has recently been associated with the response of RECQL4 to ionizing radiation [24]. These unique features of RECQL4 provide functional diversity not found in other member of the RecQ helicase family.
Emerging evidence suggests that the N-terminus of RECQL4 plays an important role in DNA replication. Sequence comparison between Xenopus RECQL4 (xRECQL4) and its homologues in other eukaryotic organisms revealed that it shares sequence homology with the yeast replication initiation factors Sld2 (Saccharomyces cerevisiae) and DRC1 (Schizosaccharomyces pombe), both of which are essential for establishment of replication forks [25,26]. Consistent with the Sld2-like function, deletion of the N-terminus of RECQL4 leads to decreased cell proliferation in vertebrate cells [25,26]. Additionally, expression of just the N-terminus can restore viability to RECLQ4-depleted chicken cells [27]. Taken together, the data suggests that the N-terminus of RECQL4 plays an important and evolutionarily conserved role in DNA replication.
The N-terminal domain is also important for subcellular localization and targeting. Using GFP-tagged RECQL4 fragments, it was shown that there are two nuclear localization regions present within the N-terminus of RECQL4, NTS1 (aa 37–66) and NTS2 (aa 363–492) [28]. The predicted mitochondrial localization sequence overlaps with NTS1 and resides in the first 84 N-terminal aa [23]. Additionally, acetylation of RECQL4 by p300 on multiple lysines within NTS2 regulates the nuclear to cytoplasm localization of RECQL4 [29]. Interestingly, NTS2 is deleted in the majority of RAPA patients [1], highlighting the importance of this sequence for the proper function of RECQL4.
Although RECQL4 is a 3′ to 5′ helicase in superfamily II and contains the characteristic helicase motifs (I, Ia, II, III, IV, V, and VI) important for coupling ATP hydrolysis to the separation of DNA strands, the helicase activity of RECQL4 was only recently demonstrated in vitro, sparking new studies into the function of RECQL4. When human RECQL4 was first isolated and purified, in vitro biochemical assays did not show any helicase activity on a variety of substrates, including a fork duplex [30]. However, by including excess single-stranded DNA (ssDNA) to trap the complementary strand after unwinding a 30-bp fork duplex, weak helicase activity was observed [31,32]. Compared with other RecQ helicases, RECQL4 shows very weak DNA unwinding activity, and it acts on very short fork DNA duplex substrates with limited substrate specificity [31,33]. It was later determined that RECQL4 preferentially anneal sDNA, rather than unwinding it, and that the strand annealing activity masked detection of helicase products [31].
The DNA unwinding ability of RECQL4 was attributed to two independent segments, the N-terminal domain and the helicase domain [31]. This finding was puzzling because the N-terminus lacks conserved homology with other known helicases. Using a helicase dead mutant, K508M, it has more recently been directly demonstrated that the ATP-dependent unwinding activity of RECQL4 comes from the conserved helicase domain [33]. It is still unknown, however, why DNA unwinding activity was observed from the N-terminal fragment in the former study, but one possibility is that the N-terminal domain possesses strand exchange activity which was interpreted as DNA unwinding activity. Moving forward, it will be important to determine the structural basis for the unwinding activity derived from the N-terminal domain and further to characterize which in vivo functions of RECQL4 are dependent upon the activity of the N-terminus and helicase domains.
RECQL4 has a narrower substrate range than the other human RecQ helicases [33]. Some of the substrates on which the RECQL4 helicase has been tested hint at the cellular activities in which RECQL4 may be involved (Figure 2). So far no helicase activity has been detected from RECQL4 on complex Holliday Junctions or G-quadruplex DNA structures in vitro [33]. This may be due to the lack of the RQC domain, which has been shown to be a high affinity G-quadruplex binding domain [34]. Interestingly, however, RECQL4 has in vitro activity on D-loop structures, including those with telomeric sequences, when excess ssDNA is included in the reaction [35]. Structurally similar to Holliday junctions, D-loops are believed to be important structures for telomeric maintenance, thus RECQL4 may promote telomeric replication.
Figure 2.
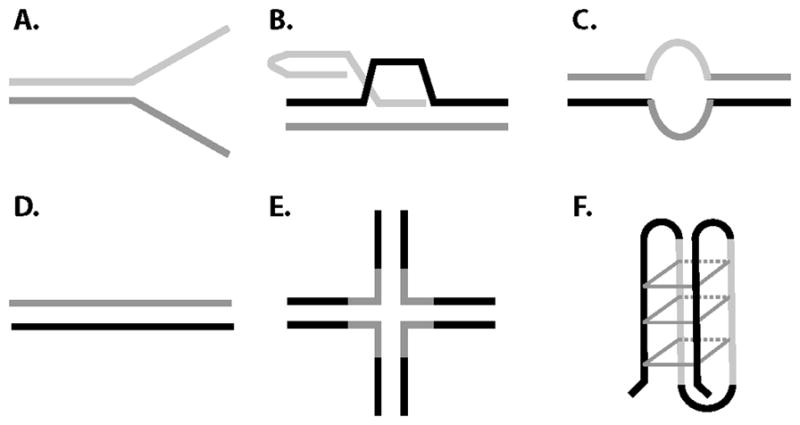
RECQL4 has a narrower substrate range than other RecQ helicases. The RECQL4 helicase can unwind forked duplexes (A), D-loops (B), and bubble (C) structures but not duplex DNA (D), Holliday junctions (E), or G-quadruplex structures (F). However, the unwinding activity from the N-terminal domain can utilize duplex DNA substrates.
Further insight into the function of RECQL4 has been inferred from the cellular phenotypes of genotoxic sensitivity in RTS type II cells. The clinical features of RTS patients are heterogeneous and this is reflected in the varied responses to different assaults at the cellular level. For example, RTS fibroblasts show hypersensitivity to hydroxyurea, camptothecin, and doxorubicin [36], but only modest sensitivity is reported following exposure to UV light, ionizing radiation, or cisplatin [21]. Interestingly, RTS cells are relatively resistant to 4-nitroquinolone oxide [36], which produces bulky DNA damage thought to be repaired by the nucleotide excision repair (NER) apparatus. This result is in contrast to similar experiments done with cells deficient for WRN and BLM, indicating that there are functional differences between the helicases. Many of these agents induce damage primarily during S-phase, and RTS cells fail to synthesize DNA after treatment with H2O2 [37], suggesting that RECQL4 is particularly important during S-phase and in DNA replication. The variability observed between genotoxic agents suggests that RECQL4 may have lesion-type specific functions in DNA repair as well.
RECQL4 protein interactions
Currently, more than ten proteins have been shown to associate with RECQL4 in vivo (Table 1), implicating it not only in DNA–related pathways, but also in posttranslational modifications, telomere maintenance, and mitochondrial DNA (mtDNA) maintenance. Similar to other RecQ proteins, multiple lines of evidence support a role for RECQL4 in DNA replication. xRECQL4, which is essential for DNA replication in Xenopus egg extracts [25], is part of the replication initiation complex and functions after pre-replication complex formation promoting replication factor loading at the origins. Specifically, the N-terminal fragment of xRECQL4 (1–596 aa) has been shown to physically interact with xCut5, a protein which controls recruitment of DNA polymerases α and ε [26].
Table 1.
Proteins interacting with human RECQL4 and its homolog
| Protein1 | Location | Detection methods2 | Interaction region on RECQL4 | Interaction function | References |
|---|---|---|---|---|---|
| Cut5 | ND | CoIP | ND | DNA replication | [28] |
| MCM10 | Nuclear | CoIP, MS, GST-PD | 1–200aa | RECQL4 helicase inhibition | [38] |
| RAD51 | Nuclear | CoLC, CoIP | ND | DNA double strand break repair | [15] |
| PARP1 | ND | PDS, CoIP | 833–1208aa | Base excision repair | [16] |
| XPA | Nuclear | CoIP, GST- PD, CoLC, CoFC | ND | Nucleotide excision repair | [18] |
| FEN1 | Nuclear | CoIP, CoLC | ND | FEN1 incision stimulation | [19] |
| POLβ | Nuclear | CoLC | ND | DNA Polβ primer extension activity stimulation | [19] |
| APE1 | Nuclear | CoLC | ND | APE1 endonuclease stimulation | [19] |
| RPA | Nucleus | CoLC | ND | RecQL4 helicase stimulation | [33] |
| BLM | Nuclear | CoIP, Y2H | 1–471 aa | BLM stimulation | [43] |
| URB1/2 | ND | CoIP, CoFC, MS | ND | Unknown | [17] |
| p300 | Nuclear | CoIP, GST- PD, CoLC | 1– 408 aa | Regulation of cellular location of RECQL4 | [31] |
| p53 | Mitochondria | CoLC, CoFC, CoIP | 270–400 aa | Silencing of nuclear location signals of RECQL4 and p53 | [22] |
| TOM20 | Mitochondria | GST-PD | 13–18 aa | Import into mitochondria | [22] |
| TFAM | Mitochondria | CoIP | ND | Unknown | [21] |
| TRF1 | Nuclear/telomere | CoLC, | ND | RECQL4 helicase stimulation and telomere maintenance | [37] |
| TRF2 | Nuclear/telomere | CoIP | ND | RECQL4 helicase stimulation and telomere maintenance | [37] |
| WRN | Nuclear/telomere | CoIP | ND | WRN stimulation on telomeric D-loop | [37] |
All RecQL4 interacting partners were found in human cells except cut5, which is a xenopus protein;
Abbreviation of detection methods as follows: ND, not determined; CoIP, co-immunoprecipitation; MS, mass spectrometry; GST-PD, GST pull-down assay; CoLC, Co-localization; CoFC, Co-fractionation; PDS, phage display screen; Y2H, yeast two hybrid assay; aa, amino acid.
Analysis of chromatin-bound RECQL4 complexes from human cells revealed multiple RECQL4-interacting DNA replication factors by mass spectrometric analysis, including MCM10, MCM2-7 helicase, CDC45, GINS, and SLD5 [38]. The N-terminus of RECQL4 (1–240 aa) directly interacts with MCM10, which mediates the association of RECQL4 with the MCM2-7 replicative DNA helicase and GINS complex in a cell-cycle dependent manner [38]. RECQL4, MCM10, and CTF4 are all required for proper loading and assembly of CDC45, MCM10, and the GINS complex onto the origins of replication [39]. Interestingly, both RECQL4 and RECQL1 have been captured at DNA replication origins, and it was shown that replication origin firing and nascent DNA synthesis were reduced after RECQL4 or RECQL1 depletion [40]. These results firmly establish a role for RECQL4 in DNA replication.
RECQL4 has also been implicated in other DNA-related processes, including DNA double-strand break repair (DSB), NER, and base excision repair (BER). Multiple approaches have been used to show that RECQL4 is involved in the repair of DSBs in human cells [16,21,41]. For example, laser confocal microscopy and live cell imaging were used to show that RECQL4 localizes to sites of laser-induced DNA damage. Human RECQL4 associates with RAD51, a key protein in the homologous recombination (HR) pathway of DSB repair in cells [16]. It also forms foci and co-localizes with RAD51 after etoposide-induced DNA damage. Furthermore, a physical interaction between RECQL4 and RAD51 was shown by co-immunoprecipitation. Another study reported the involvement of RECQL4 in NER based on the cellular co-localization of endogenous RECQL4 with XPA, a key protein in NER [19]. In vitro GST pull-down assays confirmed the direct interaction between RECQL4 and XPA [19]. UV repair is mediated by the NER pathway and in cell culture, UV-induced DNA damage is routinely used to screen for NER-dependent interactions. In HeLa cells, the RECQL4-XPA interaction was enhanced after UV irradiation. However, RTS cells are not hypersensitive to UV, so more work is needed to clarify whether RECQL4 directly participates in NER or if there is redundancy among the RecQ helicases that may mask the function of RECQL4 in NER.
The single strand break repair protein PARP1 catalyzes the addition of polymers of linear and branched chain ADP-ribose moieties (PAR) onto itself and other important target proteins, and is involved in DNA repair, transcription, and replication (reviewed in [42]). PARP1 is a DNA damage sensor, robustly activated in response to a variety of DNA damaging agents and is widely known to modulate the sensitivity of cells to γ-irradiation. Proteins post-translationally modified by PAR (as with phosphorylation and ubiquitination), can have altered catalytic function and modified protein interaction capabilities. PARP1 has been identified as a RECQL4 interacting protein in a T7 phage display screen using C-terminal fragments of RECQL4 (833–1208 aa) as bait [17]. Additionally, in vitro PARP1 was shown to PARylate the C-terminus of RECQL4 [17]. Recently, the C-terminus of RECQL4 has been suggested to be important for the response to γ-irradiation [24] so perhaps the interaction with PARP1 is important in this context. For example, after DNA damage, PARP1 creates a nucleation site at which other DNA repair proteins coalesce to facilitate repair, and it will be interesting to determine whether RECQL4’s recruitment to DNA damage differs in PARP1-deficient cells and if DNA replication re-start is impacted by these conditions.
PARP1 is also important in long patch BER, raising the possibility that RECQL4 is involved in the BER pathway, which repairs methylation, oxidation, and other relatively small DNA base and sugar lesions. After H2O2 treatment, RECQL4 co-localizes and functionally interacts with APE1, FEN,1 and DNA polymerase β, all key proteins involved in BER [20]. Biochemical experiments indicated that RECQL4 specifically stimulated the apurinic endonuclease activity of APE1, the DNA strand displacement activity of DNA polymerase β, and the incision of a 1 -or 10 -nucleotide flap DNA substrate by FEN1. Additionally, RTS cells have a dysfunctional response to oxidative stress and display elevated levels of formamido pyrimidine and 8-oxoG, DNA lesions that are markers of oxidative damage [24]. Furthermore, microarray analysis revealed that many BER pathway genes were up-regulated in RTS patient fibroblasts relative to normal cells [24]. Taken together these observations suggest a role for RECQL4 in modulating the BER pathway [20].
Recently, BLM was identified as a RECQL4-interacting protein [43]. Functionally, RECQL4 stimulated BLM helicase activity on DNA fork substrates in vitro, and the in vivo interaction between RECQL4 and BLM was enhanced during the S-phase of the cell cycle and after treatment with ionizing radiation. These results suggest there is some synergy between these RecQ helicases. In line with this, WRN and RECQL4 (see below) also functionally interact, and BLM specifically inhibits the exonuclease activity of WRN [44]. Given that there are both similar and distinct disease phenotypes in patients deficient for WRN, BLM or RECQL4, experiments that seek to identify common and unique roles for these proteins in the various biochemical pathways of genome maintenance will be of high priority.
Protein interactions also reveal a role for RECQL4 in telomere maintenance. Telomeres are the DNA-protein complexes at the ends of human chromosomes. The cells from RTS patients have elevated levels of fragile telomeric ends, and RECQL4-depleted human cells accumulate fragile sites, sister chromosome exchanges, and DSBs at telomeric sites [35]. Human RECQL4 partially localizes to telomeres and co-immunoprecipitates with the shelterin protein TRF2 [35]. TRF1 and TRF2 are specialized telomere DNA binding proteins that stabilize telomeric D-loop structures along with other shelterin proteins. Three shelterin proteins, TRF1, TRF2, and POT1, can stimulate the helicase activity of RECQL4 on a telomeric DNA substrate [35]. Additionally, RECQL4 has been shown to act synergistically with WRN, but not BLM, to resolve telomeric D -loop substrates by stimulating WRN unwinding activity. These findings unveiled a novel function for RECQL4 in telomere maintenance and extend the findings that multiple RecQ helicases function at telomeres.
In addition to its role in the nucleus, RECQL4 also functions in the mitochondria. The mitochondrial transcription factor-A (TFAM), which protects mtDNA and aids in mtDNA transcription and replication, has been reported to interact with RECQL4 [22]. RECQL4 was evaluated in vitro with polymerase γ, the replicative polymerase in mitochondria, however no productive interaction was observed [22]. In a separate study, RECQL4 was shown to be important for in vivo mtDNA recovery after ethidium bromide depletion [23], suggesting that RECQL4 has a role in mtDNA replication.
The tumor suppressor protein p53 and TOM20 are also mitochondrial interacting partners of RECQL4 [23]. Using in vitro assays, aa 293–362 of p53 and aa 270–400 of RECLQ4 were determined to be the minimal regions necessary for the interaction [23]. The interaction between RECQL4 and TOM20, a component of the mitochondrial outer membrane-locating TOM40 receptor complex, has also been characterized. It was suggested that RECQL4 and p53 enter mitochondria by interacting with the TOM complex [23]. Additional research is necessary to delineate the biological functions of the interaction between RECQL4 and p53 in mitochondria and to characterize the role of RECQL4 in mtDNA replication and repair.
Interestingly, in the absence of cellular stress, the localization of p53 to the mitochondria is dependent on RECQL4’s mitochondrial localization [23]. It is well established that p53 plays a central role in the DNA damage response signaling cascade and that its dysfunction can ultimately lead to cancer or aging [15]. However, although its presence in mitochondria has been known for some time [45], it has only recently been recognized to play a role in mitochondria regulation [46]. It has been shown that dysfunctional telomeres activate p53, which in turn alters the gene expression of PGC1α and PGC1β, master regulators of mitochondria. Interestingly, cells lacking RECQL4 have activated p53 [23] as well as telomere and mitochondrial dysfunction [22,35]. The sum of the protein interaction data suggests that under non-stressful conditions, RECQL4 exists in both the nucleoplasm and mitochondria and participates in DNA maintenance in both of these compartments.
When RECQL4 is depleted, DNA replication or repair in both the nucleus and mitochondria are altered, telomere dysfunction ensues, and p53 is activated. If the cell is not able to repair the damage, cell death may occur, leading to the deterioration of organs and premature aging phenotypes. With respect to the disease phenotypes associated with loss of RECQL4, the failure to meet the normal cellular replicative demands of affected tissues may have a direct impact on organismal growth and the gastrointestinal disturbances seen in RECQL4-related diseases. Additionally, osteosarcomas and lymphomas could arise in RTS and RAPA patients due to altered DNA repair capacity or replication stress caused by loss of RECQL4.
Aging mechanisms
Recently, several discoveries have demonstrated functional roles for RECQL4 in three major mechanisms associated with aging and cellular senescence: oxidative DNA damage repair, telomeres maintenance, and mitochondrial dysfunction (Figure 3). Oxidative damage, caused by endogenous cellular metabolism or exogenous sources, disrupts normal replication and transcription and leads to genomic instability and cellular senescence. BER is the primary mechanism to repair oxidative lesions and prevent replication fork stalling. As discussed earlier, RTS cells are hypersensitive to oxidative damage, and RECQL4 may be a modulator of the BER pathway. These findings suggest that RECQL4 has a role in promoting efficient BER and are consistent with its proposed role in promoting efficient DNA replication.
Figure 3.

Cellular localization and stress responses surrounding RECQL4. Under normal circumstances, RECQL4 can be found in the nucleoplasm and at telomeres, where it participates in DNA maintenance. Without stress, RECQL4 interacts with p53 and they shuttle between the nucleus and mitochondria. After stress, p53 and RECQL4 re-localize to the nucleus where p53 coordinates the DNA damage response including down-regulating RECQL4 expression. In both the nucleus and mitochondria, RECQL4 can localize to DNA damage and participate in modulating DNA maintenance processes.
Telomeres are hotspots for oxidative damage, which if unrepaired leads to telomere instability and shortening [47–49]. Telomere length decreases with age while instability increases, thus telomere dynamics are associated with aging and telomere length is a possible biomarker of aging [50,51]. Out of the five human RecQ helicases, only WRN and BLM were previously known to function at the telomeres (recently reviewed in [52]). However, RECQL4 has also recently been shown to have a role in telomere maintenance, as both RECQL4- knockdown and RTS patient cells exhibit abnormal telomere properties [35]. Furthermore, RECQL4 was shown to stimulate the helicase activity of WRN in vitro on telomeric D-loops, indicating possible cooperation between the helicases [35]. WRN and RECQL4 may complement each other in telomere length maintenance, particularly because WRN is capable of unwinding G-quadruplexes, which are impediments to telomere replication, whereas RECQL4 cannot [33,53].
Mitochondrial dysfunction is well documented in aging. Normal aged cells and those from patients with age-related diseases such as Alzheimer’s are marked by dysfunctional mtDNA maintenance [54]. A proteomics study gave the first clues that RECQL4 might be localized to mitochondria [55] and two independent groups have now used immunocytochemistry to confirm and extend this finding [22,23]. Although its role there is not yet fully understood, analysis of RECQL4-deficient cells revealed mitochondrial bioenergetics defects and elevated mtDNA damage [22]. The involvement of RECQL4 in DNA replication, repair, and maintenance of the nuclear and mtDNA highlight the importance of this protein in aging and senescence.
In aging research there has been great deal of interest in the idea that telomere shortening is a critical feature that leads to senescence. By contrast, the mitochondrial theory of aging posits that mitochondrial dysfunction is the cause of aging [56]. Telomere processing and mitochondrial bioenergetics have so far been separate fields with very limited interaction. That there is emerging evidence for some crosstalk between these fields of study is very exciting. Recently it has been shown that telomere dysfunction can lead to mitochondrial dysfunction [46] and vice versa [57]. It is therefore of great interest that specific proteins, like RECQL4, have now been identified that operate in both compartments.
Concluding remarks
A future challenge is to understand the biological function of RECQL4 in both compartments as well as whether and how it could function as a signaling molecule between these compartments. RECQL4 is the only known RecQ helicase to function in mitochondria, and a greater understanding of its function in this compartment should be a focus of future research. This is challenging in general because of the low abundance of DNA metabolic proteins in the mitochondria and particularly by the fact that the mitochondrial targeting sequences of RECQL4 and the Sld2-domain overlap. How one might uncouple these functional attributes is not obvious at this time. The next challenge is to determine the conditions that govern when RECQL4 is at telomeres or in the mitochondria. Such information will shed light on its signaling function between these compartments. This may involve a close collaboration with p53, which also functions at both places, and could be another signaling protein in the pathways of aging. Thus, RECQL4 may be uniquely positioned to act as a bridge between cancer and aging, functioning as a guardian of not only the nuclear genome, but also the mitochondrial genome.
Acknowledgments
We would like to thank Martin Borch Jensen and Dr. Venkateswarlu Popuri for critically reading the manuscript. We would like to thank Thomas Wynn for his artwork. This research was supported entirely by the Intramural Research Program of the NIH, National Institute on Aging, AG000726-20.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference List
- 1.Siitonen HA, et al. The mutation spectrum in RECQL4 diseases. Eur J Hum Genet. 2009;17:151–158. doi: 10.1038/ejhg.2008.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Larizza L, et al. Rothmund-Thomson syndrome. Orphanet J Rare Dis. 2010;5:2. doi: 10.1186/1750-1172-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang LL, et al. Association between osteosarcoma and deleterious mutations in the RECQL4 gene in Rothmund-Thomson syndrome. J Natl Cancer Inst. 2003;95:669–674. doi: 10.1093/jnci/95.9.669. [DOI] [PubMed] [Google Scholar]
- 4.Larizza L, et al. Rothmund-Thomson syndrome and RECQL4 defect: splitting and lumping. Cancer Lett. 2006;232:107–120. doi: 10.1016/j.canlet.2005.07.042. [DOI] [PubMed] [Google Scholar]
- 5.Kitao S, et al. Rothmund-thomson syndrome responsible gene, RECQL4: genomic structure and products. Genomics. 1999;61:268–276. doi: 10.1006/geno.1999.5959. [DOI] [PubMed] [Google Scholar]
- 6.Kitao S, et al. Cloning of two new human helicase genes of the RecQ family: biological significance of multiple species in higher eukaryotes. Genomics. 1998;54:443–452. doi: 10.1006/geno.1998.5595. [DOI] [PubMed] [Google Scholar]
- 7.Kawabe T, et al. Differential regulation of human RecQ family helicases in cell transformation and cell cycle. Oncogene. 2000;19:4764–4772. doi: 10.1038/sj.onc.1203841. [DOI] [PubMed] [Google Scholar]
- 8.Choi YW, et al. Gene expression profiles in squamous cell cervical carcinoma using array-based comparative genomic hybridization analysis. Int J Gynecol Cancer. 2007;17:687–696. doi: 10.1111/j.1525-1438.2007.00834.x. [DOI] [PubMed] [Google Scholar]
- 9.Saglam O, et al. Molecular differentiation of early and late stage laryngeal squamous cell carcinoma: an exploratory analysis. Diagn Mol Pathol. 2007;16:218–221. doi: 10.1097/PDM.0b013e3180d0aab5. [DOI] [PubMed] [Google Scholar]
- 10.Maire G, et al. Recurrent RECQL4 imbalance and increased gene expression levels are associated with structural chromosomal instability in sporadic osteosarcoma. Neoplasia. 2009;11:260–8. 3p. doi: 10.1593/neo.81384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sadikovic B, et al. Expression analysis of genes associated with human osteosarcoma tumors shows correlation of RUNX2 overexpression with poor response to chemotherapy. BMC Cancer. 2010;10:202. doi: 10.1186/1471-2407-10-202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Su Y, et al. Human RecQL4 helicase plays critical roles in prostate carcinogenesis. Cancer Res. 2010;70:9207–9217. doi: 10.1158/0008-5472.CAN-10-1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sengupta S, et al. Tumor suppressor p53 represses transcription of RECQ4 helicase. Oncogene. 2005;24:1738–1748. doi: 10.1038/sj.onc.1208380. [DOI] [PubMed] [Google Scholar]
- 14.Yamabe Y, et al. Sp1-mediated transcription of the Werner helicase gene is modulated by Rb and p53. Mol Cell Biol. 1998;18:6191–6200. doi: 10.1128/mcb.18.11.6191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet. 2012;28:128–136. doi: 10.1016/j.tig.2011.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Petkovic M, et al. The human Rothmund-Thomson syndrome gene product, RECQL4, localizes to distinct nuclear foci that coincide with proteins involved in the maintenance of genome stability. J Cell Sci. 2005;118:4261–4269. doi: 10.1242/jcs.02556. [DOI] [PubMed] [Google Scholar]
- 17.Woo LL, et al. The Rothmund-Thomson gene product RECQL4 localizes to the nucleolus in response to oxidative stress. Exp Cell Res. 2006;312:3443–3457. doi: 10.1016/j.yexcr.2006.07.023. [DOI] [PubMed] [Google Scholar]
- 18.Yin J, et al. RECQL4, mutated in the Rothmund-Thomson and RAPADILINO syndromes, interacts with ubiquitin ligases UBR1 and UBR2 of the N-end rule pathway. Hum Mol Genet. 2004;13:2421–2430. doi: 10.1093/hmg/ddh269. [DOI] [PubMed] [Google Scholar]
- 19.Fan W, Luo J. RecQ4 facilitates UV light-induced DNA damage repair through interaction with nucleotide excision repair factor xeroderma pigmentosum group A (XPA) J Biol Chem. 2008;283:29037–29044. doi: 10.1074/jbc.M801928200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schurman SH, et al. Direct and indirect roles of RECQL4 in modulating base excision repair capacity. Hum Mol Genet. 2009;18:3470–3483. doi: 10.1093/hmg/ddp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Singh DK, et al. The involvement of human RECQL4 in DNA double-strand break repair. Aging Cell. 2010;9:358–371. doi: 10.1111/j.1474-9726.2010.00562.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Croteau DL, et al. RECQL4 localizes to mitochondria and preserves mitochondrial DNA integrity. Aging Cell. 2012;11:456–466. doi: 10.1111/j.1474-9726.2012.00803.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.De S, et al. RECQL4 is essential for the transport of p53 to mitochondria in normal human cells in the absence of exogenous stress. J Cell Sci. 2012 doi: 10.1242/jcs.101501. [DOI] [PubMed] [Google Scholar]
- 24.Kohzaki M, et al. The helicase domain and C-terminus of human RecQL4 facilitate replication elongation on DNA templates damaged by ionizing radiation. Carcinogenesis. 2012 doi: 10.1093/carcin/bgs149. [DOI] [PubMed] [Google Scholar]
- 25.Sangrithi MN, et al. Initiation of DNA replication requires the RECQL4 protein mutated in Rothmund-Thomson syndrome. Cell. 2005;121:887–898. doi: 10.1016/j.cell.2005.05.015. [DOI] [PubMed] [Google Scholar]
- 26.Matsuno K, et al. The N-terminal noncatalytic region of Xenopus RecQ4 is required for chromatin binding of DNA polymerase alpha in the initiation of DNA replication. Mol Cell Biol. 2006;26:4843–4852. doi: 10.1128/MCB.02267-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abe T, et al. The N-terminal region of RECQL4 lacking the helicase domain is both essential and sufficient for the viability of vertebrate cells. Role of the N-terminal region of RECQL4 in cells. Biochim Biophys Acta. 2011;1813:473–479. doi: 10.1016/j.bbamcr.2011.01.001. [DOI] [PubMed] [Google Scholar]
- 28.Burks LM, et al. Nuclear import and retention domains in the amino terminus of RECQL4. Gene. 2007;391:26–38. doi: 10.1016/j.gene.2006.11.019. [DOI] [PubMed] [Google Scholar]
- 29.Dietschy T, et al. p300-mediated acetylation of the Rothmund-Thomson-syndrome gene product RECQL4 regulates its subcellular localization. J Cell Sci. 2009;122:1258–1267. doi: 10.1242/jcs.037747. [DOI] [PubMed] [Google Scholar]
- 30.Macris MA, et al. Biochemical characterization of the RECQ4 protein, mutated in Rothmund-Thomson syndrome. DNA Repair (Amst) 2006;5:172–180. doi: 10.1016/j.dnarep.2005.09.005. [DOI] [PubMed] [Google Scholar]
- 31.Xu X, Liu Y. Dual DNA unwinding activities of the Rothmund-Thomson syndrome protein, RECQ4. EMBO J. 2009;28:568–577. doi: 10.1038/emboj.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suzuki T, et al. DNA helicase activity in purified human RECQL4 protein. J Biochem. 2009;146:327–335. doi: 10.1093/jb/mvp074. [DOI] [PubMed] [Google Scholar]
- 33.Rossi ML, et al. Conserved helicase domain of human RecQ4 is required for strand annealing-independent DNA unwinding. DNA Repair (Amst) 2010;9:796–804. doi: 10.1016/j.dnarep.2010.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Huber MD, et al. A conserved G4 DNA binding domain in RecQ family helicases. J Mol Biol. 2006;358:1071–1080. doi: 10.1016/j.jmb.2006.01.077. [DOI] [PubMed] [Google Scholar]
- 35.Ghosh AK, et al. RECQL4, the protein mutated in Rothmund-Thomson syndrome, functions in telomere maintenance. J Biol Chem. 2012;287:196–209. doi: 10.1074/jbc.M111.295063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jin W, et al. Sensitivity of RECQL4-deficient fibroblasts from Rothmund-Thomson syndrome patients to genotoxic agents. Hum Genet. 2008;123:643–653. doi: 10.1007/s00439-008-0518-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Werner SR, et al. RECQL4-deficient cells are hypersensitive to oxidative stress/damage: Insights for osteosarcoma prevalence and heterogeneity in Rothmund-Thomson syndrome. Biochem Biophys Res Commun. 2006;345:403–409. doi: 10.1016/j.bbrc.2006.04.093. [DOI] [PubMed] [Google Scholar]
- 38.Xu X, et al. MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO J. 2009;28:3005–3014. doi: 10.1038/emboj.2009.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Im JS, et al. Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proc Natl Acad Sci U S A. 2009;106:15628–15632. doi: 10.1073/pnas.0908039106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Thangavel S, et al. Human RECQ1 and RECQ4 helicases play distinct roles in DNA replication initiation. Mol Cell Biol. 2010;30:1382–1396. doi: 10.1128/MCB.01290-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kumata Y, et al. Possible involvement of RecQL4 in the repair of double-strand DNA breaks in Xenopus egg extracts. Biochim Biophys Acta. 2007;1773:556–564. doi: 10.1016/j.bbamcr.2007.01.005. [DOI] [PubMed] [Google Scholar]
- 42.Luo X, Kraus WL. On PAR with PARP: cellular stress signaling through poly(ADP-ribose) and PARP-1. Genes Dev. 2012;26:417–432. doi: 10.1101/gad.183509.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Singh DK, et al. The human RecQ helicases BLM and RECQL4 cooperate to preserve genome stability. Nucleic Acids Res. 2012 doi: 10.1093/nar/gks349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.von Kobbe C, et al. Colocalization, physical, and functional interaction between Werner and Bloom syndrome proteins. J Biol Chem. 2002;277:22035–22044. doi: 10.1074/jbc.M200914200. [DOI] [PubMed] [Google Scholar]
- 45.de Souza-Pinto NC, et al. p53 functions in the incorporation step in DNA base excision repair in mouse liver mitochondria. Oncogene. 2004;23:6559–6568. doi: 10.1038/sj.onc.1207874. [DOI] [PubMed] [Google Scholar]
- 46.Sahin E, et al. Telomere dysfunction induces metabolic and mitochondrial compromise. Nature. 2011;470:359–365. doi: 10.1038/nature09787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.von Zglinicki T, et al. Accumulation of single-strand breaks is the major cause of telomere shortening in human fibroblasts. Free Radic Biol Med. 2000;28:64–74. doi: 10.1016/s0891-5849(99)00207-5. [DOI] [PubMed] [Google Scholar]
- 48.Kawanishi S, Oikawa S. Mechanism of telomere shortening by oxidative stress. Ann N Y Acad Sci. 2004;1019:278–284. doi: 10.1196/annals.1297.047. [DOI] [PubMed] [Google Scholar]
- 49.Kurz DJ, et al. Chronic oxidative stress compromises telomere integrity and accelerates the onset of senescence in human endothelial cells. J Cell Sci. 2004;117:2417–2426. doi: 10.1242/jcs.01097. [DOI] [PubMed] [Google Scholar]
- 50.Mather KA, et al. Is telomere length a biomarker of aging? A review. J Gerontol A Biol Sci Med Sci. 2011;66:202–213. doi: 10.1093/gerona/glq180. [DOI] [PubMed] [Google Scholar]
- 51.von Zglinicki T, Martin-Ruiz CM. Telomeres as biomarkers for ageing and age-related diseases. Curr Mol Med. 2005;5:197–203. doi: 10.2174/1566524053586545. [DOI] [PubMed] [Google Scholar]
- 52.Singh DK, et al. RecQ helicases in DNA double strand break repair and telomere maintenance. Mutat Res. 2011 doi: 10.1016/j.mrfmmm.2011.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mohaghegh P, et al. The Bloom’s and Werner’s syndrome proteins are DNA structure-specific helicases. Nucleic Acids Res. 2001;29:2843–2849. doi: 10.1093/nar/29.13.2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Coskun P, et al. A mitochondrial etiology of Alzheimer and Parkinson disease. Biochim Biophys Acta. 2012;1820:553–564. doi: 10.1016/j.bbagen.2011.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jiang Y, et al. Proteomic analysis of mitochondria in Raji cells following exposure to radiation: implications for radiotherapy response. Protein Pept Lett. 2009;16:1350–1359. doi: 10.2174/092986609789353646. [DOI] [PubMed] [Google Scholar]
- 56.Harman D. The biologic clock: the mitochondria? J Am Geriatr Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 57.Passos JF, von Zglinicki T. Mitochondria, telomeres and cell senescence. Exp Gerontol. 2005;40:466–472. doi: 10.1016/j.exger.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 58.Ichikawa K, et al. Preparation of the gene targeted knockout mice for human premature aging diseases, Werner syndrome, and Rothmund-Thomson syndrome caused by the mutation of DNA helicases. Nihon Yakurigaku Zasshi. 2002;119:219–226. doi: 10.1254/fpj.119.219. [DOI] [PubMed] [Google Scholar]
- 59.Hoki Y, et al. Growth retardation and skin abnormalities of the Recql4-deficient mouse. Hum Mol Genet. 2003;12:2293–2299. doi: 10.1093/hmg/ddg254. [DOI] [PubMed] [Google Scholar]
- 60.Mann MB, et al. Defective sister-chromatid cohesion, aneuploidy and cancer predisposition in a mouse model of type II Rothmund-Thomson syndrome. Hum Mol Genet. 2005;14:813–825. doi: 10.1093/hmg/ddi075. [DOI] [PubMed] [Google Scholar]
- 61.Sharma S, et al. RECQL, a member of the RecQ family of DNA helicases, suppresses chromosomal instability. Mol Cell Biol. 2007;27:1784–1794. doi: 10.1128/MCB.01620-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lebel M, Leder P. A deletion within the murine Werner syndrome helicase induces sensitivity to inhibitors of topoisomerase and loss of cellular proliferative capacity. Proc Natl Acad Sci U S A. 1998;95:13097–13102. doi: 10.1073/pnas.95.22.13097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chester N, et al. Stage-specific apoptosis, developmental delay, and embryonic lethality in mice homozygous for a targeted disruption in the murine Bloom’s syndrome gene. Genes Dev. 1998;12:3382–3393. doi: 10.1101/gad.12.21.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]