

Abstract
Background
Traditional cancer therapy can be successful in destroying tumors, but can also cause dangerous side effects. Therefore, many targeted therapies are in development. The transferrin receptor (TfR) functions in cellular iron uptake through its interaction with transferrin. This receptor is an attractive molecule for the targeted therapy of cancer since it is upregulated on the surface of many cancer types and is efficiently internalized. This receptor can be targeted in two ways: 1) for the delivery of therapeutic molecules into malignant cells or 2) to block the natural function of the receptor leading directly to cancer cell death.
Scope of review
In the present article we discuss the strategies used to target the TfR for the delivery of therapeutic agents into cancer cells. We provide a summary of the vast types of anti-cancer drugs that have been delivered into cancer cells employing a variety of receptor binding molecules including Tf, anti-TfR antibodies, or TfR-binding peptides alone or in combination with carrier molecules including nanoparticles and viruses.
Major conclusions
Targeting the TfR has been shown to be effective in delivering many different therapeutic agents and causing cytotoxic effects in cancer cells in vitro and in vivo.
General significance
The extensive use of TfR for targeted therapy attests to the versatility of targeting this receptor for therapeutic purposes against malignant cells. More advances in this area are expected to further improve the therapeutic potential of targeting the TfR for cancer therapy leading to an increase in the number of clinical trials of molecules targeting this receptor.
Keywords: transferrin receptor, CD71, cancer, nanoparticles, immunotoxins, delivery, conjugates, gene therapy
1. Introduction
The receptor for transferrin (Tf), referred to as TfR1 (also known as CD71), is ubiquitously expressed at low levels in most normal human tissues. A second member of the TfR family is TfR2, a protein that is highly homologous to TfR1 but whose expression is largely restricted to hepatocytes [1]. Serving as the main port of entry for iron bound Tf into cells, TfR1 is a type-II receptor that resides on the outer cell membrane and cycles into acidic endosomes into the cell in a clathrin/dynamin dependent manner [1-4]. Iron is delivered into the cell and TfR1 is recycled back to the cell surface (Figure 1-2) [5, 6]. Despite its ubiquitous expression, TfR1 is expressed on malignant cells at levels many times higher than those on normal cells and its expression can be correlated with tumor stage or cancer progression [7-12]. This high expression of the receptor on malignant cells, its ability to internalize, and the necessity of iron for cancer cell proliferation make this receptor a widely accessible portal for the delivery of drugs into malignant cells and thus, an attractive target for cancer therapy (Figure 3).
Figure 1. Schematic representation of the TfR1 and cellular uptake of iron through the Tf system via receptor-mediated endocytosis.
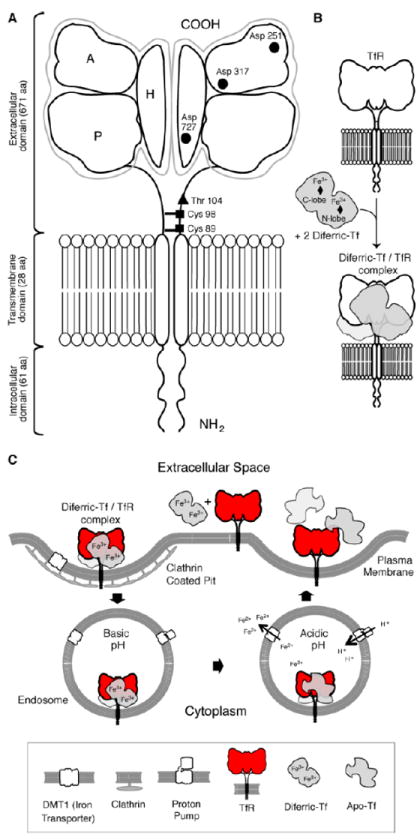
(A) TfR1 is found on the cell surface as a homodimer consisting of two monomers linked by disulfide bridges at cysteines 89 and 98 (■). The TfR contains an intracellular domain, a transmembrane domain, and a large extracellular domain. There is an O-linked glycosylation site at threonine 104 (◆) and three N-linked glycosylation sites on arginine residues 251, 317 and 727 (●). The extracellular domain of the TfR consists of three subdomains: apical (A), helical (H) and protease-like domain (P). (B) Each receptor monomer binds one Tf molecule that consists of two lobes (the N and C lobes). Each lobe of Tf binds one iron molecule and thus two diferric Tf molecules bind to the receptor with high affinity. (C) Endocytosis of the diferric Tf/TfR complex occurs via clathrin-coated pits and the complex is delivered into endosomes. Protons are pumped into the endosome resulting in a decrease in pH that stimulates a conformational change in Tf and its subsequent release of iron. The iron is then transported out of the endosome into the cytosol by the DMT1. Apotransferrin remains bound to the TfR while in the endosome and is only released once the complex reaches the cell surface. Figure is reprinted from [1] with permission from Elsevier.
Figure 2. Internalization of Tf by TfR1.

Side and top views of the TfR alone (1) or bound to Tf (2). Numbers in the figure correspond to the numbers and explanations listed on the right side of the figure.
Figure 3. Strategies for targeting therapeutic agents via TfR to malignant cells.

Targeting can be mediated by its natural ligand Tf, a specific peptide, monoclonal antibodies or single chain antibody fragments specific for the extracellular domain of the TfR. The therapeutic agent can be delivered conjugated to the compound or enclosed in a carrier. Targeting the TfR has been an option to deliver chemotherapeutic drugs, therapeutic proteins, genes in vectors, oligonucleotides or radionucleides.
Targeting of the TfR1 has occurred through the use of a variety of methods including strategies that utilize mostly its natural ligand Tf or monoclonal antibodies or their fragments (Figure 3). However, short peptides have also been used as targeting moieties. In general, cytotoxicity of Tf conjugates can be blocked by native Tf. Therefore, high levels of circulating free Tf in the blood may interfere with the effects of these Tf conjugates leading to decreased therapeutic efficacy. As Tf conjugates have the potential to interact with both TfR1 and TfR2 (which is highly expressed in the liver), they may be lethal to many normal cells in addition to the targeted malignant cells. Targeting the TfR1 through the use of monoclonal antibodies may help to circumvent these potential concerns.
There are two ways to effectively exploit the TfR for therapeutic purposes. One is through the use of molecules that are capable of antagonizing the normal function of the receptor. Work from our group and others have shown promising results for the treatment of a variety of cancers via cytotoxicity induced by the direct inhibition of TfR1 function by monoclonal antibodies. Among these are the monoclonal antibodies 42/6 [13], A24 [14], and ch128.1Av (also known as anti-hTfR1 IgG3-Av) [15, 16]. The second way of targeting the TfR is for the delivery of therapeutic agents into malignant cells. This allows the delivery of anticancer drugs to neoplastic cells by the association of the drugs to molecules with high affinity and specificity for the receptor. Even though the receptor is normally constitutively recycled, its use as a delivery vehicle is possible and has been clearly shown as discussed below. Delivery of therapeutic agents into the cytoplasm of cancer cells can be due to the alteration of the intracellular trafficking of the receptor, which can be caused by the targeting molecule or through endosomal rupture and escape caused by the therapeutic agent itself. Additionally, agents that increase vesicle pH can be used to ensure that the cargo is not degraded in low pH vesicles during intracellular trafficking. These agents can be part of the cargo itself or as additional agents that are used in combination with the targeted therapy. This review is focused on the specific targeting of TfR1 by antibodies or other ligands and how these vehicles can be employed for the delivery of a broad repertoire of anti-cancer agents (Figure 3).
2. Delivery strategies
The literature on the use of TfR1 as a target for the delivery of small molecules, proteins, nucleic acids, and even nanoparticles and viruses into many types of malignant cells continues to grow. Delivery can be achieved by the direct linkage of the therapeutic to the targeting moiety or by loading of the therapeutic into carriers such as nanoparticles linked to the targeting molecule. A wide variety of therapeutic agents have been used for TfR-targeted cancer therapy (Figure 3). They include chemotherapeutic drugs and prodrugs, bacterial toxins, plant toxins, DNA, oligonucleotides (ODN), short inhibitory RNA (siRNA), and enzymes. Targeting cancer cells through use of the TfR can enhance drug delivery by increasing intracellular drug concentration resulting in more effective tumor targeting, less non-specific toxicity, and therefore in an overall increased therapeutic efficacy.
The term “nanoparticles” encompasses numerous engineered entities, architectures, and particulate systems that usually share as a common feature a size range from a few nanometers (nm) to several hundred nm, depending on their intended use [17, 18]. These particles often self-assemble and include micelles, dendrimers, polymeric nanoparticles, cyclodextrins, and liposomes by which therapeutic agents can be entrapped, covalently bound, encapsulated, or adsorbed to overcome drug solubility issues (Figure 4) [19]. They can carry small molecules, peptides, proteins, and nucleic acids, preventing them from being recognized and inactivated by the immune system. Nanoparticles can deliver the therapeutic drugs to the tumor by passive or active targeting, as is the case of TfR targeted nanocarriers [20]. Passive targeting of nanoparticles to tumors occurs by the modulated vasculature, which allows nanocarriers to extravasate through gaps in the endothelium. The entry of the particles to the interstitial space, associated with poor lymphatic drainage from the tumor, results in higher retention times of nanoparticles in the tumor than in normal tissues, in a process known as enhanced permeability and retention (EPR) effect [20]. Significant increases in drug accumulation in the tumor tissue by the EPR effect can reach 10-fold or higher concentration with drug-loaded nanoparticles compared to free drug [21]. Active targeting requires the use of targeting moieties, such as antibodies or receptor ligands, conjugated to the surface of the nanocarrier systems for their delivery enhancement.
Figure 4. Carriers for direct targeting of therapeutic agents via TfR to malignant cells.

The various carriers can be targeted to the TfR using antibodies or their fragments, the natural ligand, or specific peptides. These carries can deliver a broad spectrum of therapeutic agents, including chemotherapeutic drugs, toxic proteins, or nucleic acids into targeted cells.
3. Targeting moiety conjugated directly to the active compound
3.1. Tf conjugates
3.1.1. Tf-chemotherapeutic drug conjugates
3.1.1.1. Tf-Doxorubicin/Adriamycin® conjugates
Doxorubicin or Adriamycin® (ADR), is an anthracycline, antineoplastic drug that has the ability to intercalate into DNA, generate free radicals, and inhibit certain enzymes such as topoisomerase II [22]. ADR is a widely used chemotherapy in treating many forms of cancer, but ultimately exhibits severe side effects, including cardiotoxicity, myelosuppression, nephrotoxicity, extravasation, and bone marrow depression due to quick diffusion throughout the body [22]. To circumvent the adverse affects of ADR, it has been chemically conjugated to Tf in an effort to deliver it directly to cancer cells expressing high levels of TfR [23-26], resulting in an effectual tissue distribution, a prolonged half-life of ADR in blood plasma, and controlled release from Tf [25]. This Tf-ADR conjugate has shown cytotoxicity in vitro against a variety of malignant human cell lines, including Lovo (colorectal adenocarcinoma), H-MESO-1 (mesothelioma), Hep2 (liver carcinoma), HL-60 (promyelocytic leukemia), K562 (erythroleukemia), HeLa (cervical adenocarcinoma), U-937 (histiocytic lymphoma), LXFL (lung carcinoma), and MDA-MB-428 (breast cancer) and the murine fibroblast cell line L929 [23-25, 27-30]. The Tf-ADR conjugate produced three to 10-fold greater in vitro cytotoxicity than free ADR in cell lines such as Lovo, Hep2, K562, HL-60, and HeLa [23, 27, 28]. Additionally, relative to free ADR, it was consistently found that less Tf-ADR conjugate was needed for an IC50 in HL60 and K562 cells [24]. The IC50 of Tf-ADR conjugate in comparison to free ADR was reduced by 57-fold for L929, 21-fold for MCF-7, and 14-fold for RT4 cells [30]. In nude mice bearing H-MESO-1 tumors, i.v. administered Tf-ADR increased the life span of the mice by 69% in comparison to 30% in mice treated with ADR alone [23].
Many studies have been conducted to evaluate the mechanism of cytotoxicity of Tf-ADR. In order to determine if the amount of ADR or Tf in the conjugate is responsible for the potency of cytotoxic effects, different compositions of the Tf-ADR conjugate were tested on HL-60 cells [28]. Conjugates composed of varying levels of Tf with a constant amount of ADR resulted in the same inhibition of HL-60 cell growth. Thus, the cytotoxicity of Tf-ADR conjugates is due to the level of ADR delivered, not from the level of Tf. In human umbilical vein endothelial cells (HUVEC), significantly less cytoxicity was observed [25]. Free ADR was more toxic than acid-sensitive conjugates of ADR, indicating that select conjugates are active against TfR-positive cells [25]. However, acid-sensitive maleimide conjugates have cytotoxicity similar to free ADR against HUVEC cells, suggesting that the chemical link between Tf and ADR is related to levels of cytoxicity. Free ADR mainly functions via DNA intercalation in the nucleus of the cell, however, the cytotoxicity of Tf-ADR may be mediated by a different mechanism. The protein conjugate was shown not to translocate to the nucleus, but to act on various enzymes within the plasma membrane, suggesting that the action of ADR was directed by the physiological interactions of Tf [26, 27, 31]. Importantly, this conjugate was also able to overcome multidrug resistance while minimizing toxicity to normal cells [28, 32, 33].
Additionally, Tf-ADR conjugates have the ability to overcome multidrug-resistant tumor cells when saturated with iron or gallium nitrate (GN), producing Fe-ADR and GN-ADR, respectively. GN is an antineoplastic drug that shares chemical properties with iron and thus binds Tf [34]. GN-ADR-Tf was able to reverse the resistance to free ADR in MCF-7 human breast cancer cells, as the IC50 decreased 100-fold with the use of GN-ADR-Tf conjugate [35]. Similarly, Fe-ADR-Tf showed a 10-fold stronger inhibition compared to free ADR. ADR was found to accumulate in the cytoplasm in resistant MCF-7 cells, however in the cells treated with the GN-ADR-Tf conjugate, ADR was found in the cytoplasm in addition to the nucleus. Thus, the reversal of resistance by the GN-ADR-Tf conjugate suggests that the localization of ADR into the nucleus is key to bypass the multi-drug resistance protein (an ATP-binding transport glycoprotein) expression, which pumps drugs out of the cytoplasm. Overall, Tf-ADR appears to have multiple mechanisms of action that may be cell-type dependent or dependent on the presence of GN within the Tf-ADR conjugate.
3.1.1.2. Tf and other chemotherapeutic drug conjugates
Tf has also been conjugated to other drugs in order to avoid the adverse side effects of these drugs in a free condition, while helping direct and localize the drug to its target. Cisplatin (Platinol-AQ®) is a platinum-based alkylating agent that is used as a treatment for various cancer types, including bladder, ovarian, and testicular cancer. Cisplatin has been chemically conjugated to Tf to produce the complex MPTC-63 [36]. This complex has been shown to be cytotoxic to human HeLa cells in only seven days compared to the control, which kept growing past seven days. MPTC-63, was also shown to be cytotoxic to feline lymphoma cells in vitro. In a Fisher rat tumor model, MPTC-63 administered i.v. (8.6 mg/cc daily for seven weeks) prevented metastatic growth of the mammary carcinoma cells in the lungs. Additionally, two out of five patients who were administered MPTC-63 showed an anti-tumor response [36]. Subsequently, another Phase I clinical trial has shown four out of 11 patients with advanced breast cancer (36%) had a positive response rate, including one complete response to MPTC-63 [37]. Only minor side effects were observed in these patients. This clinical trial also showed seven out of the eight patients had a partial response to the combination of MPTC-63 and the iron chelator deferoxamine mesylate [37]. A promising case study in a female with stage IV breast cancer, showed that treatment resulted in a complete remission of malignant ascites [38]. The treatment consisted of vaccine immunotherapy in combination with targeted chemotherapy using Tf conjugated to metals such as gallium and platinum.
Chlorambucil (Leukeran®) is an alkylating agent that interferes with DNA replication and RNA transcription. It is used to treat chronic lymphoid leukemia, non-Hodgkins lymphoma, and advanced ovarian and breast cancer. This drug was chemically conjugated to Tf. Similar to Tf-ADR, introducing a thioether forming maleimide group (a building block in organic synthesis) into the drug allowed conjugates bound to Tf via acid cleavable spacers to show cytotoxicity to MOLT4 (human leukemia) and MCF7 (human mammary carcinoma cells) [39]. Chlorambucil or the Tf-chlorambucil conjugate was used in a preliminary toxicity study in mice injected daily with 20 mg/kg. Two out of three mice treated with free chlorambucil died after subsequent injections, whereas all the mice treated with Tf-chlorambucil survived, confirming the safety of the conjugated drug [39].
Mitomycin C, or MMC (Mutamycin®), is another alkylating agent that results in DNA crosslinking. When used as a chemotherapeutic drug in patients, myelosuppression and gastrointestinal complications are common. A Tf-MCC conjugate was constructed and tested relative to free MCC against HL60, HepG2, and hepatocytes from male Wistar rats [40]. The conjugate significantly inhibited cell growth in HL60 cells, but only slightly in HepG2 cells. The level of internalization of the conjugate was larger for HepG2 relative to HL60, and the IC50 was 43% less for HepG2 than HL60. MCC cytotoxicity is specific to dividing cells and it is internalized at low levels in hepatocytes, explaining the lack of cytotoxicity observed when the conjugate was used. Daunorubicin (Cerubidine®) is an anthracycline topoisomerase inhibitor, which also blocks DNA synthesis and RNA transcription. Daunorubicin chemically conjugated to Tf was 10-fold more cytotoxic compared to free daunorubicin on NCI-H69 small cell carcinoma of the lung cells [41]. Gemcitabine (Gemzar®) is an anti-metabolite pyrimidine analog that blocks DNA synthesis. Conjugated to Tf this drug demonstrated six to 10- fold higher cytotoxicity compared to free drug against UCRU-BL13 and UCRU-BL28 human bladder cancer cells [42]. SCID mice bearing UCRU-BL28 xenografts treated with the conjugate showed partial remission and significantly smaller tumors relative to untreated mice [42]. All of the above studies demonstrate that Tf chemically conjugated to chemotherapeutic drugs enhances the cytotoxic effects of the free drug and helps to minimize potential toxicities to normal tissues.
3.1.2. Tf-toxic protein conjugates
3.1.2.1. Tf- ricin A chain (RTA) conjugates
Ricin is a highly toxic protein isolated from the plant Ricinus communis. It is a type II ribosomal inactivating protein composed of an A chain (RTA) that contains the N-glycosidase catalytic domain and a cell-binding B chain. Once in the cytoplasm, the A chain inactivates the ribosome leading to inhibition of protein synthesis and ultimately cell death. Since it lacks cell-binding activity, RTA alone is not expected to be toxic. Conjugation of Tf to RTA allows TfR-mediated delivery of RTA into cells that can restore its toxicity. The ID50 of Tf-RTA in human leukemia CEM cells was 10,000-fold less compared to the non-linked combination of Tf and RTA [43]. This toxicity was completely blocked by Tf or antibodies against RTA. Two human Burkitt’s lymphoma cell lines, Daudi and Raji, demonstrated an ID50 that was ten-fold less that the CEM cells [43]. Since these lymphoma cells lines exhibited lower TfR levels on their surface compared to CEM cells, this suggests that the density of TfR correlates with the potency of the conjugate. The efficacy of Tf-RTA was also evaluated against a 3-D tumor model of micromasses using human breast carcinoma (MCF7) and rat glioblastoma (9L) cells [44]. Tf-RTA treatment can completely block growth of these micromasses in vitro.
3.1.2.2. Tf-saporin conjugates
Saporin (SO6), derived from seeds of the plant Sapornaria officinalis, is a type I ribosome-inactivating protein that is similar to ricin but is composed of only the catalytic chain and lacks the cell-binding domain. Thus, native SO6 is expected to be non-toxic. However, like RTA, if SO6 is conjugated to a delivery vehicle, such as Tf, it becomes highly toxic. SO6 chemically conjugated to Tf was cytotoxic to K562 human erythroleukemic cells [45] and HepG2 human hepatocellular carcinoma cells [46] in vitro. Tf-SO6 inhibited protein synthesis of HepG2 in a dose-dependent manner, while free SO6 demonstrated no toxicity to HepG2 cells [46]. The Tf-SO6 conjugate has also been shown to be cytotoxic to human glioblastoma cell lines and primary glioma cancer cells isolated from patients [47, 48].
3.1.2.3. Tf-diphtheria toxin conjugates
The diphtheria toxin (DT) is secreted by the bacterium Corynebacterium diphtheriae and acts to block protein synthesis through the ADP-ribosylation of elongation factor 2. The DT is composed of three chains: an N-terminal catalytic domain, a translocation domain, and a C-terminal cell-binding domain. Tf was covalently conjugated to the DT and demonstrated high cytotoxicity to thymidine kinase-deficient mouse L (LMTK-) cells in culture, reducing protein synthesis by 50% in 24 hours [49]. A 10,000-fold greater sensitivity of mouse LMTK- cells to Tf-DT was observed relative to DT treatment alone. This cytotoxicity was blocked by ammonium chloride due to the fact that it elevates the pH of intracellular vesicles suggesting that like the native toxin, decreased pH causes a conformational change of the Tf-DT, insertion into the membrane of the vesicle, and release of the catalytic domain into the cytosol [49].
In order to further increase the delivery capability of TfR-targeted therapies, two mutant Tf molecules have been developed (K206E/R632A and K206E/K534A) that show a decreased rate of iron release [50, 51]. These mutant Tf molecules when conjugated to DT, have shown enhanced delivery and cytotoxicity against HeLa human cervical adenocarcinoma cells relative to wild-type Tf-DT [50]. The IC50 was decreased to around 1 pM compared to 1.73 pM for the wiltype Tf-DT. These mutants also showed increased internalization of Tf and cytotoxicity against U87 and U251 human glioma cells [51]. Mice with established subcutaneous (s.c.) xenograft glioma tumors treated intratumorally with mutant Tf-DT conjugate showed significant regression as the tumor bulk decreased rapidly and near-complete tumor regression was observed in all four tumors. Immunohistochemical analysis of tumors from mice sacrificed at 24 hours post-treatment verified that the toxin conjugate caused tumor growth to decrease via apoptosis.
Additionally, the DT itself has been mutated to block non-specific entry of the toxin into cells. CRM107 is a mutant form of the DT that contains two amino acid mutations in the B chain that result in the inability of the mutant toxin to bind to the cell surface [52]. Tf chemically conjugated to CRM107 (Tf-CRM107), has demonstrated in vitro cytotoxicity against many human cancer types including neuroblastoma, leukemia, ovarian cancer, and breast cancer cell lines, as well as glioblastoma and medulloblastoma-derived cell lines, medulloblastoma cells in primary culture, and other cell lines derived from tumors that are usually metastatic to the cerebrospinal fluid [52, 53]. The Tf-CRM107 conjugate was also shown to have dose-dependent growth inhibitory effects against 3D micromasses of MCF7 or rat glioblastoma 9L cells [44]. The toxicity of the DT, CRM107, and the Tf-CRM107 conjugate was compared in three guinea pigs after direct injection into the brain (percutaneously into the cisterna magna). Unconjugated CRM107 increased the maximum safe dose 100-fold compared to the wild-type DT [52]. Furthermore, Tf-CRM107 further increased the maximum safe dose 10-fold compared to CRM107 (equivalent to 1000-fold compared to wild type DT). In rhesus monkeys, direct injection of Tf-CRM107 intrathecally into the cisterna magna at concentrations up to 5000-fold higher than those used for the in vitro cytotoxicity studies in cancer cells was well tolerated [52].
A Phase I clinical trial was conducted with Tf-CRM107 to determine its effectiveness and relevance as a cancer therapy in humans [54]. In order to maintain a high concentration in the brain, the immunotoxin (IT) was delivered by intratumoral infusion to patients with malignant brain tumors. Nine out of the fifteen patients who could be evaluated (60%) responded with at least a 50% reduction in tumor volume on magnetic resonance imaging (MRI). Two of these patients had complete responses. One had no tumor for 23 months after treatment with a single infusion of Tf-CRM107, while the other had a tumor recur five months post-treatment. At higher doses of Tf-CRM107 (≥ 1.0 μg/ml), local toxicity occurred in all three patients treated. However, at lower doses (<1.0 μg/ml), Tf-CRM107 did not cause any local toxicity in patients while anti-tumor activity was present, suggesting a concentration and dose dependency in tumor response and safety. Subsequently, a Phase II clinical trial was conducted to evaluate the safety of the conjugate and observe the effect of Tf-CRM107 on refractory and recurrent glioblastoma multiforme (GBM) or anaplastic astrocytoma [55]. Fourty-four patients were enrolled in the study, with 31 receiving two infusions of Tf-CRM107 and the remaining 13 receiving one infusion, however ten were not evaluable. Out of 34 patients who received two infusions, there were five complete responders, seven partial responders, and nine showed stabilized disease upon treatment with Tf-CRM107. From the time of the first infusion treatment, the remaining 13 out of 34 patients (30%) survived past 12 months. Symptomatic progressive cerebral edema resulted in eight out of 44 patients, and seizures were seen in three patients who responded to anticonvulsant therapy. Three Phase III clinical trials were initiated, however, one of these studies was withdrawn prior to enrollment due to concerns that the drug would not meet trial criteria for efficacy (clinicaltrials.gov). Further data on the other two trials could not be found.
3.1.2.4. Tf-artemisinin conjugates
Artemisinin, isolated from the plant Artemisia annua, is an anti-malarial drug that reacts with iron to form cytotoxic free radicals. Since cancer cells require large amounts of iron for rapid proliferation, they are more susceptible to the toxic effects of artemisinin [56]. Conjugation of artemisinin with Tf (Tf-ART) allows for the delivery of iron and artemisinin by the same molecule. This conjugate and an unconjugated artemisinin analog, dihydroartemisinin (DHA), were tested for their cytotoxic effects on Molt-4 human leukemia cells and normal lymphocytes. Tf-ART and DHA had an IC50 value of 0.98 μM and 1.64 μM on Molt-4 cells respectively. In lymphocytes, the IC50 value on was 33 μM and 58.4 μM for Tf-Art and DHA, respectively, suggesting that Tf-ART is more effective in killing Molt-4, but that DHA is more potent in killing normal lymphocytes. Thus, Tf-ART shows potential to target cancer specifically. When Tf is conjugated with 16 artemisinin molecules, the conjugate showed minimal structural change of holo-Tf (iron-bound Tf) and was cytotoxic to prostate cancer cells in vitro [57]. Tf-ART induced the intrinsic apoptotic pathway via the release of cytochrome c from mitochondria. A strong correlation was shown between the number of artemisinin conjugated to Tf and the IC50 values of DU145 and PC-3 (human prostate carcinoma cell lines) [58]. The IC50 of Tf-ART (containing 4 ART), Tf-ART (6 ART), and Tf-ART (16 ART) were 18.7, 7.2, and 5.0 μM, respectively. PC3 cells were less susceptible to the toxicity of Tf-ART than DU145 cells, but were more sensitive when larger numbers of artemisinin molecules were attached to Tf. In rats bearing s.c. rat MTLn3 mammary adenocarcinoma cells, five daily injections of Tf-ART significantly retarded the growth rate of the tumors [59]. Additionally, no significant side effects were observed in these animals.
3.1.2.5. Tf-PEG-protein conjugates
Since solid tumors show hypervascular permeability and impaired lymphatic drainage, proteins conjugated to polyethylene glycol (PEG) can potentially accumulate with the tumor. However, this is via a passive process. In order to more adequately target tumor cells, these PEGylated proteins can be conjugated to Tf. The effect of a conjugate consisting of Tf and a model PEGylated protein, β-lactoglobulin B (LG), was evaluated using K562 (human erythroleukemia), KB (human epidermoid carcinoma), S-180 (murine sarcoma 180), and normal human hepatocyte LO2 cells [60]. The effects of Tf-PEG-LG (TPL) have been compared to those of LG and PEG-LG. Cell surface binding of PEG-LG on K562 and KB cells was only 10% that of Tf, suggesting that TPL dramatically improved cancer cell targeting. The pharmacokinetics of 125I-LG, 125I-PEG-LG, and 125I-TPL were evaluated in rats [60]. LG was cleared quickly from the circulation, with PEG-LG and TPL remained longer. The half-lives of TPL, PEG-LG, and LG in the plasma circulation were 17.96 h, 8.13 h, and 0.84 h, respectively. In vivo, S-180 tumor bearing mice were used to track the biodistribution of the three treatments. LG showed the lowest blood levels due to its short half-life, while TPL displayed the longest mean residence time in circulation. The accumulation of TPL in tumor was higher than in all normal tissues compared to PEG-LG, demonstrating the potential of the TPL conjugate to selectively target tumors. This delivery system was then used to evaluate the effects of an anti-cancer protein, tumor necrosis factor-α (TNF-α). This conjugate containing Tf covalently attached to PEGylated TNF-α (TPT4) bound K562 and KB cells with an affinity similar to native Tf [61]. The biodistribution and anti-tumor effects were studied in Kunming mice bearing s.c. S-180 murine sarcomas [61]. The conjugate delayed blood clearance and enhanced tumor targeting. Additionally, the inhibitory rate of tumor growth was enhanced by TPT 5- fold when compared to TNF-α alone and roughly two fold when compared to PEGylated TNF-α. Thus, the tumor targeting ability and pharmacokinetic properties of TNF-α were enhanced by conjugation to Tf.
3.1.2.6. Tf-RNases
Various types of RNases have shown in vitro cytotoxicity against human cancer cells. They are capable of blocking protein synthesis and thus behaving like potent toxins once inside the cell. Therefore, in order to deliver bovine pancreatic ribonuclease A into targeted cancer cells, this nuclease was chemically conjugated to Tf (Tf-RNase) through a disulfide bond. This conjugate was cytotoxic to K562 human erythroleukemia cells in vitro with an IC50 of 10-7 M, which was 2-fold less than the IC50 of native RNase [62]. The cytotoxicity of Tf-RNase was inhibited by excess Tf or RNase inhibitors (RI), confirming the dependence of the conjugate on both Tf targeting and RNase activity for the observed cytotoxicity. A similar effect was observed with conjugates of Tf and onconase, a protein from fertilized Rana pipiens frog eggs that is a member of the pancreatic ribonuclease A superfamily. A direct comparison of these two conjugates showed that the IC50 of Tf-RNase in K562 cells was 400 nM, whereas the IC50 of Tf-onconase was 600 nM [62]. Additionally, the Tf-onconase conjugate was 20 to 100-fold more toxic on a molar basis than unconjugated onconase. However, onconase alone was cytotoxic to these cells suggesting that the compound alone can enter the cells. The conjugation of RNases to Tf enhances their cytotoxic properties, showing potential as anti-cancer therapeutics.
Human RNases have also been explored and conjugated to Tf as potential anti-cancer therapeutics. Pancreatic RNase (hRNAse) and eosinophil-derived neurotoxin (EDN) were mutated to incorporate cysteine residues at sites of close contact to the RI but distal from their catalytic domains [63]. Tf was conjugated to the mutant human RNase (rhRNase) and the mutant EDN to specifically target cells and block RNase inhibitor binding. Tf-rhRNase was 5000-fold more toxic in vitro to U251 human glioma cells relative to wild-type hRNase [63]. Tf-EDN showed toxicity similar to hRNase, confirming that the increased resistance to RI produced greater cytotoxicity. The IC50 of Tf-rhRNase was 2 nM and 1 nM for U251 and Wehi 7.1 (mouse T lymphoma cells), respectively. These values were significantly lower compared to the IC50 of rhRNase - 80 nM and 150 nM for U251 and Wehi 7.1, respectively, suggesting that the Tf targeting through conjugation increases cell specificity and cytotoxicity. The sensitivity of two RI-sensitive RNases increased by 200-fold when RI binding was prevented, 25-fold when conjugated, and 5000-fold when the two modifications were combined. Thus, both the inhibition of RI and conjugation of RNase to Tf increase the cytotoxicity of the IT, maximizing its potential as an anti-cancer therapeutic.
3.1.3. Tf-Nucleic Acid Conjugates
Targeting the TfR through the use of Tf can also be used for the purpose of gene therapy. Conjugation of DNA or RNA to Tf through various methods serves as a means to deliver therapeutic genes or inhibitory RNA molecules directly to cancer cells. In most cases, polycations are used since they spontaneously assemble with nucleic acids and serve as alternatives to viral vectors, which may be associated with safety concerns. The type of polycation used for DNA compaction and delivery can have a substantial effect on the intracellular trafficking of the cargo, the cellular response, and cytotoxicity of targeted cells. The two most widely used polymers that have been used for targeted gene therapy are polylysine and polyethyleneimine (PEI). Polylysine is a polyamino acid that is produced from a non-toxic, naturally occurring monomer. It contains terminal amines that are protonated at physiological pH and adequately condenses DNA by electrostatically binding phosphate groups of DNA. Polylysine particles can vary in size from 30 to 300 nm in diameter. The size of polylysine can have a dramatic effect on transfection efficiency, biodistribution, and cytotoxicity [64]. The second polymer, PEI, is found in both linear and hyperbranched forms [65]. Its buffering capacity is thought to be responsible for its high transfection efficiency due to neutralization of pH in the endosome and membrane lysis due to change in ionic osmolarity. It also efficiently condenses DNA through the use of its protonated secondary amines and can be found in high (800 kDa) and low (22-25 kDa) molecular weight forms.
Conjugation of these molecules to Tf enhances tumor targeting and gene delivery, a process known as “transferrinfection” [66, 67]. Chemical conjugation of Tf to polylysine through a disulfide bond or a carbohydrate link has shown an increase in luciferase transgene expression in K562 human erythroleukemia cells [68, 69]. Delivery of the luciferase transgene using polylysine was also observed in chicken HD-3 erythroblasts when either human Tf or the chicken homologue conalbumin was used for targeted delivery [66]. Maleimide modified Tf resulted in a 10-fold increase in transfection efficiency in murine erythroleukemia cell lines (F-MEL and J2E) compared to other common transfection protocols, including electroporation, modified DEAE-dextran, synthetic lysosomes, or modified calcium phosphate procedures [70]. Taken together, these studies show that Tf conjugated to polylysine using a variety of strategies can be successfully used to deliver nucleic acids into targeted cells.
The use of peptides that contain viral-like properties has also been explored in combination with Tf-polylysine-DNA complexes. For this purpose, the influenza virus hemagglutinin HA-2 subunit that has membrane destruction properties was conjugated to polylysine and added to Tf-polylysine [71]. The presence of the HA-2 peptide in these complexes dramatically augmented Tf-polylysine-mediated gene transfer in K562 cells. Biotinylated HA-2 peptide has also been conjugated to Tf-polylysine via a streptavidin-polylysine conjugate [72]. This complex enhanced transfection efficiency of human melanoma cells and led to high transgene expression, including the secretion of high amounts of interleuken-2 (IL-2) into the tissue culture supernatants. Similar secretion levels were observed using Tf-polylysine ionically conjugated to polylysine-containing DNA complexes [72]. These studies show that virus-mimicking peptides promote vesicle lysis and release of DNA into the cytoplasm resulting in increased transgene expression.
Transferrinfection can also be mediated by the conjugation of Tf to PEI. Tf-PEI (800 kDa) showed high levels of luciferase transgene expression similar to that of an adenovirus-enhanced transferrinfection (AVET, see section 4.6.1. below for more details) [73]. This complex also resulted in high levels of IL-2 transgene expression and secretion in primary human melanoma cells, nonetheless these levels were similar to non-targeted PEI complexes [73]. However, in animals bearing s.c. M3 or B16F10 murine melanoma cells, the transfection efficiency was eight to 10-fold higher with Tf-PEI complexes injected intratumorally compared to the non-targeted PEI complexes [73]. Furthermore the addition of polyethylene glycol (PEG) to the Tf-PEI stabilized the complexes and improved the in vivo characteristics of the complexes. The presence of PEG prolonged circulation in the blood of A/J mice injected systemically via intravenous (i.v.) injection with the complexes [74]. PEG shields positive charges and thus decreased the interaction with plasma proteins, protected the DNA from nucleases, and decreased the amount of erythrocyte aggregation. This led to less toxicity and enhanced tumor targeting of either Neuro2A murine neuroblastoma or M3 murine melanoma growing s.c in syngeneic animals [74, 75].
Tf-PEI was also used to deliver therapeutic transgenes. TNF-α is a potent cytokine and has anti-cancer properties; however, non-specific toxicity often hampers its wide-spread use for cancer therapy [76]. Three different murine syngeneic models were tested using either Neuro2a murine neuroblastoma, MethA murine fibrosarcoma, or M-3 murine melanoma cells growing s.c. [77, 78]. Systemic treatment via i.v. injection with Tf-PEI complexes consisting of the TNF-α gene resulted in preferential expression of the cytokine within the tumors, without detectable levels in the serum. High serum levels of TNF-α were observed in animals treated with non-targeted complexes. Significant anti-tumor effects of Tf-PEI were observed in all three models, with complete tumor regression in the MethA model.
Moreover, Tf-PEI has been used to deliver short hairpin RNA (shRNA) that knocked down the expression of the hypoxia-inducible factor-1α (HIF-1α) in human melanoma cells [79]. HIF-1α expression is upregulated in the presence of low oxygen levels. This protein plays an important role in angiogenesis, glucose utilization, and tumor cell survival [80]. A single i.v. injection of Tf-PEI-shRNA specific for HIF-1α into nude mice bearing s.c. A375 xenografts showed an 8-fold decrease in the tumor growth rate compared to animals treated with complexes consisting of scrambled, non-targeted shRNA [79]. Furthermore, in animals bearing A2780 human melanoma tumors that do not express the TfR, no change in tumor growth was observed, showing the need for TfR targeting to achieve the anti-tumor effects of the targeted complexes.
A slightly different human TfR-targeted delivery approach utilizes the strong interaction of biotin with avidin to deliver DNA-containing PEI conjugates to tumor cells. Biotinylated Tf was mixed with avidin and biotinylated disulfide PEI complexes containing the tumor suppressor gene p53 [81]. In vitro delivery of these targeted complexes led to an increase in p53 expression and induction of apoptosis in HepG2 human hepatocellular carcinoma cells or HeLa human cervical adenocarcinoma cells. Biotinylated Tf has been used to deliver DNA without the use of PEI as well. Biotinylated Tf was added to streptavidin and biotinylated DNA plasmid, sequentially. These targeted complexes delivered functional DNA into cells expressing high levels of the hTfR (K562 human erythroleukemic, M7069 human colon cancer, and TMK-1 human gastric cancer cells) as evidenced by β-galactosidase activity [82]. However, the β-galactosidase activity was very low in human embryonic lung cells that express low levels of the hTfR. Additionally, the tissue distribution of the complexes in animals bearing s.c. K562 tumors was evaluated [82]. Mice treated i.v. with these targeted complexes carrying the green fluorescence protein (GFP) reporter gene showed disseminated expression in many tissues early after injection. However, by day three after injection of GFP, expression in the tumor was high and was maintained through day seven. GFP expression in the other tissues was weak and diminished over time. After seven days this signal was minimal. Delivery of a therapeutic gene was then tested in a metastatic tumor model. The herpes simplex virus thymidine kinase (HSV-TK) was used as the therapeutic gene. Expression of this enzyme renders cells sensitive to gancyclovir (GCV). K562 cells were injected i.v. in SCID mice. After metastatic disease had developed, animals were treated with the targeted complexes daily for four days. Then they were treated for seven days with GCV. Targeted treatment in combination with GCV significantly prolonged survival [82]. Taken together, the above studies show that conjugates containing Tf are capable of delivering a wide variety of therapeutic nucleic acids (including DNA and shRNA) and have shown anti-cancer effects both in vitro and in vivo.
3.2. Antibody and antibody fragment conjugates
3.2.1. Rat anti-murine TfR
RI7 217 and YE1/9.9 are rat monoclonal IgG2a antibodies specific for the mouse TfR. They bind to partially overlapping sites on the receptor and do not interfere with Tf binding [83]. Neither has been demonstrated to have direct anti-cancer activity, however, when chemically conjugated to RTA both were shown to have at least a 10-fold higher in vitro cytotoxicity against murine malignant cells compared to non-targeted ITs [83]. R17 217-conjugated RTA inhibited protein synthesis in BW5147 murine T-lymphoma cells, but not in CCRF-CEM human T-leukemic cells [84], demonstrating the species specificity of the antibody. At similar concentrations, the conjugate was also shown to be toxic to late erythroid progenitor cells (CFUe and BFUe). This is consistent with the high level of TfR expression in these cells. In vivo, donor bone marrow was incubated with RI7 217-RTA for four hours and was then injected into sublethally irradiated (850 r) CAF1 mice to test the sensitivity of bone marrow pluripotent stem cells to the IT using spleen colony assays. Under conditions in which the IT was toxic to CFUe, it inhibited CFUs (colony forming units) no more than 10% [84]. Therefore, the conjugate is not toxic to early non-committed hematopoietic progenitors and stem cells and thus, this population of cells should survive treatment and be able to repopulate the later progenitor cells that are affected by treatment with the IT. Both R17 217 and YE1/9.9 also demonstrated in vivo anti-cancer activity against syngeneic peritoneal P388D1 murine lymphoid tumor growth when treatment was given i.p [83]. Additionally, R17 217 prolonged survival of mice bearing murine ovarian teratocarcinoma implants.
A rat/mouse chimeric antibody (ch17 217) composed of the rat variable regions of the R17 217 antibody was developed and genetically fused to murine granulocyte-macrophage colony stimulating factor (GM-CSF) [85]. This cytokine has been used as a potent adjuvant for cancer immunotherapy since it has many anti-cancer functions including the stimulation of granulocyte and monocyte differentiation from hematopoietic stem cells, upregulation of major histocompatibility type II (MHC II) molecules, and the increase of antigen presentation in antigen presenting cells [86, 87]. The GM-CSF-containing fusion protein retained both the antibody and cytokine activities and was also shown to have in vivo anti-cancer effects [85]. It decreased the growth of metastases in two different syngeneic mouse models, including the hepatic metastases of NXS2 murine neuroblastoma cells (syngeneic to A/J) and pulmonary metastases of CT26 murine colon carcinoma cells (syngeneic to BALB/c).
3.2.2. Murine anti-rat TfR (OX26)
OX26 is a murine IgG2a anti-rat TfR antibody that has also been chemically conjugated to the RTA chain to produce an IT. This IT demonstrated anti-tumor activity in a 3 dimensional culture model system of multicellular tumor spheroids 9L of rat glioblastoma cells. High concentrations demonstrated complete growth inhibition, however, lower doses resulted in a heterogeneous response [44].
3.2.3. Murine anti-human TfR
3.2.3.1. Antibody HB21(5E9)
HB21 (also known as 5E9) is a monoclonal murine IgG1 antibody directed against the human TfR that has been shown not to inhibit the binding of Tf to the receptor [88]. It has been used, either intact or as antibody fragments, to deliver a number of toxic proteins to cancer cells. Like the DT, the Pseudomonas exotoxin (PE) is a bacterial toxin that inhibits protein synthesis by ADP-ribosylating elongation factor 2, leading to cell death. Chemical conjugation of HB21 with the PE created an IT,HB21-PE, that inhibited protein synthesis in human oral KB carcinoma cells, OVCAR-2, -3, -4, -5, and A1847 human ovarian carcinoma cells, and MCF-7 human breast carcinoma cells according to the degree of cellular binding and uptake of the IT [89]. Additionally, combination of HB21-PE with other anti-cancer agents shows enhanced cytotoxicity. Verapamil, a calcium channel blocker, enhanced the rate of protein synthesis inhibition in human ovarian carcinoma cells [89]. Calmodulin antagonists have also been shown to increase the cytotoxicity of HB21-PE [90]. Dansylcadaverine, a calmodulin antagonist, increased the activity of the IT in OVCAR-2 and -3 human ovarian carcinoma cells, and trifluoperazine dihydrochloride enhanced the cytotoxicity in OVCAR-2, -3, and -4 cells [90]. Inhibition of protein synthesis by HB21-PE in KB cells was enhanced by all three agents, and the activity of the IT was blocked by monensin, a carboxylic ionophore [90].
A truncated mutant of the PE, PE40, lacks the cell-binding domain of the toxin. An IT composed of a genetic fusion between the single chain Fv (scFv) of HB21 and the PE40 [HB21(Fv)PE40] has been constructed [91, 92]. This IT was cytotoxic to highly metastatic KM12L4 human colon carcinoma cells in vitro [91]. HB21(Fv)PE40 also had in vivo anti-tumor activity against liver metastases and s.c. KM12L4 tumors in nude mice. However, while three consecutive i.v. injections of 10 μg/dose of the IT eradicated liver metastases, it only delayed the growth of s.c. tumors. These variable responses to the IT were found to correlate with the TfR expression levels on the surface of the cells [91]. TfR expression was higher in cancer cells isolated from the liver. This shows that the expression of the receptor in KM12L4 cells is modulated by the microenvironment of the organ in which they are located. This fusion protein was also cytotoxic to human epidermoid carcinoma, breast carcinoma, and ovarian carcinoma cells among others [92]. This fusion protein was also at least 100-fold more cytotoxic than anti-TfR-LysPE40 and DT388-anti-TfR(Fv) (both of which will be discussed in the following paragraphs) [92]. Treatment of DLD1 human colon cancer cells or SKOV-3 human ovarian cancer cells with this IT adequately blocked protein synthesis, however, minimal cytotoxicity was observed. ABT-737, a BH3-only mimetic and proapoptotic Bcl-2 family member, used in combination with the anti-TfR(Fv)-PE40 was found to enhance apoptosis in KB3-1 human cervical cancer cells (which are sensitive to the effects of the IT alone) [93]. However, since the cytotoxicity of DT388-anti-TfR(Fv) was not enhanced by ABT-737, it seems that ABT-737 increases the toxicity only of PE-based ITs by promoting their translocation from the endoplasmic reticulum to the cytosol after internalization, a pathway which the DT does not utilize.
A modified form of the PE40 has a chemically reactive lysine residue included in its amino terminus (LysPE40). This modified toxin has been coupled to the HB21 antibody. The resulting IT was found to be selectively cytotoxic to various human cell lines: epidermoid carcinoma cells, KB cells, ovarian carcinoma cells, colon adenocarcinoma cells, T-cell lymphoma cells, and acute lymphoblastic leukemia cells [94]. It was most toxic to A431 human epidermoid carcinoma cells. In vivo, a treatment of four i.p. injections of 5 μg, 20 μg, 50 μg, or 150 μg of HB21-LysPE40 given over eight days caused regression of solid A431 tumors in s.c. xenografts in nude mice [94].
The diphtheria toxin 388 (DT388) has also been used to target tumor cells via the hTfR. When the C-terminus of a truncated form of the DT388 was genetically fused to the scFv of HB21, the IT showed specific cytotoxic activity in hTfR-expressing cells in a panel of human cancer cell lines [92]. However, the DT388 IT was generally less active than the PE40 IT, as discussed previously. The cholera exotoxin 40 (CET40) has also been targeted to the hTfR using HB21. The cholera exotoxin is derived from Vibrio cholerae, the bacterium that causes cholera. The cytotoxicity of a truncated version of the toxin that was genetically conjugated to HB21 was increased by greater than 10-fold by the addition of the BH3 mimetic ABT-737 in KB3-1 cells [93]. As mentioned previously, this indicates that ABT-737 increases the delivery of toxins, including the CET40 and the PE40, from the endoplasmic reticulum to the cytosol.
HB21 (5E9) has also been chemically conjugated to the plant toxin gelonin [95]. Like SO6, gelonin is a single chain ribosome-inactivating protein isolated from Gelonium multiflorum that inhibits protein synthesis. 5E9-gelonin was found to be highly toxic to a panel of human malignant cell lines, but had no effect on RADA murine thymic leukemia cells [95]. In vivo, 5E9-gelonin was administered via a single systemic i.v. injection to nude mice bearing peritoneal ascites xenografts of several human cancer cell lines [95]. It was more effective than gelonin alone and prolonged survival of the tumor-bearing animals. The IT was more effective than cyclophosphamide (a chemotherapeutic drug) and at least as effective as ADR. Intravenous injection of 5E9-gelonin also inhibited growth of s.c. nodules of the Burkitt’s lymphoma Namalwa cell line in nude mice when injected simultaneously with inoculation or when injected intratumorally [95]. Moreover, 5E9-gelonin inhibited growth of i.p. implants of Namalwa when injected three to five days after inoculation. 5E9 has also been chemically linked to pokeweed antiviral protein (PAP). PAP is a toxic protein that inhibits protein synthesis. When 5E9-PAP was tested on three human acute lymphoblastic leukemia T-cell lines (HSB-2, MOLT-3, and CEM-CCL-119), various sensitivities to the IT were observed. HSB-2 cells were more sensitive than MOLT-3 or CEM cells to the effects of 5E9-PAP. Chloroquine potentiated the inhibitory effects of the IT [96]. A toxic protein that is derived from the seeds of Luffa aegyptiaca was shown to be a ribosomal inhibitory protein (LRIP) with functional similarity to the other ribosomal inhibitory proteins that have been discussed thus far. When 5E9 was chemically linked to LRIP, the resulting IT was highly cytotoxic to HSB-2 cells in a specific, dose-dependent manner [97].
Additionally, 5E9 has been chemically conjugated to ADR via an acid-sensitive 13-acylhydrazone linker [98]. This conjugate demonstrated anti-cancer activity in vitro toward human Daudi lymphoma cells. Moreover, significant in vivo inhibition of s.c. Daudi xenograft growth in BALB/c (nu/nu) mice was observed upon i.p. treatment [98]. It was also shown that hydrolysis of the 13-acylhydrazone bond to release free ADR was required for the cytotoxic effects of 5E9-ADR.
Restrictocin is a ribosomal inhibitory protein that is unable to bind to the surface of cells. It is produced by Aspergillus restrictus and cleaves the 28S rRNA subunit to inhibit protein synthesis. Fusing the 5’ end of the DNA encoding restrictocin to the DNA encoding scFv of HB21 yields anti-TFR(scFv)-restrictocin fusion protein, while fusion at the 3’ end of restrictocin produces restrictocin-anti-TFR(scFv) [99]. Both chimeric toxins specifically inhibited protein synthesis in a dose-dependent manner in a cell-free in vitro protein synthesis assay in a wide variety of malignant human cell lines, although they were about 30-fold less active compared to recombinant restrictocin alone. The cell lines tested were K562 human erythroleukemia, HUT102 human T-cell leukemia, MCF7 human breast adenocarcinoma, COLO205 human colon adenocarcinoma, A431 human epidermoid carcinoma, A549 human lung carcinoma, and HeLa human cervical carcinoma. The ITs had the greatest effect on K562 cells, which express high levels of hTfR. However, restrictocin-anti-TfR(scFv) was found to be three to 12-fold more cytotoxic (depending on the cell line) than anti-TfR(scFv)-restrictocin in all cell lines tested [99]. Since the ITs bind hTfR and inhibit in vitro translation similarly, the differences in their activity seem to be due to intracellular events or factors. Such factors may be differences in translocation efficiency of restrictocin fragments via the Golgi apparatus and/or in the interaction of the fragments with the target RNA. Chemical incorporation of a 12-residue spacer that contains the recognition site for the protease furin between restrictocin and the HB21 scFv, in either the N-terminus or C-terminus arrangement, enhanced cytotoxicity by 2 to 30-fold in these same cell lines, depending on the cell line targeted [100]. Conversely, a furin-insensitive linker improved cytotoxicity in only the N-terminus construct (anti-TFR(scFv)-linker-restrictocin). Recombinant restrictocin has also been chemically conjugated to the full length HB21 via cleavable disulfide or non-cleavable thio-ether linkages [101]. The cleavable conjugate was found to specifically inhibit protein synthesis in a dose-dependent manner in the same panel of human cancer cell lines that were used to test the genetic fusion protein, while the non-cleavable conjugate had little cytotoxic activity in any cells lines except HUT102. This indicates that ribotoxin-based ITs may necessitate cleavable linkages in order to be maximally active, which may be due to differential processing and/or translocation after internalization.
Another toxin that has been targeted to the hTfR using HB21 is α-sarcin, a ribonucleolytic protein in the family of restrictocin that is secreted by the mold Aspergillus gignateus. α-sarcin interacts with the 28S ribosomal subunit to block protein synthesis. Chemical conjugation of recombinant α-sarcin to HB21 created an IT that specifically inhibited protein synthesis in HUT102, A549, MCF7, and U937 human histocytic lymphoma in a dose-dependent manner [102]. This effect could be blocked by the addition of excess HB21. No protein synthesis inhibition was observed in murine L929 fibroblasts indicating the need for hTfR targeting using HB21.
3.2.3.2. Antibody 454A12
454A12 is a murine IgG1 anti-hTfR antibody that has been used to target toxic proteins to cells expressing high levels of the TfR. CRM 107 is a modified diphtheria toxin where two amino acids in the B chain have been mutated. These mutations decrease the binding ability of the toxin 8000-fold but leave its translocation function unaffected [52]. When CRM 107 was chemically linked to 454A12, the resulting IT was cytotoxic to a panel of patient-derived medulloblastoma, glioblastoma, and breast carcinoma cell lines, but was not toxic to Vero cells that lack hTfR expression [52]. In the same study, 454A12 was also conjugated to a recombinant form of the RTA toxin. When the cytotoxicity of 454A12-RTA was tested in the same lines, 454A12-RTA showed similar cytotoxicity as 454A12-CRM 107 after 24 hours of incubation. However, after only 3 hours of incubation, the RTA IT was 10 to 1000-fold less toxic than the CRM 107 IT and thus 454A12-RTA demonstrated a much slower rate of cytotoxicity compared to 454A12-CRM 107 [52]. Free Tf did not have an effect on the toxicity of either of the 454A12 ITs, suggesting that 454A12 does not inhibit the binding of Tf to the receptor. However, the recombinant RTA IT displayed cytotoxic activity at very low concentrations in other human medulloblastoma (DAOY, D283MED, and D341MED), glioblastoma (U373), as well as in a neuroblastoma cell line (SH-SY5Y) [53]. Several pediatric tumor specimens, including both benign and highly malignant tumors, were also treated with low concentrations of 454A12-RTA. Interestingly, the more malignant tumors were susceptible to the IT while more slow-growing and benign tumors were not as sensitive. Another IT targeting the hTfR composed of CRM 107 and anti-TfR has also been tested for anti-cancer activity against these same cell lines. This IT was cytotoxic to the patient-dervied medulloblastoma, glioblastoma, and neuroblastoma cell lines in vitro and inhibited protein synthesis more rapidly compared to 454A12-RTA in the same cell lines [53]. Like 454A12-RTA, anti-TfR-CRM 107 had greater cytotoxicity in the more malignant pediatric tumor specimens, affecting the benign and slow-growing tumors much less. 454A12-RTA was also cytotoxic to human OVCAR-3 ovarian cancer cells, with an IC50 of 75 ng/mL [103]. This effect was potentiated by low doses of IFN-α. In vivo anti-cancer activity was also observed in established i.p. OVCAR xenografts. Intraperitoneal treatment of 454A12-RTA significantly increased survival of the animals [103]. Again, the anti-cancer activity was enhanced by IFN-α The survival rate of the IT alone was 29%, whereas the survival rate of the 454A12-RTA with IFN-α increased to 89% [103]. IFN-α alone had no anti-cancer effect in this model.
The in vitro activity of 454A12-RTA could be enhanced by combining it with other anti-cancer agents. Cytotoxicity was enhanced from two to 25-fold by verapamil depending on the concentration of verapamil in human OVCAR-3 and -4 and KB cells [90]. Dansylcadaverine also increased the cytotoxicity of the IT in OVCAR-3 and KB cells in a dose-dependent manner. Trifluoperazine, an antipsychotic of the phenothiazine chemical class, enhanced the activity of the IT in OVCAR-3 cells only. In vivo studies using 454A12 conjugated to RTA have also been completed. When nude mice with s.c. U251 MG flank tumors were treated intratumorally with 454A12-RTA four times on alternate days, mean tumor volume was reduced by 30% by day 14. Despite tumor regrowth by 10 days after the last treatment, tumor volumes were still smaller in treated mice than in control mice after 30 days [104]. Moreover, a single intraventricular injection of 120 μg or higher of 454A12-RTA into patients with leptomeningeal spread of systemic neoplasia was evaluated and showed to be tolerable with no systemic toxicities reported [105]. In four out of eight patients, a greater than 50% reduction of tumor cell counts in the lumbar cerebrospinal fluid was observed within five to seven days after treatment. The IT remained intact in the cerebrospinal fluid for up to 24 hours after injection. Even though, tumor progression was eventually observed in seven of the eight patients, this clinical trial showed that antitumor concentrations of 454A12-RTA can be attained in cancer patients. A second clinical trial was performed with i.p. therapy of this IT in twenty patients with metastatic carcinomas [106]. Only preliminary results were reported from this study and showed that 3 patients developed temporary superficial mucosities and one patient developed a fatal encephalopathy with cerebral edema. No anti-tumor effects were reported.
3.2.3.3. Antibody B3/25
B3/25 is a murine IgG1 antibody specific for the hTfR. This antibody has been used for the production of several ITs as potential therapeutics against cancer cells. The RTA subunit has been chemically conjugated to B3/25. The resulting IT specifically inhibited protein synthesis in CCRF-CEM human T leukemic cells, at levels similar but to a lesser extent than that of the intact ricin toxin [107]. The IT also inhibited protein synthesis in HeLa human cervical carcinoma cells and M21 human melanoma cells, the latter of which appeared less sensitive than CCRF-CEM [107]. In BALB/c (nu /nu) mice bearing s.c. M21 tumors, three i.v. injections of 50 μg of the antibody-RTA completely inhibited growth of M21 cells [107]. However, a single i.v. injection of 880 μg of antibody alone also blocked melanoma cell growth in this model. The B3/25 antibody alone was much less effective in preventing s.c. HeLa cell tumor growth compared to M21. B3/25 as also been covalently coupled to diphtheria fragment A, the toxic subunit of DT, producing an IT that specifically inhibited protein synthesis in CCRF-CEM cells in vitro [107].
Additionally, SO6 has been chemically linked to B3/25. This IT was cytotoxic to K562 (human erythroleukemic) and HL-60 (human promyelocytic leukemia cells) cells as evidenced by the inhibition of clonogenic cell growth and colony formation [108]. Neither the toxin alone nor a non-targeted IT demonstrated these effects. This conjugate was also toxic to normal committed human bone marrow progenitor cells (CFU-GM and BFU-E) and leukemic human hematopoietic progenitors (acute myeloid leukemia- AML- clonogenic cells) [108]. Erythroid progenitors were more sensitive to the IT than myeloid ones. An IT composed of B3/25 chemically conjugated to bovine pancreatic ribonuclease A, a protein that degrades RNA and thus inhibits translation, was produced and evaluated. This IT demonstrated cytotoxicity to K562 cells that was 100-fold higher than the ribonuclease A alone [109].
3.2.3.4. Antibody OKT9
OKT9 is a murine IgG1 anti-human TfR antibody that has been chemically conjugated with RTA. The cytotoxicity of the IT was evaluated against multicellular tumor spheroids (MTS) of MCF7 human breast carcinoma cells. While complete inhibition of MTS was observed using high concentrations of the IT, the response of individual tumor micromasses was extremely heterogenous at suboptimal doses [44]. MTS treated at these lower doses of the IT, eventually regrew to the same upper volume as control MTS. This indicates that the individual heterogeneity of MTS may significantly and unpredictably affect the cytoreductive potential of the IT.
3.2.3.5 Antibody 7D3
7D3 is also a murine IgG1 anti-human TfR antibody that has been used for IT production. When chemically conjugated to RTA, the IT inhibited protein synthesis in five human glioma cell lines (MG-1, -2, and -3, which were derived from surgical explants, and U-87 MG and U-373 MG, which are well characterized established lines) [110]. C6 rat glioma cells were not sensitive to the effects of this IT demonstrating the species specificity of the antibody. Monensin, a carboxylic ionophore, potentiated the cytotoxic effects of the IT by 16 to 842-fold depending on the cell line, with an average of 100-fold [110]. It was later discovered that monensin potentiated the effects of 7D3-RTA against H-Meso-1 human malignant mesothelioma and human colorectal cancer cells (LoVo and LS174T) [111]. Potentiation was also demonstrated in cells grown as human tumor ascites and peritoneal nodules in the peritoneum of nude mice bearing H-Meso-1 xenografts [111]. After treatment, these cells were removed from the mice and assessed for inhibition of protein synthesis. In testing the potentiation effects of monensin in vivo, it was found that IT plus monensin emulsion had greater antitumor effects than IT alone or IT plus monensin in buffer in BALB/c-nu/nu mice bearing H-Meso-1 tumors in their peritoneum. The tested treatments consisted of seven i.p. injections every other day, beginning 10-20 days after tumor cell injection. In mice bearing LS174T microscopic i.p. tumors, i.p. injection of IT plus monensin emulsion 24 hours after inoculation was more cytotoxic than IT alone [111]. In H-Meso-1 cells coincubated with 7D3-RTA and monensin, the cytotoxicity and the rate of cell killing were both higher than in cells treated only with IT. However, ADR showed no potentiation of the cytotoxicity of the IT [112]. When nude mice bearing H-Meso-1 i.p. xenografts were treated with IT plus monensin emulsion or IT plus doxorubicin, both treatments produced similar improvements of survival. Combining the IT, the monensin emulsion, and ADR further increased survival time by ten days. Thus, the combination of ITs with other therapeutic agents may enhance the antitumor properties of either agent alone [112].
3.2.3.6. Antibody 7579
The scFv of the murine monoclonal anti-human TfR antibody 7579 was genetically fused to a viral peptide/HLA-A2 complex in order to redirect cytotoxic T cells (CTL) to targeted cancer cells that express high levels of the hTfR [113]. HLA-A2 is a human class I histocompatibility molecule (MHCI) that presents antigens to CTL, which leads to the lysis of the cell displaying the antigen in the context of the HLA-A2 molecule. Either hepatitis B virus or an Epstein-Barr virus derived peptides were used to produce viral peptide/HLA-A2/scFv fusion proteins. When HLA class I-negative, TfR-expressing K562 cells were incubated with the fusion proteins and then with viral peptide-specific CTL, the fusion proteins were shown to mediate cytotoxicity of K562 cells. Partial inhibition of tumor growth and prolonged survival was observed in nude mice bearing K562 i.p. xenografts that were treated i.p. with the EBV/HLA-A2/scFv fusion protein and EBV-specific CTL [113].
3.2.3.7. Antibody 42/6
42/6 is a murine monoclonal IgA antibody that targets the human TfR and has been shown to have direct cytotoxic effects on cancer cells (reviewed in [1]). However, it has also been used as a delivery vehicle. The 42/6 antibody has been chemically conjugated to SO6. This IT showed equal cytotoxicity with a Tf-SO6 conjugate against RPMI 8226 human multiple myeloma cells [47]. The cytotoxicity of the 42/6 IT was only slightly diminished by the presence of diferric Tf, compared to the significant decrease of the toxicity mediated by the Tf-SO6 conjugate. Thus, the 42/6 IT may be advantageous over the Tf-SO6 conjugate since circulating Tf may decrease the anti-cancer efficacy of the latter. Additionally, chemical conjugation of 42/6 to bovine pancreatic ribonuclease A results in an IT that is cytotoxic to K562 cells [109].
3.2.4. Mouse/human chimeric antibodies
3.2.4.1. Antibody E6
E6 is a chimeric mouse/human IgG1 antibody specific for the human TfR that has been used as a delivery vehicle for cancer therapy. Angiogenin is a human angiogenic ribonuclease that inhibits protein synthesis. Genetic fusion of the angiogenin gene to the E6 antibody at its CH2 domains resulted in an IT that was cytotoxic to MDA-MB-231 human breast cancer cells and SF539 human glioma cells, but not to NIH3T3 mouse fibroblast cells [114]. The protein synthesis inhibition of the IT was equivalent to that of free angiogenin [114]. This fusion protein also inhibited protein synthesis in K562 cells expressing the hTfR but not in non-human-derived cells that lack the hTfR [115]. Neither E6 alone nor a combination of unfused angiogenin and the E6 antibody had toxic effects, indicating the need for the physical linkage between the two components for the observed toxicity.
The scFv region of the E6 antibody was also used to produce a fusion protein capable of targeting RNases to TfR-expressing cells. The scFv was genetically fused at its 5’ end to either human pancreatic RNase or human eosinophil-derived neurotoxin (EDN, a member of the RNase superfamily) [116, 117]. The resulting fusion proteins showed inhibition of protein synthesis in MALME human melanoma cells, ACHN human renal carcinoma cells, and MDA-MB-231 human breast carcinoma cells [117]. The fusion protein consisting of pancreatic RNase was slightly less active than the EDN-containing fusion protein on ACHN and MDA-MB-231, but the two fusion proteins exhibited similar antitumor effects in MALME cells. The fusion protein containing EDN also showed cytotoxicity to human K562 and A431 epidermoid carcinoma cells, but not to cells lacking the hTfR [116]. Excess free E6 antibody inhibited this effect. Several angiogenin-containing fusion proteins were developed using various linkers that connect the variable light and heavy chains of the scFv, as well as with or without a linker connecting the scFv and angiogenin [118]. All fusion proteins were cytotoxic to MDA-MB-231, ACHN, and HT-29 (human colon adenocarcinoma) cells, however, various sensitivities were observed. These results show that a spacer between the scFv and angiogenin is needed for optimal activity and that the linker can affect cellular binding and cellular cytotoxicity [118].
3.2.4.2. Antibody-Avidin fusion proteins
An antibody-avidin fusion protein consisting of avidin genetically fused to the CH3 domains of a mouse/human chimeric IgG3 molecule specific for the rat TfR has been described and was designed as a universal delivery system due to the high affinity interaction of avidin with biotin [15]. This antibody-avidin fusion protein contains the variable regions of the murine antibody OX26 and was surprisingly shown to have intrinsic pro-apoptotic activity. This fusion protein is also capable of delivering active biotinylated therapeutic agents into cells expressing the rat TfR [15]. Two non-toxic enyzmes (FCU1-P67 or PNP-P67) have been conjugated with this antibody-avidin fusion protein and used in an antibody-directed enzyme prodrug therapy (ADEPT) therapeutic strategy [119]. This method relies on the direct targeting of the enzyme in TfR-expressing cancer cells. Within the tumor microenvironment, the prodrug would be converted by the enzyme into toxic metabolites that lead to cancer cell death. FCU1-P67 is a fusion protein of the yeast enzyme FCU1 (a genetically engineered chimeric protein consisting of cytosine deaminase and uracil phosphoribosyltransferase) and the carboxyl-terminal domain of human propionyl-CoA carboxylase alpha subunit (P67). P67 serves as a human biotin acceptor domain, as it is recognized by BirA (a biotin holoenzyme synthetase) that mono-biotinylates the protein and through the conjugation to the antibody-avidin fusion protein can be targeted to TfR-expressing cells. The cytosine deaminase portion of the FCU1 enyzme cleaves the relatively non-toxic prodrug 5-fluorocytosine resulting in the formation of the toxic metabolite 5-fluorouracil, which is then metabolized by uracil phosphoribosyltransferase into 5-fluorouridine 5’-monophosphate, a second toxic metabolite. When biotinylated FCU1-P67 was conjugated to the antibody-avidin fusion protein, the IT effectively eliminated Y3-Ag1.2.3 rat myeloma cells in vitro only in the presence of 5-fluorocytosine in a dose-dependent manner [119]. Similarly, a second enzyme PNP-P67 was also used using this strategy. PNP is the Escherichia coli enzyme purine nucleoside phosphorylase [119]. Like FCU1, PNP was genetically fused to P67. PNP cleaves the prodrug 2-fluoro-2’-deoxyadenosine (F-dAdo) to produce the cytotoxic drug 2-fluoroadenine (F-Ade). Conjugation of the antibody-avidin fusion protein to biotinylated PNP-P67 resulted in dose-dependent cytotoxicity to Y3-Ag1.2.3 cells in the presence of F-dAdo [119]. No cytotoxic effects were observed when the biotinylated enzymes were conjugated to a non-targeted antibody-avidin fusion protein.
A similar antibody-avidin fusion protein has been produced that targets the human TfR. It consists of a mouse/human chimeric IgG3 with avidin genetically fused to the CH3 domains of the antibody. Similar to the fusion protein that targets the rat TfR, this fusion protein also has intrinsic in vitro cytotoxic activity against a variety of malignant human cells [15, 16, 120] as well as in vivo anti-cancer activity [121]. It also has delivery capabilities and has been conjugated to biotinylated SO6 [122]. The resulting IT demonstrated a stronger cytotoxic effect of the antibody-avidin fusion protein alone in cells that were sensitive to the fusion protein alone (IM-9 human EBV-transformed lymphoblastoid cells). The conjugate also overcame the resistance of U266 human myeloma cells to the cytotoxic effects of the antibody fusion protein alone. This study shows that this antibody-avidin fusion protein may be a useful therapeutic tool due to its functions as a direct anti-cancer agent as well as a universal delivery system.
3.3. TfR-targeted peptide conjugates
Two peptides targeting the human TfR that do not interfere with Tf binding to its receptor have been synthesized. These two peptides were covalently conjugated to artemisinin to form targeted conjugates [123]. These conjugates allow artemisinin to be internalized with receptor-bound Tf. The iron present on Tf interacts with artemisinin to generate toxic free radicals that can lead to oxidative stress-induced cell death. These conjugates demonstrated anti-cancer activity against Molt-4 human leukemia cells with an IC50 of about 1 μM [123]. They were not toxic to normal lymphocytes (IC50 > 10 mM), demonstrating cancer cell selectivity of the conjugates.
4. Targeting moiety conjugated to carriers loaded with the active compound
4.1. Polymeric micelles
Polymeric micelles are nanoscopic structures characterized by a core-shell structure and are the product of self-assembly of amphiphilic copolymers in aqueous media. These nano-structures tend to be smaller than 100 nm and have the capacity to hold hydrophobic drugs at their cores [124, 125]. The shell of the micelles is formed by the hydrophilic portion of the amphiphilic co-polymers, which favors the dispersion of the system in aqueous media [126] and the increase of circulation time in vivo, favoring a preferential accumulation in tumor [127]. The self-assembly of the micelles takes place above a concentration called the critical micelle concentration (CMC). Amphiphilic copolymers generally have a much lower CMC value than that presented by low molecular weight surfactants [128]. This feature makes polymeric micelle structures very stable to dilution and thus they can circulate in the bloodstream with negligible dissociation [129]. The architecture of these nanoscopic structures can increase the solubility [130], control the release [131], reduce toxicity [132], and improve drug selectivity for the tumor site [133].
The binding of ligands such as Tf to the surface of the micelles has been used for the direct targeting of these structures as carriers of antineoplastic drugs or genetic material to tumors [134]. Polyion micelles for TfR mediated targeting to tumors were prepared by synthesizing a copolymer of poly(ethylene glycol)-poly(ethylene imine) biotin (biotin-PEG-PEI) that form micellar structures to which were bound biotin-conjugated Tf to form a complex using avidin as the linker [135]. These complex micelles had a diameter of 75-103 nm and were used for the delivery of antisense DNA into cancer cells. The loaded nanoparticle significantly improved the incorporation and release of antisense oligonucleotides against human multidrug resistance protein-1 (MDR-1) to inhibit expression in vitro of the efflux transporter P-glycoprotein (Pgp), which is responsible for multidrug resistance in human oral epidermoid carcinoma KBV cells and human breast carcinoma MCF-7ADR cells [135]. Using a similar strategy, Tf was coupled via biotin-avidin to the surface of micelles of PEG-PLAbiotin (Tf-PEG-PLA) with the aim of targeting tumors on the central nervous system. The diameters of the nanoparticles were 95–110 nm, and flow cytometry measurements showed that the cellular uptake of the Tf-PEG-PLA micelles was significantly higher (92.8 ± 2.5%) than the PEG-PLA micelles without Tf (68.3 ± 3.1%) in rat C6 glioma cells in vitro. Observations by fluorescence microscopy of brain slices in an intra-cranial rat tumor model of C6 glioma showed that PEG-PLA micelles functionalized with Tf could target the tumor cells in vivo [136], although further studies are needed to confirm that Tf functionalized PEG–PLA nanoparticles are effective in protecting against brain tumor growth.
4.2. Dendrimers
Dendrimers are three-dimensional, non-immunogenic, hyper-branched, polymeric structures defined by a central core with radially-oriented stems that form globular or semi-globular nanoparticles [137]. These synthetic macromolecules are also known as arborols, cauliflower or cascade polymers because of their particular structure [138]. The basic architecture of dendrimers consists of three parts: 1) a multifunctional central core; 2) branching units surrounding the core; and 3) the surface, with multiple functional groups. The central core is surrounded by layers called generations (G), which are incorporated stepwise in focal points during their synthesis [139]. The different layers are synthesized in an iterative manner from monomers, allowing the control of the dimensions of the dendrimer [140, 141]. In general there are two approaches for the synthesis of dendrimers: divergent and convergent. Divergent synthesis starts at the core and the dendrimer is built up radially, generation by generation, with the number of Gs proportional to its size. In contrast, the convergent approach starts from the surface and concludes at the core [142]. These structures have a size between 2-10 nm and the volume can be easily modified according to the number of Gs incorporated, i.e.: the polyamidoamine PAMAM G-2 dendrimer has a diameter of 2.3 nm, while that of PAMAM G-5 is 5.3 nm [137].
Today, dendrimers have a variety of applications in cancer therapy, including the encapsulation or conjugation of antineoplasic drugs [143, 144], and the transport of nucleic acids (Table 1) [145]. Dendrimers can hold different substances within their layers via complexes with functional groups called endo-receptors as well as on its periphery conjugated to other groups called exo-receptors [138, 146]. In the case of the poly(propylene imine) (PPI) dendrimers, the presence of multiple amino groups on its surface provide the proper platform for the binding of specific ligands that can be used for the targeting of anticancer compounds to tumor cells [147]. Koppu et al. developed dendrimers of diaminobutiric poly(propylene imine) (DAB) G-3 conjugated to Tf (DAB-Tf) using dimethylsuberimidate (DMS) as a crosslinking agent as a therapeutic gene delivery system of plasmid DNA against human tumors [148]. Cell lines A431 (human epithelial carcinoma cell line) and T98G (human glioma cell line) were used to determine the efficacy of transfection of the DAB-Tf system with a plasmid coding for β-galactosidase. The dendrimer with Tf exhibited 1.3-fold higher expression compared to the dendrimer without Tf. The i.v. injection of DAB-Tf with the vector coding for the enzyme into female BALB/c immunodeficient mice bearing s.c. A431 tumors achieved higher expression of the enzyme in the tumor compared to the other organs. Moreover, when DAB-Tf was complexed with a plasmid coding for TNF-α (DAB-Tf-complex) and injected i.v. it achieved complete tumor response in 90% of cases, compared to the DAB-complex without Tf, which resulted in complete tumor response in 40% of cases [148].
Table 1.
Examples of TfR-targeted carriers used to deliver therapeutic agents into cancer cells.
| System | Composition | Ligand | Size (nm) | Cargo | Cell/Tumor model | Comments | Ref |
|---|---|---|---|---|---|---|---|
| Liposomes | Chol/DSPC/DSPE–PEG2000–COOH | holo-Tf | 70 | ADR | Human hepatocellularcarcinoma cell line (HepG2) | The IC50 of Tf-SL-ADR was 5.22 μM, SL-ADR was 20.4 μM and free ADR was 166.2 μM. | 185 |
|
| |||||||
| Male ICR mice bearing s.c. HepG2 cells | The average weight of tumors in the group of mice treated with Tf-SL-ADR was 0.33g, with SL-ADR 1.17 g, and with free ADR 1.38 g. The dose equivalent of 3 mg/kg of ADR was used in all treatments. | ||||||
|
| |||||||
| SPC/Chol/ mPEG-PE |
holo-Tf | 129 | Cisplatin (cDDP) | human ovarian carcinoma cell lines A2780 and A2780cis (cDDP resistant) | IC50 at 48h = 8.28 μM compared to the free cDDP (IC50 at 48h = 14.47 μM) in drug resistant cell only. | 193 | |
|
| |||||||
| DPC/chol/ MBPE |
Murine IgG1 anti-human TfR (OKT9) | N/D | ADR | K562 human erythroleukemia cell line | Free ADR and OKT9-liposome loaded with ADR showed similar IC50 0.45 and 0.35 μg/ml respectively. | 195 | |
|
| |||||||
| ADR-resistant subline K562/ADM | 3.5 times lower IC50 (8 μg/mL) than free ADR (28 μg/mL). | ||||||
|
| |||||||
| DC-Chol/egg PC/PEG–DSPE | holo-Tf | 126–156 | Oligonucleotide (G3139 a Bcl-2 antisense) | K562 human erythroleukemia cells | Exhibited significantly greater Bcl-2 dowregulation (up to 70%) compared to 8% by free G3139 and 40% by non- targeted LP-G3139. | 190-191 | |
| Decreased the IC50 of daunorubicin from 1.77 μM to 0.18 μM (a 9.8-fold decrease). | |||||||
|
| |||||||
| Athymic nude mice bearing s.c. K562 tumors | Greater accumulation in the tumor and was found to be more effective in tumor growth inhibition and extending survival than free G3139. | ||||||
|
| |||||||
| DOTAP/DDAB/DOPE | Murine IgG1 anti-human TfR scFv (5E9) | 92.9 | luciferase gene | Human prostate cell line DU145 human breast cancer cell line MDA-MB-435 |
4-10 fold more efficient transfection activity compared to the untargeted liposomes. | 201-202 | |
|
| |||||||
| p53 expression plasmid (pCMVp53) |
Nude mice bearing s.c. DU145 human prostate cancer cells | Displayed a high level of the expression of exogenous wild type 53 preferentially in the tumors. | |||||
|
| |||||||
| Nude mice bearing s.c. JSQ-3 human head and neck cancer cells | Tumor specific expression observed after systemic gene delivery. | ||||||
|
| |||||||
| Chol/DSPC/DODAP/ | Holo-Tf | 175.5 | siRNA and ODN (anti-BCR-ABL) | LAMA-84 leukemia cell line | Cell internalization of fluorescently labeled siRNA (Cy3-siRNA) was only detected with Tf-liposomes. For non-targeted liposome no Cys3-siRNA inside the cell was observed. | 204 | |
|
| |||||||
| K562 human erythroleukemia cell line | Treatment with Tf-liposomes loaded with 2μM siRNA or ODN resulted in higher levels of toxicity. Non-targeted liposomes or empty targeted liposomes at the same lipid concentration did not induce toxicity. | ||||||
|
| |||||||
| PC/chol/ DOPE-DPPTPA/mPEG-DSPE |
Holo-Tf | 118.3 | ADR | KB human carcinoma cell line identified as a subline derived from HeLa cervical cancer cells | Tf-liposomes loaded with ADR and free ADR showed similar IC50, 13.91 and 14.38 μM respectively, but both were lower than non-targeted liposomes (43.74 μM) | 187 | |
|
| |||||||
| DSPC/Chol /DSPE-PEG | Holo-Tf | 180-200 | cDDP | Nude mice bearing i.p. MKN45P human gastric cancer cell line | Tf-PEG-liposome loaded with cDDP injected at day 1 and 4 at a dose of 5 mg/kg significantly extended survival of mice compared to non-targeted PEG-liposomes, free cDDP or bare liposomes. | 186 | |
|
| |||||||
| DOTAP/ DOPE |
Holo-Tf | 63.8 | p53 expression plasmid (pCMVp53) |
Nude mice bearing DU145 human head and neck cancer cells in a s.c. xenograft model | Targeted liposomes loaded the plasmid plus radiation-showed significant improvement in survival with complete tumor regression and no sign of recurrence even 6 months after treatment, compared to targeted and loaded liposomes, radiation alone, or non-targeted liposomes. | 189 | |
|
| |||||||
| Dendrimers | Polypropylenimine-G3 | Tf | 287 | Plasmid DNA coding for TNFα | A431 human epidermoid carcinoma cell line and T98G glioblastoma in vitro. A431 in vivo in nude mice s.c. xenograft model. | In vitro IC50 of 7.09±0.88 μg/mL compared to nontargeted DAB polyplexes (IC50 of 9.47±1.15 μg/mL) in A431 cells. In vitro IC50 of 21.72± 2.68 μg/mL compared to nontargeted DAB polyplexes (IC50 of 21.72± 2.68 μg/mL) in T98G cells. In the A431 s.c. model 50 μg Tf-DAB polyplexes injected i.v. elicited tumor regression and long term survival with 90% complete response and 10% partial response after one month. | 147 |
|
| |||||||
| PEG-Poly(amidoamine)-G4 (PAMAM) | Tf and wheat germ agglutinin (WGA) | 199 | ADR | Murine C6 glioma cells | In vitro cytotoxicity showed an of IC50 = 0.271 μM compared to free ADR (IC50 = 0.315 μM). | 150 | |
|
| |||||||
| PEG-Poly(amidoamine)-G5 (PAMAM) | Peptide HAIYPRH (T7) | 10 | ADR | Human Bel-7402 hepatocellular carcinoma cell line | In vitro cytotoxicity in the presence of soluble Tf (IC50 231.5 nM) than in the absence of Tf (IC50 676.7 nM). This was close to that of free ADR (IC50 191.5 nM). | 148 | |
|
| |||||||
| BALB/c nude mice bearing s.c. Bel-7402 xenograft tumors | Significantly enhanced ADR accumulation in the tumor by approximately 1.7-fold compared to that of unmodified ones and by approximately 5.3-fold compared to that of free ADR. For in vivo antitumor studies, tumor growth of mice treated i.v. with 2 mg ADR/kg was significantly inhibited compared to that of mice treated with non-targeted dendrimer or saline, but not to 5 mg free ADR/kg. | ||||||
|
| |||||||
| PEG-Poly(amidoamine)-G5 (PAMAM) | Peptide HAIYPRH (T7) | 200 | Therapeutic gene encoding human tumor necrosis factor-related apoptosis-inducing ligand (pORF-hTRAIL) and ADR. | BALB/c nude mice bearing Bel-7402 s.c. tumors | Higher efficiency in cellular uptake and gene expression than unmodified co-delivery system in Bel-7402 cells. The co-delivery system induced apoptosis of tumor cells in vitro and inhibited tumor growth in vivo more efficiently in comparison with single ADR or pORF-hTRAIL delivery system. In mice bearing Bel-7402 xenografts, lower doses (0.16 mg/kg ADR) effectively inhibited tumor growth comparable to high doses (5 mg/kg) of free ADR (77% versus 69%). | 149 | |
|
| |||||||
| Nanoparticles | PLGA; D-α-tocopherol polyethylene glycol 1000 succinate (TPGS) | Holo-Tf | 187.0 | 7α-(4’-amino) phenylthio-1,4-androstadiene-3,17-dione (7α-APTADD) | Human breast cancer cell line SKBR-3 | IC50 of the targeted 7α-APTADD loaded Tf-nanoparticles was in the range of 0.77–1.21 nM and the IC50 of the 7α-APTADD loaded non-targeted nanoparticles was in the range of 1.90–3.41nM. | 160 |
|
| |||||||
| PLA-PEG-biotin (BP); PLA-PEG (LP) | Biotinilated Tf | 106.8 | Paclitaxel (PTX) (Taxol®) |
BT4C rat glioma cell line | Significantly increased the anti-tumoral activity compared to free PTX and non-targeted nanoparticles in vitro. | 164 | |
|
| |||||||
| Poly(aminopoly (ethylene glycol)-cyanoacrylate-co-hexadecyl cyanoacrylate) (H2N-PEG-PHDCA), MePEG-PHDCA and poly(hexadecyl cyanoacrylate) (PHDCA) |
Holo-Tf | 109.8 | Poly(ethylene) glycol-hydroxycamptothecin conjugate (PEG-HCPT) | Mice bearing murine sarcoma S180 cell line s.c. tumors | For mice treated i.v. with 1 mg/kg × 7 Tf-PEG-NP (PEG-HCPT), the mean tumor weight was 0.03 g which was significantly lower compared to 0.12 g of PEG-NP (PEG-HCPT), 0.18 g of non-NP (PEG-HCPT), 0.23 g of PEG-HCPT solution, 0.31 g of HCPT alone, and 0.47 g for saline. | 166 | |
|
| |||||||
| PLGA | Holo-Tf | 222 | PTX | Human breast cancer cells MCF-7/Adr resistant to ADR | At a concentration of 1 g/ml the targeted particle showed a 70% antiproliferative effect, while non-targeted particles showed 55% and free PTX showed 20%. | 162 | |
|
| |||||||
| Poly(aminopoly (ethylene glycol) cyanoacrylate-co-hexadecyl cyanoacrylate) (poly(H2 NPEGCA-co-HDCA)) | Holo-Tf | 101.4 | PTX | Mice bearing s.c. murine sarcoma S-180 cells | For mice treated i.v. with 20 mg/kg × 5 Tf-NP (PTX), the tumor burden was much smaller compared with free PTX or non-targeted NPs. In addition, the life span of tumor-bearing mice was significantly increased when they were treated with Tf-NPs. | 165 | |
|
| |||||||
| Poly(lactide)-d-α-Tocopheryl polyethylene glycol succinate (PLA-TPGS) | Holo-Tf | 137.6 | Docetaxel | C6 rat glioma cell line | In vitro cytotoxicity: the IC50 of docetaxel loaded PLA-TPGS/Tf was5.10±0.44 μg/mL, for PLA-TPGS 5.96±1.15 μg/mL, for PLGA = 6.53±1.26 μg/mL, and for Taxotere® = 16.77±3.93 μg/mL. | 167 | |
|
| |||||||
| PLGA | Holo-Tf | 220 | PTX | Human prostate cáncer cell line PC3 | The IC50 for PTX loaded Tf-conjugated NPs (PTX-NPs-Tf) was 0.6×10-3 μM, for non-targeted Tx-NPs was 3.1×10-3 μM, and for PTX in solution was 3.5×10 -3 μM. | 161 | |
|
| |||||||
| Athymic nude mice bearing s.c. PC3 tumors C6 rat glioma cell line | Animals that received a single-dose 24 mg/kg intratumoral injection of PTX-NPs-Tf demonstrated complete tumor regression and significantly extended survival rate compared to those that received either Tx-NPs or Tx-Cremophor® EL. | ||||||
|
| |||||||
| PLGA | Holo-Tf | 182.8 | PTX | C6 rat glioma cell line | Cytotoxicity was significantly higher and the observed in the order o Tf-PTX-PLGA-NPs > P85-PTX-PLGA-NPs > PTX-PLGA-NPs in comparison to PTX. | 163 | |
|
| |||||||
| Rats bearing s.c. C6 glioma tumors | In vivo biodistribution in male Sprague-Dawley rats bearing C6 glioma showed higher tumor PTX concentrations in animals administered with PTX-NPs compared to PTX. | ||||||
N/D = not determined
In other studies, PAMAM G-5 dendrimers modified with PEG (PAMAM-PEG) were linked covalently to the peptide HAIYPRH (T7), which exhibits an elevated affinity for the TfR, resulting in PAMAM-PEG-T7 nanoparticles. This system was loaded with ADR, formulating PAMAM-PEG-T7/ADR nanoparticles for the targeting of ADR to tumors overexpressing TfR [149]. PAMAM-PEG-T7/ADR enhanced the toxicity to human hepatocellular carcinoma Bel-7402 cells in vitro, and favored the accumulation 1.7-fold in s.c. Bel-7402 xenograft tumors in vivo compared to the nanoparticles without the ligand, and 5.3-fold compared to free ADR. In addition, systemic application of the dendrimer conjugated to T7 achieved lower concentrations of ADR in liver, kidney, head, spleen and lung and significantly inhibited the tumor growth of s.c. xenograft tumors than its counterpart without T7 [149]. The system mentioned above was modified to deliver, simultaneously, a plasmid encoding for a gene that induces apoptosis (pORF-hTRAIL) and ADR (PAMAM-PEG-T7/pORF-hTRAILeADR) [150]. The nanoparticles with the T7 peptide showed higher efficacy in cellular uptake and gene expression in Bel-7402 cells in vitro and accumulated in tumor more efficiently than their counterpart without the peptide. In addition, the PAMAM-PEG-T7/pORF-hTRAILeADR co-delivery system ADR dose of 0.16 mg/kg inhibited 77% of tumor growth in the Bel-7402 s.c. xenograft model in vivo compared to 69% by 5mg/kg of free ADR. These results suggest the presence of a synergistic activity in the co-delivery system between pORF-hTRAIL and ADR targeted to TfR by T7, and that these nanoparticles may show an advantage in the selective destruction of cancer cells with lower secondary effects [150]. Other dendrimer systems conjugated with Tf and wheat germ agglutinin and loaded with ADR have shown in vitro cytotoxicity against the murine C6 glioma cell line [151], but the size of the Tf-conjugated systems were 10- fold higher the those using the T7 peptide as targeting moiety (199 nm versus 10 nm).
4.3. Single Polymers and Polymeric nanoparticles
Single polymers and polymeric nanoparticles are emerging as promising drug delivery vehicles because of their multimodular structure that enables them to actively target discrete cells through multiple barriers and to simultaneously carry multiple drugs of various chemical natures (Table 1). In recent years, nanoparticles have matured from simple devices to multifunctional, biodegradable, non-toxic, and non-immunogenic constructs, capable of delivering synergistically functioning drugs in vivo. These polymeric nanoparticles have a size between 10 and 1000 nm and are constructed with natural or synthetic polymers [152]. One of the essential requirements for their use in the clinic is that polymeric materials are biocompatible and biodegradable. After administration, the polymers should be reduced to monomers and then metabolized and eliminated by the body’s natural pathways [152]. The choice of biomaterials with different physicochemical properties (molecular weight, dispersion index, hydrophobicity, and crystallinity) allows the modulation and control of the drug delivery rate within the system. Due to their structure and composition, polymeric nanoparticles have a greater stability when in contact with biological fluids than colloidal systems [153].
Depending on the different methods of preparation and the properties of material used, it is possible to generate either single macromolecules, or by self association form nanocapsules or nanospheres [154]. The nanocapsules are systems in which the drug is adsorbed on the surface of the particle or placed in an aqueous or oily interior surrounded by the polymeric layer [155]. In contrast, the structure of the nanospheres is constituted by a spherical matrix, where the drug can be dissolved, entrapped, chemically bound or adsorbed on the surface [156]. One of the salient features of these structures is the ability to add functional groups on its surface in order to attach ligands with the possibility of actively targeting the nanoparticles to malignant cells or tissues [157].
Polysaccharides, including chitosan (CS), are natural biodegradable polymers most commonly used in the preparation of single polymeric nanodrugs [158]. CS is a macromolecule of animal origin obtained from chitin [159]. Another, recently introduced biopolymer poly(malic) acid (PLMA) is an aliphatic polyester that is a non-self associating, polyfunctional polymer in which functional groups can be added all along the molecule [160]. In the case of synthetic biodegradable polymers, the family of aliphatic polyesters stands out such as poly(lactic acid) (PLA), poly(lactic-co-glycolic) acid (PLGA), or poly(ε-caprolactone) (PCL) [153], all of which are self-associating polymers in which functional groups can only be added only at their termini. Some of these compounds are approved by the FDA for use in drug delivery systems and have widespread uses in the biomedical and biomaterial areas. Moreover, these polymeric compounds loaded with anticancer drugs have been conjugated to different ligands to target TfR in order to increase the effectiveness of the encapsulated drug. PLGA nanoparticles loaded with the aromatase inhibitor 7α-APTADD were significantly more effective preventing proliferation of the human breast cancer cell line SK-BR-3 than non-targeted nanoparticles, suggesting that Tf-conjugated lipid-coated PLGA nanoparticles are potential vehicles for improving the efficiency and specificity of therapeutic delivery of aromatase inhibitors [161].
Particulate nanodrugs consisting of PLGA loaded with paclitaxel (PTX) were conjugated to Tf (PTX-NPs-Tf) using an epoxy compound (Denacol-EX-521) with a diameter of 220 nm [162]. PTX-NPs-Tf activity evaluated in human prostate cancer PC3 cells in vitro showed a 70% inhibition of proliferation at 1 ng/ml, while at the same concentration the nanoparticles without ligand (PTX-NPs) exhibited 25% inhibition, and paclitaxel in solution (PTX-Cr) resulted in a 35% inhibition [162]. In addition, in vivo studies using nude mice in a s.c. tumor model with PC3 cells showed complete tumor regression after an intratumoral dose of 24 mg/kg PTX-NPs-Tf, and resulted in significantly increased survival compared to those receiving PTX-NPs or PTX-Cr [162]. In other studies, encapsulated PTX-NPs-Tf showed a significant anti-proliferative activity in vitro compared to the unconjugated nanoparticles or PTX in solution against the human breast cancer cell lines MCF-7 and MCF-7/ADR drug resistant [163]. This activity is positively correlated to greater cellular uptake and reduced exocytosis compared to the unconjugated nanoparticles, thereby achieving higher intracellular retention and drug levels [163]. Other studies with paclitaxel loaded PLGA nanocarriers conjugated to Tf (Tf-PTX-PLGA-NPs) with an average size of 170 nm showed that nanoparticles conjugated to Tf exhibited higher intracellular uptake to rat C6 glioma cell compared to nanoparticles without Tf [164]. In addition, the biodistribution studies in rats implanted s.c. with the C6 cells showed that higher intratumoral concentrations of paclitaxel were observed in animals treated i.v. with Tf-PTX-PLGA-NPs compared to the nanoparticles without Tf or PTX in solution after 24 hours of injection [164]. Pulkkinen et al. observed that another PTX loaded nanopaticle using Tf as targeting moiety were able improve significantly improve the anti-tumoral activiy in the BT4C rat glioma model in vivo than non targeted nanoparticles or free PTX [165]. These results are consistent with those obtained in another study using PEG nanoparticles of poly(cyanoacrylate) conjugated to Tf and loaded with PTX (PEG-NP-Tf) and with an approximate size of 100 nm [166]. Intravenous administration of PEG-NP-Tf reached the highest intratumoral drug concentrations compared to the PTX solution and NPs without ligand. Moreover, the lifespan for mice treated with PEG-NP-Tf increased significantly and 50% of the animals had complete tumor regression [166]. Other Tf-targeted nanoparticles loaded with poly(ethylene) glycol-hydroxycamptothecin conjugate exhibited significant improvement in the reduction of tumor weight compared to non targeted formulations in the in vivo model S180 murine sarcoma [167]. Gan et al. observed that Tf-conjugated poly(lactide)-d-α-tocopheryl polyethylene glycol succinate diblock copolymer nanoparticles loaded with docetaxel could be more efficient eliciting cytotoxicity against C6 glioma than other non targeted formulations [168].
Polymeric pegylated Tf-conjugated particulate nanodrugs of poly(cyanoacrylate) (PEG-NPs-Tf) with a size under 200 nm can be used to encapsulate and deliver plasmid DNA (pDNA) [169]. In vitro studies showed a higher degree of cell attachment of these NP at 4°C compared to NP without ligand against human K562 cell tumor cells overexpressing the TfR. The presence of free Tf significantly decreased the binding of PEG-NPs-Tf to the cells demonstrating that the binding of the ligand was receptor-specific [169].
In addition to Tf, other ligands can be used to modify the surface of these nanocarriers such as anti-TfR monoclonal antibodies used as a targeting strategy [170]. Fluorescein labeled CS nanospheres conjugated with PEG obtained with the PRINT technology (Particle Replication In Non-wetting Templates) were bioconjugated either with the OKT9 murine anti-human TfR antibody (NPs-OKT9) or with human Tf (NPs-hTf) with a size of 200 nm [157]. In both cases greater than 80% uptake was observed in several human tumor cell lines (HeLa, Ramos, H460, SKOV-3, HepG2, and LNCaP) compared to bovine Tf conjugated nanoparticles (NPs-bTf) or IgG1 (NPs-IgG1). The targeting efficiency was dependent on nanocarrier concentration, ligand density, dosing time, and level of cell surface receptor expression. The formulations NPs and NP-hTf-OKT9 were not toxic in vitro to most cell lines except for the Ramos human B-cell lymphoma cell line. For these cells a strong correlation was found between the viability and the amount of ligand (OKT-9 or hTf) that can be conjugated to the surface of the nanoparticles, with lower cell viability associated with higher percentage of ligand conjugate, suggesting that the polyvalency of the moiety targeting TfR plays a role in the toxicity in some malignancies [157].
The Polycefin family of naturally-derived polymeric drug delivery is another example [160, 171]. This type of vehicle is built by hierarchic conjugation chemistry of functional groups onto the scaffold of PMLA, a polycarboxy polyester obtained from the microorganism Physarum polycephalum. The blood brain barrier (BBB) and the blood-tumor barrier (BTB) can be obstacles for the successful delivery of drugs for the treatment of brain cancer [172]. Multi-targeted polycefin-based single polymer nanodrugs have been designed as a strategy to overcome these barriers [171, 173, 174]. Components of these conjugates include: (1) a pH-dependent endosomal escape unit to allow exiting into the cytoplasm and prevent subsequent degradation in lysosomes, (2) a tracking fluorescent dye, (3) moieties to optimize solubility, (4) the therapeutic payload (ODN or drug), and (5) monoclonal antibodies targeting the rat TfR (OX26) to target the rat endothelial cells for transport across the BBB and BTB (Figure 5). These nanoconjugates were shown to cross the BTB by transcytosis and to specifically target the tumor cells where they were internalized by receptor-mediated endocytosis [160]. When ODN directed against the α4 and β1 subunits of human laminin-411, an adhesion molecule that is overexpressed in the tumor neovasculature, were attached as the therapeutic payload, intracranial treatment with these nanoconugates reduced microvessel density and increased survival in mice challenged with human U87MG glioblastoma cells [175].
Figure 5. Schematic drawing of Polycefin molecule, a customized drug prepared to treat malignant tissue.
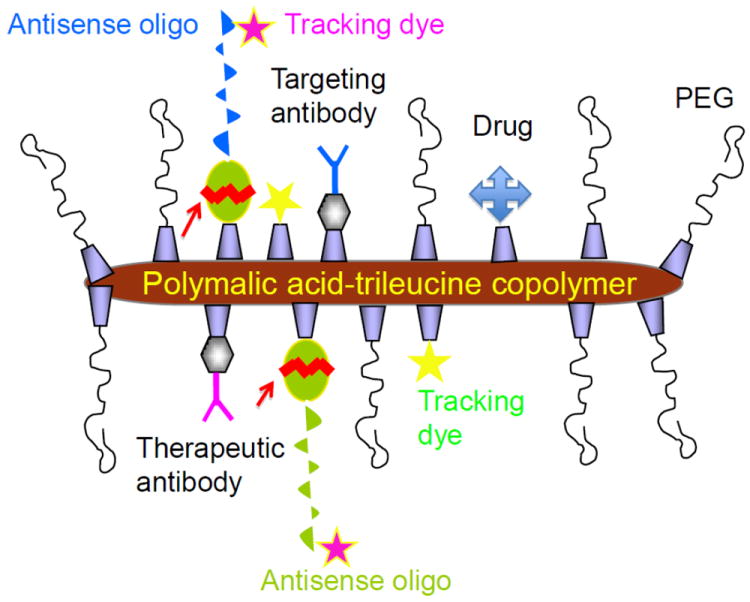
Polymalic acid-trileucine copolymer is the platform carrying the other covalently bound functional groups and trileucine that mediates pH-dependent delivery through endodomal membrane into recipient cell cytoplasm. Functional (optional) groups: multiple same or different antisense oligos or siRNA (arrows, cleavage from the platform by action of glutathione in the cytoplasm), multiple same or different chemotherapeutic drugs (linked through glutathione or pH-responsive spacers for cleavage from the platform), same or different mAb or other proteins/peptides for specific tumor and/or recipient cell targeting, same or different tracking fluorescent dyes, protecting PEG (dispensible depending on the composition of the cargo).
Polycefin variants containing two different targeting antibodies have also been evaluated for the treatment of brain cancers (Figure 5). Tandemly coupled antibodies targeting the mouse TfR (to target the murine endothelial cells) and an anti-nucleosome antibody 2C5 (to target the human cancer cells) were covalently attached to the polymer and were shown to have enhanced human tumor targeting ability in murine models [176]. Another polycephin variant containing an anti-human TfR antibody for human cancer cell targeting in tandem with the anti-mouse TfR antibody has also been produced. After eight rounds of intravenous injection every three days in xenograft models, tumors of treated mice were 90% smaller than those of control mice and their blood vessels resembled those in healthy brain tissue [177].
Polycefin nanoconjugates targeting human breast cancer have also been developed [178]. These conjugates were synthesized for repetitive systemic treatments of HER2/neu-positive human breast tumors in a xenogeneic mouse model. Various moieties were covalently attached to PMLA, including a combination of (1) ODN directed against the HER2/neu mRNA, to block the synthesis of the oncoprotein HER2/neu; (2) anti-HER2/neu antibody trastuzumab (Herceptin®), to target breast cancer cells and inhibit receptor activity simultaneously; and (3) murine transferrin receptor antibody, to target the tumor vasculature and mediate extravasation of the nanobiopolymer through the host endothelial system. The multi-targeted nanoconjugates significantly inhibited the growth of HER2/neu-positive breast cancer cells in vitro and in vivo by enhanced apoptosis and inhibition of HER2/neu receptor signaling with suppression of Akt phosphorylation [178]. In vivo imaging analysis and confocal microscopy demonstrated selective accumulation of the nanodrug in tumor cells via an active delivery mechanism. Systemic treatment of human breast tumor-bearing nude mice resulted in more than 90% inhibition of tumor growth and tumor regression, as compared to partial (50%) tumor growth inhibition in mice treated with trastuzumab or AON, either free or attached to PMLA [178]. These findings offer a preclinical proof-of-concept for use of the PMLA nanoplatform for combination cancer therapy.
4.4. Liposomes
Liposomes are colloidal systems composed of amphiphilic phospholipids that self-assemble spontaneously into bilayers forming spherical vesicles, which are closed due to the interaction between the phosphate groups of phospholipids and the water molecules [179]. These vesicles are composed of natural or synthetic phospholipids and have a diameter that can range between 0.02 and 10 μm [180]. Because of their structure, liposomes can carry hydrophilic and hydrophobic drugs. Hydrophilic compounds are dissolved in the aqueous interior of the vesicles, while hydrophobic compounds are placed in the lipid bilayer, and charged drugs can be associated to the surface lipids.
This type of system can increase efficiency and reduce unwanted cytotoxic effects produced by the administration of chemotherapy [20]. Several non-PEGylated liposomal formulations (DaunoXome®, Myocet®, VincaXome®, DepoCyt®) and other PEGylated formulations (Doxil®, Caelyx®) with antineoplastic drugs are available in the market for clinical use. The surface modification of liposomes with PEG can sterically stabilize them and avoid the mononuclear phagocytic system (MPS), thus extending their circulation time in blood [181]. Furthermore, to increase their specificity, these systems can be conjugated with molecules that recognize selectively overexpressed receptors on the surface of cancer cells [20], including the TfR (Table 1). For example, PEGylated liposomes conjugated on the surface with Tf (Tf-PEG-liposomes) could attach to mouse Colon 26 cells in vitro and were internalized by receptor-mediated endocytosis. In the mouse s.c. tumor model Colon 26 was observed that the Tf-PEG-liposomes had a prolonged blood circulation, low uptake by the reticuloendothelial system (RES) and showed enhanced internalized into malignant cells in vivo [182]. The internalization of the complex ligand-TfR via endocytosis can be used to promote the uptake of liposomes and the release of the drug in the cell cytoplasm [183]. In an in vitro study in rat glioma C6 cells, a significant increase in cellular uptake of Tf-conjugated liposomes loaded with ADR compared to liposomes without the ligand was observed [184]. Fluorescence studies and confocal microscopy confirmed than liposomes covalently linked to Tf had a greater association to human T-lymphoblastic leukemia CEM cells in vitro than liposome formulations without the ligand [185]. Li et al. observed that liposomes decorated with Tf and loaded with ADR (Tf-SL-ADR) were able to enhance the intracellular uptake of ADR in the HepG2 human hepatoma cell line compared to the unconjugated particle [186]. Moreover, in an in vivo s.c. HepG2 xenograft model, i.v. administration of Tf-SL-ADR resulted in a significant increase in the concentrations of drug in the tumor site compared to the free drug (ADR). On the other hand, the ADR levels in heart and kidney were lower for Tf-SL-ADR compared with those of ADR in solution [186]. The administration of cisplatin loaded PEGylated liposomes linked to Tf to immunosupressed mice in a model of peritoneal dissemination of MKN45P human gastric cancer cells showed higher levels of the drug in tumor cells and increased survival compared to animals treated with cisplatin in solution or in liposomes without ligand [187]. In contrast, Xu et al. reported that a different formulation (PC/chol/DOPE-DPPTPA/mPEG-DSPE) of Tf-conjugated liposome loaded with ADR showed limited advantage over free ADR in KB human cervical carcinoma cells in vitro, but it was better than the non-targeted liposome [188]. However, Anabousi et al. observed that A549 human alveolar basal epithelial cells exhibit overexpression of TfR compared to primary cell cultures of alveolar type II epithelium [189]. Tf-conjugated liposomes loaded with ADR showed greater cytotoxicity against A549 cell line compared to a primary cell culture of human alveolar non-cancerous cells ATI/ATII. This in vitro data highlight the potential of these systems to specifically eliminate the tumor cells that overexpress TfR [189].
Tf-conjugated liposomes loaded with genetic material have shown promise as a targeting strategy. A Tf-conjugated liposome complex loaded with DNA encoding for the tumor supressor protein p53 have shown efficacy in vitro and in vivo in the DU145 human head and neck cancer model [190]. A different Tf-conjugated liposome system loaded with a Bcl-2 specific oligonucleotides antisense G3139 showed improved targeting and internalization into K562 cancer cells in vitro and in vivo, extending the survival and improving tumor growth inhibition compared to the antisense alone [191, 192].
One of the major problems of chemotherapy is the development of resistance at the cellular level, which is mainly associated with overexpression in the plasma membrane of the efflux pump Pgp [155]. To evade this mechanism of resistance Tf-conjugated liposomes containing the Pgp inhibitor verapamil and ADR (Tf-L-ADR/VER) were developed and showed an efficient uptake in the TfR+ K562 human erythromyeloblastoid leukemia cell line [193]. In vitro studies in K562 cells resistant to ADR (K562/ADR) Tf-LADR/VER showed 5.2 and 2.8-fold greater cytotoxicity (IC50 = 4.18 μM) than liposomes without ligand (L-ADR/VER) (IC50 =21.7 μM) and than Tf-conjugated liposomes loaded with ADR alone (Tf-L-ADR) (IC50= 11.5 μM) respectively, suggesting that co-encapsulated ADR and verapamil could be an effective strategy for selective targeting and reversal of drug resistance. In addition, Krieger et al. observed that liposomes loaded with cisplatin and decorated with Tf were able to overcome resistance of ovarian cancer cells to cisplatin in vitro [194].
Another strategy for active targeting with liposomes is the use of anti-TfR specific antibodies that can be attached to the surface of liposomes loaded with chemotherapeutic drugs, called immunoliposomes. Huwyler et al. reported the efficacy of immunoliposomes conjugated with the anti-rat TfR OX26 monoclonal antibody as a daunomycin delivery system, presenting evidence of endocytosis of fluorescent immunoliposomes by RG2 rat glioma cells [195]. In other studies, liposomes conjugated to anti-human TfR monoclonal antibody OKT9 and loaded with ADR (OKT9-CIL) were used in vitro in K562/ADM human leukemia cells resistant to ADR. OKT9-CIL was internalized into K562/ADM cells and caused accumulation of intracellular ADR, resulting in an increase of ADR cytotoxicity in ADR-resistant tumor cells compared to free drug [196].
In some types of carcinomas, photodynamic therapy is used after photosensitizers are administered systemically or topically with the intent to accumulate selectively in neoplastic tissue before being irradiated. The encapsulation of photosensitizing agents such as aluminum phthalocyanine tetrasulfonate in PEGylated liposomes bound to Tf (Tf-Lip-AlPcS4) demonstrated the ability of active targeting via TfR in human AY-27 transitional-cell carcinoma cells, favoring the accumulation of tumor cells both in vitro and in vivo [197].
Gene therapy is seen as a promising strategy for cancer treatment and requires two main entities, the therapeutic gene and the vector responsible for its transportation and arrival at the target cell in an intact form [198]. Cationic liposomes can complex with DNA and form lipoplexes through a combination of electrostatic interactions. However, these vectors lack tumor specificity and exhibit low transfection efficiency in vivo compared to viral vectors [199]. The ability to bind TfR with a suitable ligand such as a single-chain Fv antibody fragment allows the targeting and internalization of the complex by malignant cells, where the DNA escapes from the endocytic pathway to the nucleus [199]. During preparation, cationic lipids such as dioleoyltrimethylammonium -propane (DOTAP) or dimethyldioctadecylammonium bromide (DDAB) can be fused to neutral lipids such as cholesterol and/or dioleolyphosphatidylethanolamine (DOPE) [200]. Additionally, Xu et al. showed that cationic liposomes bound to Tf are also adequate systems for active targeting of genes in tumor models in vivo [200]. Gene expression was predominantly observed in tumor tissues after i.v. administration of Tf-liposome-plasmid encoding for p53 (Lip-T-pSVb). No evidence of gene expression was observed in normal tissues [200]. In human tumor cells of the nasal vestibule (JSQ-3), that were resistant to radiation, it was observed that the addition of Tf-lipoplex produced a significant increase in transfection efficiency of 70-80% of the pro-apoptotic p53 compared to lipoplex without Tf (5-20%), resulting in the reversion of the radiation resistant phenotype of the JSQ-3 cells [201]. More recently, Xu and colleagues developed cationic immunoliposomes by incorporating into their liposomal structure a variable fragment (Fv) of anti-transferrin receptor (TfRscFv) for the release of the p53 gene into prostate tumor cells [202]. The TfRscFv has the advantage over the Tf molecule in that scFv has a much smaller size than Tf, resulting in a smaller immunolipoplex with better penetration into solid tumors. The liposomes bound to TfRscFv increased binding to tumor cells and the transfection efficiency significantly both in vitro and in vivo [202, 203]. Intravenous administration of immunoliposomes with TfRscFv, in mice xenotransplanted with JSQ-3 cells revealed a consistent delivery to the tumor site together with an efficient expression of the p53 tumor suppressor gene compared with liposomes without ligand [202]. In another study, using a different method of bioconjugation, i.v. administration of PEG-conjugated immunoliposomes and TfRscFv resulted in gene expression levels higher than those mediated by the same system without PEG [204]. In addition, high levels of exogenous gene expression in the PANC-1 human pancreatic cancer mouse xenograft model and low expression of p53 in liver indicates the preferential tumor targeting of the system with PEG.
A different approach in gene therapy is the silencing of genes that play an important role in the malignancy of cancer cells. Encapsulation of siRNA or ODN fragments specifically designed to silence the BCR-ABL oncogene in Tf-conjugated liposomes effectively decreased cell viability in two human leukemia cell lines K562 and LAMA-84, no significant effects were observed in the nonmalignant human fibroblast cell line BJ [205]. A decreased level of mRNA encoding BCR-ABL was observed only with liposomes conjugated to Tf.
Among the TfR targeting liposomal systems, two are under clinical investigation. MBP-426 is a liposome conjugated with Tf for the delivery of oxaliplatin that has completed a Phase I clinical trial (2006, NCT00355888). This formulation is also in a Phase Ib/II clinical trial in second line patients with gastric, gastroesophageal, or esophageal adenocarcinoma (2009, NCT00964080). The other system under clinical evaluation is the SGT-53 immunoliposomes using antibody fragments as ligands and is in phase I clinical trial. SGT-53 is a liposome encapsulating plasmid DNA coding for wild type p53, forming a complex that is targeted to tumor cells by means of an anti-TfR scFv (TfRscFv) attached to the surface of the liposome (2007, NCT0040613).
4.5. Cyclodextrins
Cyclodextrins (CDs) are cyclic macromolecules formed by glucopyranose units linked by glycosidic bonds α-1, 4. The most commonly used are composed of 6, 7, or 8 glucopyranose units and are called α-cyclodextrin (α-CD), β-cyclodextrin (β-CD) and γ-cyclodextrin (γ-CD), respectively [206]. They have a molecular structure that is “toroidal” and rigid with primary and secondary hydroxyl groups oriented toward the surface. The presence of free hydroxyl groups on the outside surface of the rings makes the CDs soluble in water, which is the result of the interaction capacity of these hydroxyl groups to aqueous media. The internal cavity of the CDs is hydrophobic, so these compounds are able to accommodate smaller hydrophobic molecules to form inclusion complexes [207].
The CD-based polymers are formed by a short polycationic segment through which nucleic acids (polyanionic) can come together through electrostatic interactions. This union can provide protection against nucleic acid degradation by nucleases prior to intracellular delivery [208]. CDs containing polycations (CDP) have proven to be very good carriers of DNases and nucleic acids such as plasmid DNA and siRNA [209]. These systems present instability in biological fluids such as blood. One way of stabilizing them is to decorate their surface with PEG through the formation of a conjugate with adamantane (AD). Adamantane in the conjugate PEG-AD has the ability to form inclusion complexes with the β-CDs on the surface of the nanoparticles [208]. In addition, PEGylation (PEG-AD) allows binding of the ligands at the opposite end of that of the AD, providing the capacity to interact with specific receptors on the cell surface. For example, Bellocq et al. used apo-transferrin as a ligand for formation of the complex Tf-PEG-AD, linking via oxidation with sodium periodate or by binding to amino groups of lysine [210]. Two human cell lines (HT29 and HeLa-A2780) have been used to demonstrate that the targeting of these polymers based on β-CD to the TfR is required for proper recruitment and retention of nanoparticles in tumor cells [211].
Additionally, CD particles have been used to deliver siRNA into tumor cells. Imidazole-modified CD were mixed with Tf-adamantine-PEG5000 (Tf-AD-PEG) [212] and the siRNA targeting the EWS-FLI1 fusion protein (formed by a translocation that is commonly found in Ewing’s tumors). This fusion protein has been shown to be a transcription factor involved in tumorigenesis. TC71 human Ewing’s sarcoma cells expressing luciferase were injected i.v. into Nod/SCID mice, a model that mimics metastatic Ewing’s family tumors [212]. Three consecutive i.v. injections of the targeted complexes on days 35-37 after tumor injection reduced EWS-FLI1 expression in established tumors and transiently decreased tumor growth [212]. Additionally, when treatments were given twice weekly for four weeks starting on the day of tumor cell inoculation, the targeted complexes dramatically inhibited tumor engraftment. Twenty percent of animals developed tumors compared to 90-100% in the control groups, which consisted of treatment with siRNA alone, treatment with targeted complexes consisting of a scrambled siRNA, or untreated animals. Non-targeted complexes (without Tf) consisting of the siRNA specific for EWS-FLI1 showed a delayed progression of tumor engraftment, however, tumor growth rate was similar to the other control groups. Furthermore, these animals went on to develop large tumors. Safety of the Tf-AD-PEG targeted complexes was evaluated in immunocompetent mice and no significant changes in blood cell counts, liver or kidney function, and the pathology of major organs were observed. This study suggests the safety and efficacy of this non-viral, targeted delivery system. In a subsequent study, β-CDP loaded with siRNA and decorated with Tf, was administered to non-human primates and was well tolerated in i.v. doses of three and nine mg/kg siRNA, while the higher dose of 27 mg/kg indicators resulted in kidney toxicity [213].
This class of drugs is currently on clinical evaluation with CALA-01, an injectable formulation developed by Calando Pharmaceuticals, Inc., which is currently in a Phase I clinical trial entitled “Dose-Escalating Study in Adults With Solid Tumors Refractory to Standard-of-Care Therapies” (Identifier NCT00689065). This formulation is composed of a linear polymer based Tf decorated CD loaded with siRNA. The siRNA used inhibits tumor growth by reducing the expression of the M2 subunit of ribonucleotide reductase and has shown evidence of eliciting specific gene inhibition in humans [214].
4.6. Viral Vectors
The goals of gene therapy strategies are to specifically deliver the transgene to cancer cells and to maintain expression as needed for therapeutic purposes [215]. Viral vectors are often used in gene therapy due to their high efficiency of gene transfer. However, the main problem for specific gene transfer is the broad natural tropism of the viruses, which results in toxicity to normal, non-targeted cells. To overcome this problem, many strategies have been employed to modify the surface of viral particles [215]. These strategies include pseudotyping (changing the tropism of a virus by replacing the viral attachment proteins with those of a different virus), using adapter molecules (that bind both the viral attachment protein as well as the targeted cancer cell), and genetic incorporation of ligands that target a tumor-associated antigen on the surface of the cancer cell.
4.6.1. Tf-targeted viruses
The TfR has been used as a target for such modified viral vectors. Modified adenoviral particles have been the most extensively studied TfR-targeted vectors. Adenoviruses are non-enveloped double stranded DNA viruses. One strategy to modify the adenoviral surface is termed adenovirus-enhanced transferrinfection (AVET) and makes use of the strong biotin-(strept)avidin interaction to produce DNA-containing targeted viral particles [73]. For AVET complex formation, biotinylated adenovirus is mixed with strepavidin-containing polylysine. DNA is then added to this mixture to compact the DNA and link it to the biotinylated virus. In the final step, Tf-conjugated to polylysine is added to the DNA complex for targeting purposes. These AVET complexes resulted in high murine IL-2 expression in vitro and secretion by murine M3 melanoma cells as well as by five primary human melanoma cell lines [73]. Although the level of secretion is variable among the cell lines, the highest level of secretion was observed in cells treated with the AVET complexes when compared to other Tf-targeted polylysine complexes. In this model, IL-2 would serve to increase the immunogenicity of the tumor. Additionally, the highest luciferase expression was observed with the intratumoral injection of the AVET complexes into s.c. M3 or B16F10 murine melanomas [73]. The transfection efficiency of AVET complexes after intratumoral injection into s.c. growing Neuro2a murine neuroblastoma cells or M3 murine melanoma cells was 10 and 100-fold higher, respectively, compared to naked DNA [75]. The AVET complexes containing the luciferase gene yielded gene expression in K562 human erythroleukemic cells similar to that caused by a Tf-polylysine conjugate that was combined with chloroquine, an agent that increases vesicle pH [216]. Taken together these results show that the AVET complexes are efficient gene delivery vectors and that the virus within the complex acts as an endosomolytic agent, helps to prevents DNA degradation, and enhances transgene expression.
When used as allogeneic melanoma cell vaccines in animal models, murine melanoma cells transfected with AVET complexes carrying the murine IL-2 gene induced cross protection against syngeneic melanoma tumors in DBA/2 mice [217]. This vaccine strategy was effective in both a prophylactic vaccination setting where protection was observed from subsequent tumor challenge as well as in a therapeutic vaccination setting against pre-existing tumor deposits [217]. In a Phase I clinical trial, these AVET complexes were used to exogenously transfect human melanoma cells with the human IL-2 gene for the use as an autologous vaccination strategy in patients with advanced disease [218]. This trial showed that the complexes were well tolerated overall. No partial or complete responses were observed, however, there was an increase in melanoma cell-specific delayed-type hypersensitivity reactions in eight of 15 patients, the occurrence of vitiligo in three of 15 patients, and signs of tumor regression in three of 15 patients, which suggest the presence of a vaccine-induced immune response.
A similar strategy to modify the adenoviral surface to enhance tumor targeting also uses the strong interaction of biotin and streptavidin to create molecular conjugate vectors (MCV) [219, 220]. These MCV are constructed by chemically linking polylysine to the adenovirus. DNA encoding the transgene is then added. Subsequently, streptavidin linked polylysine is added to the complex followed by biotinylated Tf, resulting in complexes that were termed “Trans-MCV.” These complexes were modified slightly by using a recombinant adenovirus in which the transgene is within the viral genome (Trans-recMCV) [219]. Using luciferase as the transgene, Trans-recMCV were shown to have a 5-fold higher transfection efficiency in human K562 erythroleukemic cells compared to Trans-MCV. Luciferase expression was prolonged to 14-20 days compared to 4-8 days with Trans-CMV. Moreover, luciferase expression could be detected in 35-50% of the single cell clones for up to 6 months. However, in other human leukemia cell lines (MBO2, MO-7e, KG1, TF1, THP1, and U-937) this increased transfection efficiency was not observed [220].
A different strategy is direct modification of the adenovirus capsid for targeting purposes. Solvent-exposed positions of the adenoviral capsid were genetically altered to cysteines, allowing for the controlled chemical conjugation of Tf to the adenovirus through the corresponding thiol groups [221]. A thiol-reactive maleimide group was introduced onto Apo-Tf (not bound to iron) through the use of N-hydroxysuccinimide-polyethylene glycol 3400-maleimide (NHS-PEG3400-mal). The NHS moiety covalently introduces a reactive maleimide group, which is separated from the protein by the PEG spacer. The maleimide-modified Tf was then chemically conjugated to the genetically modified adenovirus that also expresses enhanced green fluorescence protein (EGFP). This targeted viral vector showed a 30-fold increase in transduction efficiency in K562 human erythroleukemic cells [221]. This study also showed that the spacer length and the chemical nature of coupling did not influence the targeting efficiency. Additionally, this targeting effect was completely blocked by the addition of excess free Tf, which demonstrates that TfR targeting was required for the increase in transduction efficiency.
Lentiviruses are of the Retroviridae family and can deliver a significant amount of genetic information into the DNA of the host cell. They have the unique ability among retroviruses of being able to replicate in non-dividing cells, so they are one of the most efficient methods of gene delivery. Lentiviruses have a broad natural tropism, thus pseudotyping and the use of the targeting molecule human Tf have been reported. Recombinant lentiviral vectors pseudotyped with the baculovirus major envelope glycoprotein gp64 that display streptavidin or avidin fused to the transmembrane anchor of the vesicular stomatitis virus G protein (VSV-G) have been constructed [222]. Targeting of BT4C rat glioma and human D54 glioblastoma cells occurred by the pre-incubation of the cells with biotinylated Tf prior to transduction with the recombinant lentiviral particles expressing EGFP. In BT4C, transduction efficiencies increased by 20-30% with avidin containing viruses and 50-60% with streptavidin containing targeted, recombinant lentiviral vectors [222]. In the human glioblastoma cells, the efficiencies increased by 20% and 40% respectively.
The opposite strategy, in which the viral surface is biotinylated and conjugated to an avidin-containing molecule targeting the TfR, has also been reported. A lentiviral vector was pseudotyped with a mutated form of the Sindbis virus protein. The mutations in the envelope protein of the Sindbis envelope have been shown to help eliminate the broad tropism of the virus [223]. To further target the virus, the biotin-adaptor peptide (BAP) was inserted into the receptor binding region of the mutated envelope protein allowing biotinylation at specific sites [224]. Biotinylated-Tf was then bridged to the biotinylated lentiviral vector through the use of neutravidin, which is a mutant form of avidin that shows less non-specific cell-binding compared to avidin. Like avidin, neutravidin has four biotin binding sites and thus efficiently links the biotinylated-Tf with the biotinylated lentivirus. This targeted viral vector encoding the firefly luciferase as a reporter gene showed an increase in transduction efficiency in Jurkat human T-cell leukemia cells [224]. Additionally, biotinylated lentivirus was conjugated to an antibody-avidin fusion protein targeting either the rat or the human TfR (see section 3.2.4.2.) [224]. Due to a lack of species cross-reactivity of the two antibody-avidin fusion proteins, the specificity of delivery of the biotinylated lentiviral particles was demonstrated using either human Jurkat or rat myeloma Y3-Ag 1.2.3 cells. These studies show that the biotinylated, pseudotyped lentiviral vector is a versatile targeting system that may be used with a wide variety of molecules targeting various cell surface antigens, including the TfR.
Another TfR-targeted viral vector for gene delivery is based on the Sendai virus. This virus is a negative sense, single-stranded RNA virus of the Paramyxoviridae family. The targeted molecule consists of an inactivated hemagglutinating virus of Japan (Sendai virus) envelope (HVJ-E) [225]. HVJ-E by itself has been shown to have apoptotic activity, however, the wild type Sendai virus envelope protein hemagglutinin-neuraminidase recognizes many different cell types that may lead to off target effects. Therefore, this protein was eliminated and replaced with a chimeric protein consisting of the viral F protein and Tf to increase tumor targeting [225]. This targeted viral vector increased the infection rate of HeLa human cervical adenocarcinoma cells3-fold in vitro. This effect was blocked by excess free Tf. Additionally, in animals bearing s.c. HeLa xenograft tumors, systemic injection of the Tf-modified viral vector containing quantum dots demonstrated a 32-fold increase in tumor targeting efficiency compared to the wild type HVJ-E.
TfR-targeted virus-like particles have also been developed and explored as potential gene delivery vectors [226]. These particles were made using the RNA phage MS2. Antisense ODN were covalently coupled to the capsid assembly-initiation signal, the translational operator RNA. Incubation of this complex with disassembled phage-coat protein subunits triggers encapsulation of the ODN into protein shells. Targeting of the TfR occurred through the conjugation of the capsid protein to Tf via a thiol-maleimide linkage. The targeted virus-like particles were used to deliver an ODN targeting p120-FB2, a nucleolar protein known to induce apoposis in cancer cells, to HL-60 human protelocytic leukemia or T24 human bladder cancer cells. These synthetic virons demonstrated a three to four fold increase in the rate of internalization and an 8-fold increase in cytotoxicity compared to ODN alone [226]. This study shows that the targeted synthetic virons can enhance the efficacy and stability of the ODN.
4.6.2. Targeting mediated by a TfR-specific peptide
In addition to the ligand (Tf) and antibodies, targeting of the TfR can also occur through specific peptides. A fiber-modified adenovirus carrying a TfR-specific peptide (Tf-peptide) has shown increased transfection efficiencies in human ovarian cancer cells [227]. In this study, β-galactosidase was inserted into the viral genome and used as the reporter gene. A five-fold increase in expression was observed in cells that expressed high levels of the TfR (OVCAR3 human ovarian cancer cells). No improved efficiency was observed in cells that had lower TfR expression levels (SK-OV-3 human ovarian cancer cells). The correlation between transfection efficiency and TfR expression was also shown in other cell lines: human HeLa cells (high TfR levels) and 293 (low TfR levels). This TfR-targeted peptide does not interfere with the binding of Tf to its receptor [227] and thus is a good candidate for the targeted gene therapy of cancer.
4.6.3. Targeting mediated by natural tropism
The above studies describe targeted viral vectors for which the viral surfaces have been modified in order to achieve targeting of the TfR. However, there are viruses that have a natural tropism for the TfRs, such as the New World arenaviruses [228] and the canine parvovirus [229], which may be used directly as platforms for targeted anti-tumor agents. In fact, canine parvovirus-like particles have been developed and evaluated for their delivery capability into TfR-expressing cells [230]. The parvovirus is a dog viral pathogen that utilizes either the canine or human TfRs for cell entry. It is a non-enveloped, single stranded DNA virus that codes for three polypeptides, VP1-3. The virus-like particles consisted of the VP2 polypeptide that was fluorescently labeled via chemical conjugation. These particles showed uptake in HeLa human cervical adenocarcinoma cells, HT-29 human colon adenocarincoma cells, and MDA-MB231 human breast adenocarcinoma cells (all TfR expressing cells) [230]. Uptake was also observed in chinese hamster ovary (CHO) cells that express hTfR1 (CHO-TRVb1), but not the parental CHO cell line that lacks hTfR. Similar to the wild-type virus, the viral particles were also shown to localize to endosomes and lysosomes after internalization. This study shows that the canine virus-like particles are a novel platform that can be further explored for tumor-targeted delivery.
5. Conclusions
In summary, TfR displays multiple desirable characteristics for use in the targeting of cytotoxic agents to cancer tissue. Although conventional chemotherapeutics are often successful in destroying cancer cells, their non-targeted nature renders them also toxic to normal cells, which can lead to the development of many dangerous and often life-threatening side effects. Although the TfR has been explored for some time as a targeting molecule for a variety of malignancies, improvements in the technology of the targeting moieties, therapeutic agents, and carrier strategies have renewed interest in the TfR as a target for the treatment of cancer. Targeting the TfR can occur through the use of its natural ligand Tf, specific peptides, monoclonal antibodies or their fragments. Delivered compounds include chemotherapeutics, toxins, polymers, gene therapy vectors, and nanoparticles with different configurations and varieties of cargos, all of which can be delivered into cancer cells. These targeted therapies can cause cytotoxic effects including growth inhibition and/or induction of apoptosis in a variety of malignancies in vitro and in vivo, and some are currently in clinical evaluation. Moreover, the TfR could be targeted for different therapeutic approaches, including the in vitro purging of cancer cells for autologous transplantation and for in vivo passive immunotherapy through agents that are directly cytotoxic or through the delivery of anti-cancer agents through receptor-mediated endocytosis. The advances in all these fronts show the enormous potential for the development of cancer therapies with higher efficacy and minimal toxicity via targeting of TfR.
Highlights.
Summary of delivery strategies targeting the transferrin receptor
Summary of complexes with targeting moiety directly conjugated to the therapeutic
Summary of approaches where targeted carriers are loaded with the therapeutic
Acknowledgments
This work was supported in part by the NIH/NCI grants R01CA136841, R01CA107023, R01CA123495, R01CA57152, U01CA151815, K01CA138559, the Howard Hughes Medical Institute Gilliam Fellowship, and the Whitcome Fellowship of the Molecular Biology Institute at UCLA. GH and DAC are members of the National Council for Scientific and Technological Research (CONICET), Argentina. EB is supported by the PFDT fellowship from the National Agency for Promotion of Science and Technology (ANPCyT-FONARSEC PICT-PRH 2008-00315), Argentina.
Abbreviations
- AD
adamantane
- ADEPT
antibody-directed enzyme prodrug therapy
- ADR
doxorubicin/adriamycin
- AV
avidin
- AVET
adenovirus-enhanced transferrinfection
- ART
artemisinin
- BAP
biotin-adapter peptide
- BBB
blood-brain barrier
- BFU-e
blast forming unit-erythroid
- BTB
blood-tumor barrier
- CD
cyclodextrins
- CDP
cyclodextrin-containing polycations
- CET40
cholera exotoxin 40
- CFU-e
colony forming unit-erythroid
- CHO
Chinese hamster ovary cells
- CMC
critical micelle concentration
- CS
chitosan
- CTL
cytotoxic T lymphocyte
- DAB
diaminobutiric poly(propylene imine)
- DDAB
dimethyldioctadecylammonium bromide
- DHA
dihydroartemisinin
- DMS
dimethylsuberimidate
- DOPE
dioleolyphosphatidylethanolamine
- DOTAP
dioleoyltrimethylammonium -propane
- DT
diphtheria toxin
- EDN
eosinophil-derived neurotoxin
- EGFP
enhanced green fluorescence protein
- EPR
enhanced permeability and retention
- GBM
glioblastoma multiforme
- GCV
gancyclovir
- GFP
green fluorescence protein
- GM-CSF
granulocyte-macrophage colony stimulating factor
- GN
gallium nitrate
- G generation
one of the layers in a dendrimer
- HIF-1α
hypoxia inducible factor–1α
- HSV-TK
herpes simplex virus thymidine kinase
- HUVEC
human umbilical vein endothelial cells
- HVJ-E
hemagglutinating virus of Japan (Sendai virus) envelope
- IC50
concentration in which growth of 50% of the cells is inhibited
- ID50
dose in which growth of 50% of the cells is inhibited
- IFN-α
interferon-α
- IT
immunotoxin
- i.p.
intraperitoneal
- i.v.
intravenous
- LG
β-lactoglobulin
- LMTK-
thymidine kinase-deficient mouse L cells
- LRIP
Luffa aegyptiaca ribosomal inactivating protein
- Lys PE40
modified PE40 that has a chemically reactive lysine residue in its amino terminus
- MCC
mitomycin c
- MCV
molecular conjugate vectors
- MDR-1
multidrug resistance protein-1
- MHC
major histocompatibility complex
- MPS
mononuclear phagocytic system
- NP
nanoparticle
- nm
nanometer
- MTS
multicellular tumor spheroids
- ODN
oligonucleotides
- PAMAM
polyamidoamine
- PAP
pokweed antiviral protein
- PCL
poly(ε-caprolactone)
- pDNA
plasmid DNA
- PE
Pseudomonas exotoxin
- PE40
Truncated mutant of PE (lacks the cell binding domain)
- PEG
polyethylene glycol
- PEI
polyethylene imine
- Pgp
P-glycoprotein
- PLA
poly(lactic) acid
- PLGA
poly(lactic-co-glycolic) acid
- PLMA
poly(malic) acid
- PNP
purine nucleoside phosphorylase
- PPI
poly(propylene imine)
- Pt
platinum
- PTX
paclitaxel
- RES
reticuloendothelial system
- rhRNase
recombinant human RNase
- RI
RNase inhibitors
- RIP
ribosomal inactivating protein
- RTA
ricin toxin chain A
- s.c.
subcutaneous
- scFv
single chain variable region of an antibody
- shRNA
short hairpin RNA
- SO6
saporin-6
- siRNA
short inhibitory RNA
- Tf
transferrin
- TfR
transferrin receptor
- TNFα
tumor necrosis factor α
- TPL
Tf-PEG-β-lactoglobulin
- VSV-G
vesicular stomatitis virus G protein
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Daniels TR, Delgado T, Rodriguez JA, Helguera G, Penichet ML. The transferrin receptor part I: Biology and targeting with cytotoxic antibodies for the treatment of cancer. Clin Immunol. 2006;121:144–158. doi: 10.1016/j.clim.2006.06.010. [DOI] [PubMed] [Google Scholar]
- 2.Cheng Y, Zak O, Aisen P, Harrison SC, Walz T. Structure of the human transferrin receptor-transferrin complex. Cell. 2004;116:565–576. doi: 10.1016/s0092-8674(04)00130-8. [DOI] [PubMed] [Google Scholar]
- 3.Fuchs H, Lucken U, Tauber R, Engel A, Gessner R. Structural model of phospholipid-reconstituted human transferrin receptor derived by electron microscopy. Structure. 1998;6:1235–1243. doi: 10.1016/s0969-2126(98)00124-5. [DOI] [PubMed] [Google Scholar]
- 4.Lebron JA, Bennett MJ, Vaughn DE, Chirino AJ, Snow PM, Mintier GA, Feder JN, Bjorkman PJ. Crystal structure of the hemochromatosis protein HFE and characterization of its interaction with transferrin receptor. Cell. 1998;93:111–123. doi: 10.1016/s0092-8674(00)81151-4. [DOI] [PubMed] [Google Scholar]
- 5.Hemadi M, Kahn PH, Miquel G, El Hage Chahine JM. Transferrin’s mechanism of interaction with receptor 1. Biochemistry. 2004;43:1736–1745. doi: 10.1021/bi030142g. [DOI] [PubMed] [Google Scholar]
- 6.Ciechanover A, Schwartz AL, Lodish HF. Sorting and recycling of cell surface receptors and endocytosed ligands: the asialoglycoprotein and transferrin receptors. J Cell Biochem. 1983;23:107–130. doi: 10.1002/jcb.240230111. [DOI] [PubMed] [Google Scholar]
- 7.Yang DC, Wang F, Elliott RL, Head JF. Expression of transferrin receptor and ferritin H-chain mRNA are associated with clinical and histopathological prognostic indicators in breast cancer. Anticancer Res. 2001;21:541–549. [PubMed] [Google Scholar]
- 8.Prior R, Reifenberger G, Wechsler W. Transferrin receptor expression in tumours of the human nervous system: relation to tumour type, grading and tumour growth fraction. Virchows Arch A Pathol Anat Histopathol. 1990;416:491–496. doi: 10.1007/BF01600299. [DOI] [PubMed] [Google Scholar]
- 9.Das Gupta A, Shah VI. Correlation of transferrin receptor expression with histologic grade and immunophenotype in chronic lymphocytic leukemia and non-Hodgkin’s lymphoma. Hematol Pathol. 1990;4:37–41. [PubMed] [Google Scholar]
- 10.Habeshaw JA, Lister TA, Stansfeld AG, Greaves MF. Correlation of transferrin receptor expression with histological class and outcome in non-Hodgkin lymphoma. Lancet. 1983;1:498–501. doi: 10.1016/s0140-6736(83)92191-8. [DOI] [PubMed] [Google Scholar]
- 11.Kondo K, Noguchi M, Mukai K, Matsuno Y, Sato Y, Shimosato Y, Monden Y. Transferrin receptor expression in adenocarcinoma of the lung as a histopathologic indicator of prognosis. Chest. 1990;97:1367–1371. doi: 10.1378/chest.97.6.1367. [DOI] [PubMed] [Google Scholar]
- 12.Seymour GJ, Walsh MD, Lavin MF, Strutton G, Gardiner RA. Transferrin receptor expression by human bladder transitional cell carcinomas. Urol Res. 1987;15:341–344. doi: 10.1007/BF00265663. [DOI] [PubMed] [Google Scholar]
- 13.White S, Taetle R, Seligman PA, Rutherford M, Trowbridge IS. Combinations of anti-transferrin receptor monoclonal antibodies inhibit human tumor cell growth in vitro and in vivo: evidence for synergistic antiproliferative effects. Cancer Res. 1990;50:6295–6301. [PubMed] [Google Scholar]
- 14.Callens C, Moura IC, Lepelletier Y, Coulon S, Renand A, Dussiot M, Ghez D, Benhamou M, Monteiro RC, Bazarbachi A, Hermine O. Recent advances in adult T-cell leukemia therapy: focus on a new anti-transferrin receptor monoclonal antibody. Leukemia. 2008;22:42–48. doi: 10.1038/sj.leu.2404958. [DOI] [PubMed] [Google Scholar]
- 15.Ng PP, Dela Cruz JS, Sorour DN, Stinebaugh JM, Shin SU, Shin DS, Morrison SL, Penichet ML. An anti-transferrin receptor-avidin fusion protein exhibits both strong proapoptotic activity and the ability to deliver various molecules into cancer cells. Proc Natl Acad Sci U S A. 2002;99:10706–10711. doi: 10.1073/pnas.162362999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ng PP, Helguera G, Daniels TR, Lomas SZ, Rodriguez JA, Schiller G, Bonavida B, Morrison SL, Penichet ML. Molecular events contributing to cell death in malignant human hematopoietic cells elicited by an IgG3-avidin fusion protein targeting the transferrin receptor. Blood. 2006;108:2745–2754. doi: 10.1182/blood-2006-04-020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang X, Yang L, Chen ZG, Shin DM. Application of nanotechnology in cancer therapy and imaging. CA Cancer J Clin. 2008;58:97–110. doi: 10.3322/CA.2007.0003. [DOI] [PubMed] [Google Scholar]
- 18.Haley B, Frenkel E. Nanoparticles for drug delivery in cancer treatment. Urol Oncol. 2008;26:57–64. doi: 10.1016/j.urolonc.2007.03.015. [DOI] [PubMed] [Google Scholar]
- 19.Moghimi SM, Hunter AC, Murray JC. Nanomedicine: current status and future prospects. Faseb J. 2005;19:311–330. doi: 10.1096/fj.04-2747rev. [DOI] [PubMed] [Google Scholar]
- 20.Sapra P, Allen TM. Ligand-targeted liposomal anticancer drugs. Prog Lipid Res. 2003;42:439–462. doi: 10.1016/s0163-7827(03)00032-8. [DOI] [PubMed] [Google Scholar]
- 21.Northfelt DW, Martin FJ, Working P, Volberding PA, Russell J, Newman M, Amantea MA, Kaplan LD. Doxorubicin encapsulated in liposomes containing surface-bound polyethylene glycol: pharmacokinetics, tumor localization, and safety in patients with AIDS-related Kaposi’s sarcoma. J Clin Pharmacol. 1996;36:55–63. doi: 10.1002/j.1552-4604.1996.tb04152.x. [DOI] [PubMed] [Google Scholar]
- 22.Perry MC. Continuous Intravenous Infusion Chemotherapy. In: Pine JW, editor. The Chemotherapy Source Book. 4. Lippincott Williams and Wilkins; Philadelphia, PA: 2008. pp. 79–87. [Google Scholar]
- 23.Singh M, Atwal H, Micetich R. Transferrin directed delivery of adriamycin to human cells. Anticancer Res. 1998;18:1423–1427. [PubMed] [Google Scholar]
- 24.Berczi A, Barabas K, Sizensky JA, Faulk WP. Adriamycin conjugates of human transferrin bind transferrin receptors and kill K562 and HL60 cells. Arch Biochem Biophys. 1993;300:356–363. doi: 10.1006/abbi.1993.1048. [DOI] [PubMed] [Google Scholar]
- 25.Kratz F, Beyer U, Roth T, Tarasova N, Collery P, Lechenault F, Cazabat A, Schumacher P, Unger C, Falken U. Transferrin conjugates of doxorubicin: synthesis, characterization, cellular uptake, and in vitro efficacy. J Pharm Sci. 1998;87:338–346. doi: 10.1021/js970246a. [DOI] [PubMed] [Google Scholar]
- 26.Barabas K, Sizensky JA, Faulk WP. Evidence in support of the plasma membrane as the target for transferrin-adriamycin conjugates in K562 cells. Am J Reprod Immunol. 1991;25:120–123. doi: 10.1111/j.1600-0897.1991.tb01078.x. [DOI] [PubMed] [Google Scholar]
- 27.Sun IL, Sun EE, Crane FL, Morre DJ, Faulk WP. Inhibition of transplasma membrane electron transport by transferrin-adriamycin conjugates. Biochim Biophys Acta. 1992;1105:84–88. doi: 10.1016/0005-2736(92)90165-i. [DOI] [PubMed] [Google Scholar]
- 28.Fritzer M, Barabas K, Szuts V, Berczi A, Szekeres T, Faulk WP, Goldenberg H. Cytotoxicity of a transferrin-adriamycin conjugate to anthracycline-resistant cells. Int J Cancer. 1992;52:619–623. doi: 10.1002/ijc.2910520421. [DOI] [PubMed] [Google Scholar]
- 29.Hatano T, Ohkawa K, Matsuda M. Cytotoxic effect of the protein-doxorubicin conjugates on the multidrug-resistant human myelogenous leukemia cell line, K562, in vitro. Tumour Biol. 1993;14:288–294. doi: 10.1159/000217841. [DOI] [PubMed] [Google Scholar]
- 30.Lai BT, Gao JP, Lanks KW. Mechanism of action and spectrum of cell lines sensitive to a doxorubicin-transferrin conjugate. Cancer Chemother Pharmacol. 1998;41:155–160. doi: 10.1007/s002800050722. [DOI] [PubMed] [Google Scholar]
- 31.Barabas K, Sizensky JA, Faulk WP. Transferrin conjugates of adriamycin are cytotoxic without intercalating nuclear DNA. J Biol Chem. 1992;267:9437–9442. [PubMed] [Google Scholar]
- 32.Fritzer M, Szekeres T, Szuts V, Jarayam HN, Goldenberg H. Cytotoxic effects of a doxorubicin-transferrin conjugate in multidrug-resistant KB cells. Biochem Pharmacol. 1996;51:489–493. doi: 10.1016/0006-2952(95)02225-2. [DOI] [PubMed] [Google Scholar]
- 33.Lubgan D, Jozwiak Z, Grabenbauer GG, Distel LV. Doxorubicin-transferrin conjugate selectively overcomes multidrug resistance in leukaemia cells. Cell Mol Biol Lett. 2009;14:113–127. doi: 10.2478/s11658-008-0037-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chitambar CR. Gallium compounds as antineoplastic agents. Curr Opin Oncol. 2004;16:547–552. doi: 10.1097/01.cco.0000142071.22226.d2. [DOI] [PubMed] [Google Scholar]
- 35.Wang F, Jiang X, Yang DC, Elliott RL, Head JF. Doxorubicin-gallium-transferrin conjugate overcomes multidrug resistance: evidence for drug accumulation in the nucleus of drug resistant MCF-7/ADR cells. Anticancer Res. 2000;20:799–808. [PubMed] [Google Scholar]
- 36.Elliott RL, Stjernholm R, Elliott MC. Preliminary evaluation of platinum transferrin (MPTC-63) as a potential nontoxic treatment for breast cancer. Cancer Detect Prev. 1988;12:469–480. [PubMed] [Google Scholar]
- 37.Head JF, Wang F, Elliott RL. Antineoplastic drugs that interfere with iron metabolism in cancer cells. Adv Enzyme Regul. 1997;37:147–169. doi: 10.1016/s0065-2571(96)00010-6. [DOI] [PubMed] [Google Scholar]
- 38.Elliott RL, Head JF. Complete resolution of malignant ascites in stage IV breast cancer by peritoneal drainage and innovative chemoimmunotherapy: a case report. Cancer Biother Radiopharm. 2006;21:138–145. doi: 10.1089/cbr.2006.21.138. [DOI] [PubMed] [Google Scholar]
- 39.Beyer U, Roth T, Schumacher P, Maier G, Unold A, Frahm AW, Fiebig HH, Unger C, Kratz F. Synthesis and in vitro efficacy of transferrin conjugates of the anticancer drug chlorambucil. J Med Chem. 1998;41:2701–2708. doi: 10.1021/jm9704661. [DOI] [PubMed] [Google Scholar]
- 40.Tanaka T, Shiramoto S, Miyashita M, Fujishima Y, Kaneo Y. Tumor targeting based on the effect of enhanced permeability and retention (EPR) and the mechanism of receptor-mediated endocytosis (RME) Int J Pharm. 2004;277:39–61. doi: 10.1016/j.ijpharm.2003.09.050. [DOI] [PubMed] [Google Scholar]
- 41.Bejaoui N, Page M, Noel C. Cytotoxicity of transferrin-daunorubicin conjugates on small cell carcinoma of the lung (SCCL) cell line NCI-H69. Anticancer Res. 1991;11:2211–2213. [PubMed] [Google Scholar]
- 42.Mayers GL, Raghavan D, Hitt S, Glaves D. Proceedings of the American Association for Cancer Research. Vol. 89. New Orleans, Louisiana, USA: 1998. Transferrin-gemcitabine conjugate: application to chemotherapy; p. 63. [Google Scholar]
- 43.Raso V, Basala M. A highly cytotoxic human transferrin-ricin A chain conjugate used to select receptor-modified cells. J Biol Chem. 1984;259:1143–1149. [PubMed] [Google Scholar]
- 44.Chignola R, Foroni R, Franceschi A, Pasti M, Candiani C, Anselmi C, Fracasso G, Tridente G, Colombatti M. Heterogeneous response of individual multicellular tumour spheroids to immunotoxins and ricin toxin. Br J Cancer. 1995;72:607–614. doi: 10.1038/bjc.1995.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bergamaschi G, Cazzola M, Dezza L, Savino E, Consonni L, Lappi D. Killing of K562 cells with conjugates between human transferrin and a ribosome-inactivating protein (SO-6) Br J Haematol. 1988;68:379–384. doi: 10.1111/j.1365-2141.1988.tb04218.x. [DOI] [PubMed] [Google Scholar]
- 46.Ippoliti R, Lendaro E, D’Agostino I, Fiani ML, Guidarini D, Vestri S, Benedetti PA, Brunori M. A chimeric saporin-transferrin conjugate compared to ricin toxin: role of the carrier in intracellular transport and toxicity. Faseb J. 1995;9:1220–1225. doi: 10.1096/fasebj.9.12.7672515. [DOI] [PubMed] [Google Scholar]
- 47.Gosselaar PH, van-Dijk AJ, de-Gast GC, Polito L, Bolognesi A, Vooijs WC, Verheul AF, Krouwer HG, Marx JJ. Transferrin toxin but not transferrin receptor immunotoxin is influenced by free transferrin and iron saturation. Eur J Clin Invest. 2002;32(Suppl 1):61–69. doi: 10.1046/j.1365-2362.2002.0320s1061.x. [DOI] [PubMed] [Google Scholar]
- 48.Cimini A, Mei S, Benedetti E, Laurenti G, Koutris I, Cinque B, Cifone MG, Galzio R, Pitari G, Leandro LD, Giansanti F, Lombardi A, Fabbrini MS, Ippoliti R. Distinct cellular responses induced by saporin and a trasferrin-saporin conjugate in two different human glioblastoma cell lines. J Cell Physiol. 2011 doi: 10.1002/jcp.22805. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- 49.O’Keefe DO, Draper RK. Characterization of a transferrin-diphtheria toxin conjugate. J Biol Chem. 1985;260:932–937. [PubMed] [Google Scholar]
- 50.Yoon DJ, Chu DS, Ng CW, Pham EA, Mason AB, Hudson DM, Smith VC, MacGillivray RT, Kamei DT. Genetically engineering transferrin to improve its in vitro ability to deliver cytotoxins. J Control Release. 2009;133:178–184. doi: 10.1016/j.jconrel.2008.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yoon DJ, Kwan BH, Chao FC, Nicolaides TP, Phillips JJ, Lam GY, Mason AB, Weiss WA, Kamei DT. Intratumoral therapy of glioblastoma multiforme using genetically engineered transferrin for drug delivery. Cancer Res. 2010;70:4520–4527. doi: 10.1158/0008-5472.CAN-09-4311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnson VG, Wrobel C, Wilson D, Zovickian J, Greenfield L, Oldfield EH, Youle R. Improved tumor-specific immunotoxins in the treatment of CNS and leptomeningeal neoplasia. J Neurosurg. 1989;70:240–248. doi: 10.3171/jns.1989.70.2.0240. [DOI] [PubMed] [Google Scholar]
- 53.Martell LA, Agrawal A, Ross DA, Muraszko KM. Efficacy of transferrin receptor-targeted immunotoxins in brain tumor cell lines and pediatric brain tumors. Cancer Res. 1993;53:1348–1353. [PubMed] [Google Scholar]
- 54.Laske DW, Youle RJ, Oldfield EH. Tumor regression with regional distribution of the targeted toxin TF-CRM107 in patients with malignant brain tumors. Nat Med. 1997;3:1362–1368. doi: 10.1038/nm1297-1362. [DOI] [PubMed] [Google Scholar]
- 55.Weaver M, Laske DW. Transferrin receptor ligand-targeted toxin conjugate (Tf-CRM107) for therapy of malignant gliomas. J Neurooncol. 2003;65:3–13. doi: 10.1023/a:1026246500788. [DOI] [PubMed] [Google Scholar]
- 56.Lai H, Sasaki T, Singh NP. Targeted treatment of cancer with artemisinin and artemisinin-tagged iron-carrying compounds. Expert Opin Ther Targets. 2005;9:995–1007. doi: 10.1517/14728222.9.5.995. [DOI] [PubMed] [Google Scholar]
- 57.Nakase I, Lai H, Singh NP, Sasaki T. Anticancer properties of artemisinin derivatives and their targeted delivery by transferrin conjugation. Int J Pharm. 2008;354:28–33. doi: 10.1016/j.ijpharm.2007.09.003. [DOI] [PubMed] [Google Scholar]
- 58.Nakase I, Gallis B, Takatani-Nakase T, Oh S, Lacoste E, Singh NP, Goodlett DR, Tanaka S, Futaki S, Lai H, Sasaki T. Transferrin receptor-dependent cytotoxicity of artemisinin-transferrin conjugates on prostate cancer cells and induction of apoptosis. Cancer Lett. 2009;274:290–298. doi: 10.1016/j.canlet.2008.09.023. [DOI] [PubMed] [Google Scholar]
- 59.Lai H, Nakase I, Lacoste E, Singh NP, Sasaki T. Artemisinin-transferrin conjugate retards growth of breast tumors in the rat. Anticancer Res. 2009;29:3807–3810. [PubMed] [Google Scholar]
- 60.Jiang Y, Tang G, Hong M, Zhu S, Fang C, Shi B, Pei Y. Active tumor-targeted delivery of PEG-protein via transferrin-transferrin-receptor system. J Drug Target. 2007;15:672–683. doi: 10.1080/10611860701603414. [DOI] [PubMed] [Google Scholar]
- 61.Jiang YY, Liu C, Hong MH, Zhu SJ, Pei YY. Tumor cell targeting of transferrin-PEG-TNF-alpha conjugate via a receptor-mediated delivery system: design, synthesis, and biological evaluation. Bioconjug Chem. 2007;18:41–49. doi: 10.1021/bc060135f. [DOI] [PubMed] [Google Scholar]
- 62.Rybak SM, Newton DL, Mikulski SM, Viera A, Youle R. Cytotoxic Onconase and Ribonuclease A Chimeras: Comparison and In Vitro Characterization. Drug Delivery. 1993;1:3–10. [Google Scholar]
- 63.Suzuki M, Saxena SK, Boix E, Prill RJ, Vasandani VM, Ladner JE, Sung C, Youle RJ. Engineering receptor-mediated cytotoxicity into human ribonucleases by steric blockade of inhibitor interaction. Nat Biotechnol. 1999;17:265–270. doi: 10.1038/7010. [DOI] [PubMed] [Google Scholar]
- 64.Ward CM, Read ML, Seymour LW. Systemic circulation of poly(L-lysine)/DNA vectors is influenced by polycation molecular weight and type of DNA: differential circulation in mice and rats and the implications for human gene therapy. Blood. 2001;97:2221–2229. doi: 10.1182/blood.v97.8.2221. [DOI] [PubMed] [Google Scholar]
- 65.Tang MX, Szoka FC. The influence of polymer structure on the interactions of cationic polymers with DNA and morphology of the resulting complexes. Gene Ther. 1997;4:823–832. doi: 10.1038/sj.gt.3300454. [DOI] [PubMed] [Google Scholar]
- 66.Wagner E, Zenke M, Cotten M, Beug H, Birnstiel ML. Transferrin-polycation conjugates as carriers for DNA uptake into cells. Proc Natl Acad Sci U S A. 1990;87:3410–3414. doi: 10.1073/pnas.87.9.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zatloukal K, Wagner E, Cotten M, Phillips S, Plank C, Steinlein P, Curiel DT, Birnstiel ML. Transferrinfection: a highly efficient way to express gene constructs in eukaryotic cells. Ann N Y Acad Sci. 1992;660:136–153. doi: 10.1111/j.1749-6632.1992.tb21066.x. [DOI] [PubMed] [Google Scholar]
- 68.Cotten M, Langle-Rouault F, Kirlappos H, Wagner E, Mechtler K, Zenke M, Beug H, Birnstiel ML. Transferrin-polycation-mediated introduction of DNA into human leukemic cells: stimulation by agents that affect the survival of transfected DNA or modulate transferrin receptor levels. Proc Natl Acad Sci U S A. 1990;87:4033–4037. doi: 10.1073/pnas.87.11.4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wagner E, Cotten M, Mechtler K, Kirlappos H, Birnstiel ML. DNA-binding transferrin conjugates as functional gene-delivery agents: synthesis by linkage of polylysine or ethidium homodimer to the transferrin carbohydrate moiety. Bioconjug Chem. 1991;2:226–231. doi: 10.1021/bc00010a006. [DOI] [PubMed] [Google Scholar]
- 70.Taxman DJ, Lee ES, Wojchowski DM. Receptor-targeted transfection using stable maleimido-transferrin/thio-poly-L-lysine conjugates. Anal Biochem. 1993;213:97–103. doi: 10.1006/abio.1993.1391. [DOI] [PubMed] [Google Scholar]
- 71.Wagner E, Plank C, Zatloukal K, Cotten M, Birnstiel ML. Influenza virus hemagglutinin HA-2 N-terminal fusogenic peptides augment gene transfer by transferrin-polylysine-DNA complexes: toward a synthetic virus-like gene-transfer vehicle. Proc Natl Acad Sci U S A. 1992;89:7934–7938. doi: 10.1073/pnas.89.17.7934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Plank C, Oberhauser B, Mechtler K, Koch C, Wagner E. The influence of endosome-disruptive peptides on gene transfer using synthetic virus-like gene transfer systems. J Biol Chem. 1994;269:12918–12924. [PubMed] [Google Scholar]
- 73.Wightman L, Patzelt E, Wagner E, Kircheis R. Development of transferrin-polycation/DNA based vectors for gene delivery to melanoma cells. J Drug Target. 1999;7:293–303. doi: 10.3109/10611869909085512. [DOI] [PubMed] [Google Scholar]
- 74.Ogris M, Brunner S, Schuller S, Kircheis R, Wagner E. PEGylated DNA/transferrin-PEI complexes: reduced interaction with blood components, extended circulation in blood and potential for systemic gene delivery. Gene Ther. 1999;6:595–605. doi: 10.1038/sj.gt.3300900. [DOI] [PubMed] [Google Scholar]
- 75.Kircheis R, Schuller S, Brunner S, Ogris M, Heider KH, Zauner W, Wagner E. Polycation-based DNA complexes for tumor-targeted gene delivery in vivo. J Gene Med. 1999;1:111–120. doi: 10.1002/(SICI)1521-2254(199903/04)1:2<111::AID-JGM22>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- 76.Hallahan DE, Vokes EE, Rubin SJ, O’Brien S, Samuels B, Vijaykumar S, Kufe DW, Phillips R, Weichselbaum RR. Phase I dose-escalation study of tumor necrosis factor-alpha and concomitant radiation therapy. Cancer J Sci Am. 1995;1:204–209. [PubMed] [Google Scholar]
- 77.Kircheis R, Ostermann E, Wolschek MF, Lichtenberger C, Magin-Lachmann C, Wightman L, Kursa M, Wagner E. Tumor-targeted gene delivery of tumor necrosis factor-alpha induces tumor necrosis and tumor regression without systemic toxicity. Cancer Gene Ther. 2002;9:673–680. doi: 10.1038/sj.cgt.7700487. [DOI] [PubMed] [Google Scholar]
- 78.Kursa M, Walker GF, Roessler V, Ogris M, Roedl W, Kircheis R, Wagner E. Novel shielded transferrin-polyethylene glycol-polyethylenimine/DNA complexes for systemic tumor-targeted gene transfer. Bioconjug Chem. 2003;14:222–231. doi: 10.1021/bc0256087. [DOI] [PubMed] [Google Scholar]
- 79.Liu Y, Tao J, Li Y, Yang J, Yu Y, Wang M, Xu X, Huang C, Huang W, Dong J, Li L, Liu J, Shen G, Tu Y. Targeting hypoxia-inducible factor-1alpha with Tf-PEI-shRNA complex via transferrin receptor-mediated endocytosis inhibits melanoma growth. Mol Ther. 2009;17:269–277. doi: 10.1038/mt.2008.266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Koh MY, Spivak-Kroizman TR, Powis G. HIF-1alpha and cancer therapy. Recent Results Cancer Res. 2010;180:15–34. doi: 10.1007/978-3-540-78281-0_3. [DOI] [PubMed] [Google Scholar]
- 81.Zeng X, Sun YX, Qu W, Zhang XZ, Zhuo RX. Biotinylated transferrin/avidin/biotinylated disulfide containing PEI bioconjugates mediated p53 gene delivery system for tumor targeted transfection. Biomaterials. 2010;31:4771–4780. doi: 10.1016/j.biomaterials.2010.02.039. [DOI] [PubMed] [Google Scholar]
- 82.Sato Y, Yamauchi N, Takahashi M, Sasaki K, Fukaura J, Neda H, Fujii S, Hirayama M, Itoh Y, Koshita Y, Kogawa K, Kato J, Sakamaki S, Niitsu Y. In vivo gene delivery to tumor cells by transferrin-streptavidin-DNA conjugate. Faseb J. 2000;14:2108–2118. doi: 10.1096/fj.99-1052com. [DOI] [PubMed] [Google Scholar]
- 83.Bjorn MJ, Groetsema G. Immunotoxins to the murine transferrin receptor: intracavitary therapy of mice bearing syngeneic peritoneal tumors. Cancer Res. 1987;47:6639–6645. [PubMed] [Google Scholar]
- 84.Lesley J, Domingo DL, Schulte R, Trowbridge IS. Effect of an anti-murine transferrin receptor-ricin A conjugate on bone marrow stem and progenitor cells treated in vitro. Exp Cell Res. 1984;150:400–407. doi: 10.1016/0014-4827(84)90583-4. [DOI] [PubMed] [Google Scholar]
- 85.Dreier T, Lode HN, Xiang R, Dolman CS, Reisfeld RA, Kang AS. Recombinant immunocytokines targeting the mouse transferrin receptor: construction and biological activities. Bioconjug Chem. 1998;9:482–489. doi: 10.1021/bc980020e. [DOI] [PubMed] [Google Scholar]
- 86.Shi Y, Liu CH, Roberts AI, Das J, Xu G, Ren G, Zhang Y, Zhang L, Yuan ZR, Tan HS, Das G, Devadas S. Granulocyte-macrophage colony-stimulating factor (GM-CSF) and T-cell responses: what we do and don’t know. Cell Res. 2006;16:126–133. doi: 10.1038/sj.cr.7310017. [DOI] [PubMed] [Google Scholar]
- 87.Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, Muramatsu S, Steinman RM. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–1702. doi: 10.1084/jem.176.6.1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Haynes BF, Hemler M, Cotner T, Mann DL, Eisenbarth GS, Strominger JL, Fauci AS. Characterization of a monoclonal antibody (5E9) that defines a human cell surface antigen of cell activation. J Immunol. 1981;127:347–351. [PubMed] [Google Scholar]
- 89.Pirker R, FitzGerald DJ, Hamilton TC, Ozols RF, Willingham MC, Pastan I. Anti-transferrin receptor antibody linked to Pseudomonas exotoxin as a model immunotoxin in human ovarian carcinoma cell lines. Cancer Res. 1985;45:751–757. [PubMed] [Google Scholar]
- 90.Pirker R, FitzGerald DJ, Willingham MC, Pastan I. Enhancement of the activity of immunotoxins made with either ricin A chain or Pseudomonas exotoxin in human ovarian and epidermoid carcinoma cell lines. Cancer Res. 1988;48:3919–3923. [PubMed] [Google Scholar]
- 91.Shinohara H, Fan D, Ozawa S, Yano S, Van Arsdell M, Viner JL, Beers R, Pastan I, Fidler IJ. Site-specific expression of transferrin receptor by human colon cancer cells directly correlates with eradication by antitransferrin recombinant immunotoxin. Int J Oncol. 2000;17:643–651. doi: 10.3892/ijo.17.4.643. [DOI] [PubMed] [Google Scholar]
- 92.Batra JK, Fitzgerald DJ, Chaudhary VK, Pastan I. Single-chain immunotoxins directed at the human transferrin receptor containing Pseudomonas exotoxin A or diphtheria toxin: anti-TFR(Fv)-PE40 and DT388-anti-TFR(Fv) Mol Cell Biol. 1991;11:2200–2205. doi: 10.1128/mcb.11.4.2200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Traini R, Ben-Josef G, Pastrana DV, Moskatel E, Sharma AK, Antignani A, Fitzgerald DJ. ABT-737 overcomes resistance to immunotoxin-mediated apoptosis and enhances the delivery of pseudomonas exotoxin-based proteins to the cell cytosol. Mol Cancer Ther. 2010;9:2007–2015. doi: 10.1158/1535-7163.MCT-10-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Batra JK, Jinno Y, Chaudhary VK, Kondo T, Willingham MC, FitzGerald DJ, Pastan I. Antitumor activity in mice of an immunotoxin made with anti-transferrin receptor and a recombinant form of Pseudomonas exotoxin. Proc Natl Acad Sci U S A. 1989;86:8545–8549. doi: 10.1073/pnas.86.21.8545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Scott CF, Jr, Goldmacher VS, Lambert JM, Jackson JV, McIntyre GD. An immunotoxin composed of a monoclonal antitransferrin receptor antibody linked by a disulfide bond to the ribosome-inactivating protein gelonin: potent in vitro and in vivo effects against human tumors. J Natl Cancer Inst. 1987;79:1163–1172. [PubMed] [Google Scholar]
- 96.Ramakrishnan S, Houston LL. Inhibition of human acute lymphoblastic leukemia cells by immunotoxins: potentiation by chloroquine. Science. 1984;223:58–61. doi: 10.1126/science.6318313. [DOI] [PubMed] [Google Scholar]
- 97.Ramakrishnan S, Enghlid JJ, Bryant HL, Jr, Xu FJ. Characterization of a translation inhibitory protein from Luffa aegyptiaca. Biochem Biophys Res Commun. 1989;160:509–516. doi: 10.1016/0006-291x(89)92462-5. [DOI] [PubMed] [Google Scholar]
- 98.Braslawsky GR, Kadow K, Knipe J, McGoff K, Edson M, Kaneko T, Greenfield RS. Adriamycin(hydrazone)-antibody conjugates require internalization and intracellular acid hydrolysis for antitumor activity. Cancer Immunol Immunother. 1991;33:367–374. doi: 10.1007/BF01741596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Rathore D, Batra JK. Construction, expression and characterization of chimaeric toxins containing the ribonucleolytic toxin restrictocin: intracellular mechanism of action. Biochem J. 1997;324(Pt 3):815–822. doi: 10.1042/bj3240815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Goyal A, Batra JK. Inclusion of a furin-sensitive spacer enhances the cytotoxicity of ribotoxin restrictocin containing recombinant single-chain immunotoxins. Biochem J. 2000;345(Pt 2):247–254. [PMC free article] [PubMed] [Google Scholar]
- 101.Rathore D, Batra JK. Generation of active immunotoxins containing recombinant restrictocin. Biochem Biophys Res Commun. 1996;222:58–63. doi: 10.1006/bbrc.1996.0697. [DOI] [PubMed] [Google Scholar]
- 102.Rathore D, Nayak SK, Batra JK. Overproduction of fungal ribotoxin alpha-sarcin in Escherichia coli: generation of an active immunotoxin. Gene. 1997;190:31–35. doi: 10.1016/s0378-1119(96)00696-8. [DOI] [PubMed] [Google Scholar]
- 103.Pearson JW, Hedrick E, Fogler WE, Bull RL, Ferris DK, Riggs CW, Wiltrout RH, Sivam G, Morgan AC, Groves E, et al. Enhanced therapeutic efficacy against an ovarian tumor xenograft of immunotoxins used in conjunction with recombinant alpha-interferon. Cancer Res. 1990;50:6379–6388. [PubMed] [Google Scholar]
- 104.Laske DW, Ilercil O, Akbasak A, Youle RJ, Oldfield EH. Efficacy of direct intratumoral therapy with targeted protein toxins for solid human gliomas in nude mice. J Neurosurg. 1994;80:520–526. doi: 10.3171/jns.1994.80.3.0520. [DOI] [PubMed] [Google Scholar]
- 105.Laske DW, Muraszko KM, Oldfield EH, DeVroom HL, Sung C, Dedrick RL, Simon TR, Colandrea J, Copeland C, Katz D, Greenfield L, Groves ES, Houston LL, Youle RJ. Intraventricular immunotoxin therapy for leptomeningeal neoplasia. Neurosurgery. 1997;41:1039–1049. doi: 10.1097/00006123-199711000-00005. discussion 1049-1051. [DOI] [PubMed] [Google Scholar]
- 106.Ghetie V, Vitetta E. Immunotoxins in the therapy of cancer: from bench to clinic. Pharmacol Ther. 1994;63:209–234. doi: 10.1016/0163-7258(94)90025-6. [DOI] [PubMed] [Google Scholar]
- 107.Trowbridge IS, Domingo DL. Anti-transferrin receptor monoclonal antibody and toxin-antibody conjugates affect growth of human tumour cells. Nature. 1981;294:171–173. doi: 10.1038/294171a0. [DOI] [PubMed] [Google Scholar]
- 108.Cazzola M, Bergamaschi G, Dezza L, D’Uva R, Ponchio L, Rosti V, Ascari E. Cytotoxic activity of an anti-transferrin receptor immunotoxin on normal and leukemic human hematopoietic progenitors. Cancer Res. 1991;51:536–541. [PubMed] [Google Scholar]
- 109.Rybak SM, Saxena SK, Ackerman EJ, Youle RJ. Cytotoxic potential of ribonuclease and ribonuclease hybrid proteins. J Biol Chem. 1991;266:21202–21207. [PubMed] [Google Scholar]
- 110.Recht LD, Griffin TW, Raso V, Salimi AR. Potent cytotoxicity of an antihuman transferrin receptor-ricin A-chain immunotoxin on human glioma cells in vitro. Cancer Res. 1990;50:6696–6700. [PubMed] [Google Scholar]
- 111.Griffin T, Raso V. Monensin in lipid emulsion for the potentiation of ricin A chain immunotoxins. Cancer Res. 1991;51:4316–4322. [PubMed] [Google Scholar]
- 112.Griffin TW, Stocl M, Collins J, Fernandes J, Maher VE. Combined antitumor therapy with the chemotherapeutic drug doxorubicin and an anti-transferrin receptor immunotoxin: in vitro and in vivo studies. J Immunother. 1992;1991(11):12–18. doi: 10.1097/00002371-199201000-00002. [DOI] [PubMed] [Google Scholar]
- 113.Li J, Weng X, Liang Z, Zhong M, Chen X, Lu S, Sun W, Song Y, Wu X, Shen G. Viral specific cytotoxic T cells inhibit the growth of TfR-expressing tumor cells with antibody targeted viral peptide/HLA-A2 complex. Cell Immunol. 2010;263:154–160. doi: 10.1016/j.cellimm.2010.03.008. [DOI] [PubMed] [Google Scholar]
- 114.Newton DL, Pollock D, DiTullio P, Echelard Y, Harvey M, Wilburn B, Williams J, Hoogenboom HR, Raus JC, Meade HM, Rybak SM. Antitransferrin receptor antibody-RNase fusion protein expressed in the mammary gland of transgenic mice. J Immunol Methods. 1999;231:159–167. doi: 10.1016/s0022-1759(99)00154-4. [DOI] [PubMed] [Google Scholar]
- 115.Rybak SM, Hoogenboom HR, Meade HM, Raus JC, Schwartz D, Youle RJ. Humanization of immunotoxins. Proc Natl Acad Sci U S A. 1992;89:3165–3169. doi: 10.1073/pnas.89.8.3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Newton DL, Nicholls PJ, Rybak SM, Youle RJ. Expression and characterization of recombinant human eosinophil-derived neurotoxin and eosinophil-derived neurotoxin-anti-transferrin receptor sFv. J Biol Chem. 1994;269:26739–26745. [PubMed] [Google Scholar]
- 117.Zewe M, Rybak SM, Dubel S, Coy JF, Welschof M, Newton DL, Little M. Cloning and cytotoxicity of a human pancreatic RNase immunofusion. Immunotechnology. 1997;3:127–136. doi: 10.1016/s1380-2933(97)00070-5. [DOI] [PubMed] [Google Scholar]
- 118.Newton DL, Xue Y, Olson KA, Fett JW, Rybak SM. Angiogenin single-chain immunofusions: influence of peptide linkers and spacers between fusion protein domains. Biochemistry. 1996;35:545–553. doi: 10.1021/bi951650w. [DOI] [PubMed] [Google Scholar]
- 119.Asai T, Trinh R, Ng PP, Penichet ML, Wims LA, Morrison SL. A human biotin acceptor domain allows site-specific conjugation of an enzyme to an antibody-avidin fusion protein for targeted drug delivery. Biomol Eng. 2005;21:145–155. doi: 10.1016/j.bioeng.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 120.Ortiz-Sanchez E, Daniels TR, Helguera G, Martinez-Maza O, Bonavida B, Penichet ML. Enhanced cytotoxicity of an anti-transferrin receptor IgG3-avidin fusion protein in combination with gambogic acid against human malignant hematopoietic cells: functional relevance of iron, the receptor, and reactive oxygen species. Leukemia. 2009;23:59–70. doi: 10.1038/leu.2008.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Daniels TR, Ortiz-Sanchez E, Luria-Perez R, Quintero R, Helguera G, Bonavida B, Martinez-Maza O, Penichet ML. An Antibody-based Multifaceted Approach Targeting the Human Transferrin Receptor for the Treatment of B-cell Malignancies. J Immunother. 2011;34:500–508. doi: 10.1097/CJI.0b013e318222ffc8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Daniels TR, Ng PP, Delgado T, Lynch MR, Schiller G, Helguera G, Penichet ML. Conjugation of an anti transferrin receptor IgG3-avidin fusion protein with biotinylated saporin results in significant enhancement of its cytotoxicity against malignant hematopoietic cells. Mol Cancer Ther. 2007;6:2995–3008. doi: 10.1158/1535-7163.MCT-07-0330. [DOI] [PubMed] [Google Scholar]
- 123.Oh S, Kim BJ, Singh NP, Lai H, Sasaki T. Synthesis and anti-cancer activity of covalent conjugates of artemisinin and a transferrin-receptor targeting peptide. Cancer Lett. 2009;274:33–39. doi: 10.1016/j.canlet.2008.08.031. [DOI] [PubMed] [Google Scholar]
- 124.Lukyanov AN, Torchilin VP. Micelles from lipid derivatives of water-soluble polymers as delivery systems for poorly soluble drugs. Adv Drug Deliv Rev. 2004;56:1273–1289. doi: 10.1016/j.addr.2003.12.004. [DOI] [PubMed] [Google Scholar]
- 125.Chiappetta DA, Sosnik A. Poly(ethylene oxide)-poly(propylene oxide) block copolymer micelles as drug delivery agents: improved hydrosolubility, stability and bioavailability of drugs. Eur J Pharm Biopharm. 2007;66:303–317. doi: 10.1016/j.ejpb.2007.03.022. [DOI] [PubMed] [Google Scholar]
- 126.Savic R, Luo L, Eisenberg A, Maysinger D. Micellar nanocontainers distribute to defined cytoplasmic organelles. Science. 2003;300:615–618. doi: 10.1126/science.1078192. [DOI] [PubMed] [Google Scholar]
- 127.Khan D. The use of nanocarriers for drug delivery in cancer therapy. J Cancer Sci Ther. 2010;2:058–062. [Google Scholar]
- 128.Jones M, Leroux J. Polymeric micelles - a new generation of colloidal drug carriers. Eur J Pharm Biopharm. 1999;48:101–111. doi: 10.1016/s0939-6411(99)00039-9. [DOI] [PubMed] [Google Scholar]
- 129.Blanco E, Kessinger CW, Sumer BD, Gao J. Multifunctional micellar nanomedicine for cancer therapy. Exp Biol Med (Maywood) 2009;234:123–131. doi: 10.3181/0808-MR-250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Huh KM, Min HS, Lee SC, Lee HJ, Kim S, Park K. A new hydrotropic block copolymer micelle system for aqueous solubilization of paclitaxel. J Control Release. 2008;126:122–129. doi: 10.1016/j.jconrel.2007.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Licciardi M, Giammona G, Du J, Armes SP, Tang Y, Lewis AL. New folate-functionalized biocompatible block copolymer micelles as potential anti-cancer drug delivery systems. Polymer. 2006;47:2946–2955. [Google Scholar]
- 132.Danhier F, Magotteaux N, Ucakar B, Lecouturier N, Brewster M, Preat V. Novel self-assembling PEG-p-(CL-co-TMC) polymeric micelles as safe and effective delivery system for paclitaxel. Eur J Pharm Biopharm. 2009;73:230–238. doi: 10.1016/j.ejpb.2009.06.015. [DOI] [PubMed] [Google Scholar]
- 133.Yokoyama M, Okano T, Sakurai Y, Fukushima S, Okamoto K, Kataoka K. Selective delivery of adriamycin to a solid tumor using a polymeric micelle carrier system. J Drug Target. 1999;7:171–186. doi: 10.3109/10611869909085500. [DOI] [PubMed] [Google Scholar]
- 134.Gaucher G, Dufresne MH, Sant VP, Kang N, Maysinger D, Leroux JC. Block copolymer micelles: preparation, characterization and application in drug delivery. J Control Release. 2005;109:169–188. doi: 10.1016/j.jconrel.2005.09.034. [DOI] [PubMed] [Google Scholar]
- 135.Vinogradov S, Batrakova E, Li S, Kabanov A. Polyion complex micelles with protein-modified corona for receptor-mediated delivery of oligonucleotides into cells. Bioconjug Chem. 1999;10:851–860. doi: 10.1021/bc990037c. [DOI] [PubMed] [Google Scholar]
- 136.Ren WH, Chang J, Yan CH, Qian XM, Long LX, He B, Yuan XB, Kang CS, Betbeder D, Sheng J, Pu PY. Development of transferrin functionalized poly(ethylene glycol)/poly(lactic acid) amphiphilic block copolymeric micelles as a potential delivery system targeting brain glioma. J Mater Sci Mater Med. 2010;21:2673–2681. doi: 10.1007/s10856-010-4106-5. [DOI] [PubMed] [Google Scholar]
- 137.Agarwal A, Saraf S, Asthana A, Gupta U, Gajbhiye V, Jain NK. Ligand based dendritic systems for tumor targeting. Int J Pharm. 2008;350:3–13. doi: 10.1016/j.ijpharm.2007.09.024. [DOI] [PubMed] [Google Scholar]
- 138.Zeng F, Zimmerman SC. Dendrimers in Supramolecular Chemistry: From Molecular Recognition to Self-Assembly. Chem Rev. 1997;97:1681–1712. doi: 10.1021/cr9603892. [DOI] [PubMed] [Google Scholar]
- 139.Boas U, Heegaard PM. Dendrimers in drug research. Chem Soc Rev. 2004;33:43–63. doi: 10.1039/b309043b. [DOI] [PubMed] [Google Scholar]
- 140.Aulenta F, Hayes W, Rannard S. Dendrimers: a new class of nanoscopic containers and delivery devices. Eur Polymer J. 2003;39:1741. [Google Scholar]
- 141.Tomalia DA. The dendritic state. Materials Today. 2005:34–46. [Google Scholar]
- 142.Liu M, Frechet JM. Designing dendrimers for drug delivery. Pharm Sci Technolo Today. 1999;2:393–401. doi: 10.1016/s1461-5347(99)00203-5. [DOI] [PubMed] [Google Scholar]
- 143.Bhadra D, Bhadra S, Jain S, Jain NK. A PEGylated dendritic nanoparticulate carrier of fluorouracil. Int J Pharm. 2003;257:111–124. doi: 10.1016/s0378-5173(03)00132-7. [DOI] [PubMed] [Google Scholar]
- 144.Lee CC, Gillies ER, Fox ME, Guillaudeu SJ, Frechet JM, Dy EE, Szoka FC. A single dose of doxorubicin-functionalized bow-tie dendrimer cures mice bearing C-26 colon carcinomas. Proc Natl Acad Sci U S A. 2006;103:16649–16654. doi: 10.1073/pnas.0607705103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Vincent L, Varet J, Pille JY, Bompais H, Opolon P, Maksimenko A, Malvy C, Mirshahi M, Lu H, Vannier JP, Soria C, Li H. Efficacy of dendrimer-mediated angiostatin and TIMP-2 gene delivery on inhibition of tumor growth and angiogenesis: in vitro and in vivo studies. Int J Cancer. 2003;105:419–429. doi: 10.1002/ijc.11105. [DOI] [PubMed] [Google Scholar]
- 146.Nanjwade BK, Bechra HM, Derkar GK, Manvi FV, Nanjwade VK. Dendrimers: emerging polymers for drug-delivery systems. Eur J Pharm Sci. 2009;38:185–196. doi: 10.1016/j.ejps.2009.07.008. [DOI] [PubMed] [Google Scholar]
- 147.Gupta U, Dwivedi SK, Bid HK, Konwar R, Jain NK. Ligand anchored dendrimers based nanoconstructs for effective targeting to cancer cells. Int J Pharm. 2010;393:185–196. doi: 10.1016/j.ijpharm.2010.04.002. [DOI] [PubMed] [Google Scholar]
- 148.Koppu S, Oh YJ, Edrada-Ebel R, Blatchford DR, Tetley L, Tate RJ, Dufes C. Tumor regression after systemic administration of a novel tumor-targeted gene delivery system carrying a therapeutic plasmid DNA. J Control Release. 2010;143:215–221. doi: 10.1016/j.jconrel.2009.11.015. [DOI] [PubMed] [Google Scholar]
- 149.Han L, Huang R, Liu S, Huang S, Jiang C. Peptide-conjugated PAMAM for targeted doxorubicin delivery to transferrin receptor overexpressed tumors. Mol Pharm. 2010;7:2156–2165. doi: 10.1021/mp100185f. [DOI] [PubMed] [Google Scholar]
- 150.Han L, Huang R, Li J, Liu S, Huang S, Jiang C. Plasmid pORF-hTRAIL and doxorubicin co-delivery targeting to tumor using peptide-conjugated polyamidoamine dendrimer. Biomaterials. 2011;32:1242–1252. doi: 10.1016/j.biomaterials.2010.09.070. [DOI] [PubMed] [Google Scholar]
- 151.He H, Li Y, Jia XR, Du J, Ying X, Lu WL, Lou JN, Wei Y. PEGylated Poly(amidoamine) dendrimer-based dual-targeting carrier for treating brain tumors. Biomaterials. 2011;32:478–487. doi: 10.1016/j.biomaterials.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 152.Pinto Reis C, Neufeld RJ, Ribeiro AJ, Veiga F. Nanoencapsulation I. Methods for preparation of drug-loaded polymeric nanoparticles. Nanomedicine. 2006;2:8–21. doi: 10.1016/j.nano.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 153.Jain RA. The manufacturing techniques of various drug loaded biodegradable poly(lactide-co-glycolide) (PLGA) devices. Biomaterials. 2000;21:2475–2490. doi: 10.1016/s0142-9612(00)00115-0. [DOI] [PubMed] [Google Scholar]
- 154.Soppimath KS, Aminabhavi TM, Kulkarni AR, Rudzinski WE. Biodegradable polymeric nanoparticles as drug delivery devices. J Control Release. 2001;70:1–20. doi: 10.1016/s0168-3659(00)00339-4. [DOI] [PubMed] [Google Scholar]
- 155.Brigger I, Dubernet C, Couvreur P. Nanoparticles in cancer therapy and diagnosis. Adv Drug Deliv Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 156.Letchford K, Burt H. A review of the formation and classification of amphiphilic block copolymer nanoparticulate structures: micelles, nanospheres, nanocapsules and polymersomes. Eur J Pharm Biopharm. 2007;65:259–269. doi: 10.1016/j.ejpb.2006.11.009. [DOI] [PubMed] [Google Scholar]
- 157.Wang J, Tian S, Petros RA, Napier ME, Desimone JM. The complex role of multivalency in nanoparticles targeting the transferrin receptor for cancer therapies. J Am Chem Soc. 2010;132:11306–11313. doi: 10.1021/ja1043177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Liu Z, Jiao Y, Wang Y, Zhou C, Zhang Z. Polysaccharides-based nanoparticles as drug delivery systems. Adv Drug Deliv Rev. 2008;60:1650–1662. doi: 10.1016/j.addr.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 159.Nagpal K, Singh SK, Mishra DN. Chitosan nanoparticles: a promising system in novel drug delivery. Chem Pharm Bull (Tokyo) 2010;58:1423–1430. doi: 10.1248/cpb.58.1423. [DOI] [PubMed] [Google Scholar]
- 160.Lee BS, Fujita M, Khazenzon NM, Wawrowsky KA, Wachsmann-Hogiu S, Farkas DL, Black KL, Ljubimova JY, Holler E. Polycefin, a new prototype of a multifunctional nanoconjugate based on poly(beta-L-malic acid) for drug delivery. Bioconjug Chem. 2006;17:317–326. doi: 10.1021/bc0502457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Zheng Y, Yu B, Weecharangsan W, Piao L, Darby M, Mao Y, Koynova R, Yang X, Li H, Xu S, Lee LJ, Sugimoto Y, Brueggemeier RW, Lee RJ. Transferrin-conjugated lipid-coated PLGA nanoparticles for targeted delivery of aromatase inhibitor 7alpha-APTADD to breast cancer cells. Int J Pharm. 2011;390:234–241. doi: 10.1016/j.ijpharm.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sahoo SK, Ma W, Labhasetwar V. Efficacy of transferrin-conjugated paclitaxel-loaded nanoparticles in a murine model of prostate cancer. Int J Cancer. 2004;112:335–340. doi: 10.1002/ijc.20405. [DOI] [PubMed] [Google Scholar]
- 163.Sahoo SK, Labhasetwar V. Enhanced antiproliferative activity of transferrin-conjugated paclitaxel-loaded nanoparticles is mediated via sustained intracellular drug retention. Mol Pharm. 2005;2:373–383. doi: 10.1021/mp050032z. [DOI] [PubMed] [Google Scholar]
- 164.Shah N, Chaudhari K, Dantuluri P, Murthy RS, Das S. Paclitaxel-loaded PLGA nanoparticles surface modified with transferrin and Pluronic((R))P85, an in vitro cell line and in vivo biodistribution studies on rat model. J Drug Target. 2009;17:533–542. doi: 10.1080/10611860903046628. [DOI] [PubMed] [Google Scholar]
- 165.Pulkkinen M, Pikkarainen J, Wirth T, Tarvainen T, Haapa-aho V, Korhonen H, Seppala J, Jarvinen K. Three-step tumor targeting of paclitaxel using biotinylated PLA-PEG nanoparticles and avidin-biotin technology: Formulation development and in vitro anticancer activity. Eur J Pharm Biopharm. 2008;70:66–74. doi: 10.1016/j.ejpb.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 166.Xu Z, Gu W, Huang J, Sui H, Zhou Z, Yang Y, Yan Z, Li Y. In vitro and in vivo evaluation of actively targetable nanoparticles for paclitaxel delivery. Int J Pharm. 2005;288:361–368. doi: 10.1016/j.ijpharm.2004.10.009. [DOI] [PubMed] [Google Scholar]
- 167.Hong M, Zhu S, Jiang Y, Tang G, Sun C, Fang C, Shi B, Pei Y. Novel anti-tumor strategy: PEG-hydroxycamptothecin conjugate loaded transferrin-PEG-nanoparticles. J Control Release. 2010;141:22–29. doi: 10.1016/j.jconrel.2009.08.024. [DOI] [PubMed] [Google Scholar]
- 168.Gan CW, Feng SS. Transferrin-conjugated nanoparticles of poly(lactide)-D-alpha-tocopheryl polyethylene glycol succinate diblock copolymer for targeted drug delivery across the blood-brain barrier. Biomaterials. 2010;31:7748–7757. doi: 10.1016/j.biomaterials.2010.06.053. [DOI] [PubMed] [Google Scholar]
- 169.Li Y, Ogris M, Wagner E, Pelisek J, Ruffer M. Nanoparticles bearing polyethyleneglycol-coupled transferrin as gene carriers: preparation and in vitro evaluation. Int J Pharm. 2003;259:93–101. doi: 10.1016/s0378-5173(03)00211-4. [DOI] [PubMed] [Google Scholar]
- 170.Aktas Y, Yemisci M, Andrieux K, Gursoy RN, Alonso MJ, Fernandez-Megia E, Novoa-Carballal R, Quinoa E, Riguera R, Sargon MF, Celik HH, Demir AS, Hincal AA, Dalkara T, Capan Y, Couvreur P. Development and brain delivery of chitosan-PEG nanoparticles functionalized with the monoclonal antibody OX26. Bioconjug Chem. 2005;16:1503–1511. doi: 10.1021/bc050217o. [DOI] [PubMed] [Google Scholar]
- 171.Kateb B, Chiu K, Black KL, Yamamoto V, Khalsa B, Ljubimova JY, Ding H, Patil R, Portilla-Arias JA, Modo M, Moore DF, Farahani K, Okun MS, Prakash N, Neman J, Ahdoot D, Grundfest W, Nikzad S, Heiss JD. Nanoplatforms for constructing new approaches to cancer treatment, imaging, and drug delivery: what should be the policy? Neuroimage. 2011;54(Suppl 1):S106–124. doi: 10.1016/j.neuroimage.2010.01.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Groothuis DR. The blood-brain and blood-tumor barriers: a review of strategies for increasing drug delivery. Neuro Oncol. 2000;2:45–59. doi: 10.1093/neuonc/2.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ljubimova JY, Fujita M, Khazenzon NM, Lee BS, Wachsmann-Hogiu S, Farkas DL, Black KL, Holler E. Nanoconjugate based on polymalic acid for tumor targeting. Chem Biol Interact. 2008;171:195–203. doi: 10.1016/j.cbi.2007.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Patil R, Portilla-Arias J, Ding H, Inoue S, Konda B, Hu J, Wawrowsky KA, Shin PK, Black KL, Holler E, Ljubimova JY. Temozolomide delivery to tumor cells by a multifunctional nano vehicle based on poly(beta-L-malic acid) Pharm Res. 2010;27:2317–2329. doi: 10.1007/s11095-010-0091-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Fujita M, Khazenzon NM, Ljubimov AV, Lee BS, Virtanen I, Holler E, Black KL, Ljubimova JY. Inhibition of laminin-8 in vivo using a novel poly(malic acid)-based carrier reduces glioma angiogenesis. Angiogenesis. 2006;9:183–191. doi: 10.1007/s10456-006-9046-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Fujita M, Lee BS, Khazenzon NM, Penichet ML, Wawrowsky KA, Patil R, Ding H, Holler E, Black KL, Ljubimova JY. Brain tumor tandem targeting using a combination of monoclonal antibodies attached to biopoly(beta-L-malic acid) J Control Release. 2007;122:356–363. doi: 10.1016/j.jconrel.2007.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Ding H, Inoue S, Ljubimov AV, Patil R, Portilla-Arias J, Hu J, Konda B, Wawrowsky KA, Fujita M, Karabalin N, Sasaki T, Black KL, Holler E, Ljubimova JY. Inhibition of brain tumor growth by intravenous poly (beta-L-malic acid) nanobioconjugate with pH-dependent drug release [corrected] Proc Natl Acad Sci U S A. 2010;107:18143–18148. doi: 10.1073/pnas.1003919107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Inoue S, Ding H, Portilla-Arias J, Hu J, Konda B, Fujita M, Espinoza A, Suhane S, Riley M, Gates M, Patil R, Penichet ML, Ljubimov AV, Black KL, Holler E, Ljubimova JY. Polymalic acid-based nanobiopolymer provides efficient systemic breast cancer treatment by inhibiting both HER2/neu receptor synthesis and activity. Cancer Res. 2011;71:1454–1464. doi: 10.1158/0008-5472.CAN-10-3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Malam Y, Loizidou M, Seifalian AM. Liposomes and nanoparticles: nanosized vehicles for drug delivery in cancer. Trends Pharmacol Sci. 2009;30:592–599. doi: 10.1016/j.tips.2009.08.004. [DOI] [PubMed] [Google Scholar]
- 180.Drulis-Kawa Z, Dorotkiewicz-Jach A. Liposomes as delivery systems for antibiotics. Int J Pharm. 2010;387:187–198. doi: 10.1016/j.ijpharm.2009.11.033. [DOI] [PubMed] [Google Scholar]
- 181.Misra R, Acharya S, Sahoo SK. Cancer nanotechnology: application of nanotechnology in cancer therapy. Drug Discov Today. 2010;15:842–850. doi: 10.1016/j.drudis.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 182.Ishida O, Maruyama K, Tanahashi H, Iwatsuru M, Sasaki K, Eriguchi M, Yanagie H. Liposomes bearing polyethyleneglycol-coupled transferrin with intracellular targeting property to the solid tumors in vivo. Pharm Res. 2001;18:1042–1048. doi: 10.1023/a:1010960900254. [DOI] [PubMed] [Google Scholar]
- 183.Kobayashi T, Ishida T, Okada Y, Ise S, Harashima H, Kiwada H. Effect of transferrin receptor-targeted liposomal doxorubicin in P-glycoprotein-mediated drug resistant tumor cells. Int J Pharm. 2007;329:94–102. doi: 10.1016/j.ijpharm.2006.08.039. [DOI] [PubMed] [Google Scholar]
- 184.Eavarone DA, Yu X, Bellamkonda RV. Targeted drug delivery to C6 glioma by transferrin-coupled liposomes. J Biomed Mater Res. 2000;51:10–14. doi: 10.1002/(sici)1097-4636(200007)51:1<10::aid-jbm2>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 185.Fonseca C, Moreira JN, Ciudad CJ, Pedroso de Lima MC, Simoes S. Targeting of sterically stabilised pH-sensitive liposomes to human T-leukaemia cells. Eur J Pharm Biopharm. 2005;59:359–366. doi: 10.1016/j.ejpb.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 186.Li X, Ding L, Xu Y, Wang Y, Ping Q. Targeted delivery of doxorubicin using stealth liposomes modified with transferrin. Int J Pharm. 2009;373:116–123. doi: 10.1016/j.ijpharm.2009.01.023. [DOI] [PubMed] [Google Scholar]
- 187.Iinuma H, Maruyama K, Okinaga K, Sasaki K, Sekine T, Ishida O, Ogiwara N, Johkura K, Yonemura Y. Intracellular targeting therapy of cisplatin-encapsulated transferrin-polyethylene glycol liposome on peritoneal dissemination of gastric cancer. Int J Cancer. 2002;99:130–137. doi: 10.1002/ijc.10242. [DOI] [PubMed] [Google Scholar]
- 188.Xu S, Liu Y, Tai HC, Zhu J, Ding H, Lee RJ. Synthesis of transferrin (Tf) conjugated liposomes via Staudinger ligation. Int J Pharm. 2011;404:205–210. doi: 10.1016/j.ijpharm.2010.10.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Anabousi S, Bakowsky U, Schneider M, Huwer H, Lehr CM, Ehrhardt C. In vitro assessment of transferrin-conjugated liposomes as drug delivery systems for inhalation therapy of lung cancer. Eur J Pharm Sci. 2006;29:367–374. doi: 10.1016/j.ejps.2006.07.004. [DOI] [PubMed] [Google Scholar]
- 190.Xu L, Frederik P, Pirollo KF, Tang WH, Rait A, Xiang LM, Huang W, Cruz I, Yin Y, Chang EH. Self-assembly of a virus-mimicking nanostructure system for efficient tumor-targeted gene delivery. Hum Gene Ther. 2002;13:469–481. doi: 10.1089/10430340252792594. [DOI] [PubMed] [Google Scholar]
- 191.Chiu SJ, Liu S, Perrotti D, Marcucci G, Lee RJ. Efficient delivery of a Bcl-2-specific antisense oligodeoxyribonucleotide (G3139) via transferrin receptor-targeted liposomes. J Control Release. 2006;112:199–207. doi: 10.1016/j.jconrel.2006.02.011. [DOI] [PubMed] [Google Scholar]
- 192.Zhang X, Koh CG, Yu B, Liu S, Piao L, Marcucci G, Lee RJ, Lee LJ. Transferrin receptor targeted lipopolyplexes for delivery of antisense oligonucleotide g3139 in a murine k562 xenograft model. Pharm Res. 2009;26:1516–1524. doi: 10.1007/s11095-009-9864-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Wu J, Lu Y, Lee A, Pan X, Yang X, Zhao X, Lee RJ. Reversal of multidrug resistance by transferrin-conjugated liposomes co-encapsulating doxorubicin and verapamil. J Pharm Pharm Sci. 2007;10:350–357. [PubMed] [Google Scholar]
- 194.Krieger ML, Eckstein N, Schneider V, Koch M, Royer HD, Jaehde U, Bendas G. Overcoming cisplatin resistance of ovarian cancer cells by targeted liposomes in vitro. Int J Pharm. 2010;389:10–17. doi: 10.1016/j.ijpharm.2009.12.061. [DOI] [PubMed] [Google Scholar]
- 195.Huwyler J, Yang J, Pardridge WM. Receptor mediated delivery of daunomycin using immunoliposomes: pharmacokinetics and tissue distribution in the rat. J Pharmacol Exp Ther. 1997;282:1541–1546. [PubMed] [Google Scholar]
- 196.Suzuki S, Inoue K, Hongoh A, Hashimoto Y, Yamazoe Y. Modulation of doxorubicin resistance in a doxorubicin-resistant human leukaemia cell by an immunoliposome targeting transferring receptor. Br J Cancer. 1997;76:83–89. doi: 10.1038/bjc.1997.340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Derycke AS, Kamuhabwa A, Gijsens A, Roskams T, De Vos D, Kasran A, Huwyler J, Missiaen L, de Witte PA. Transferrin-conjugated liposome targeting of photosensitizer AlPcS4 to rat bladder carcinoma cells. J Natl Cancer Inst. 2004;96:1620–1630. doi: 10.1093/jnci/djh314. [DOI] [PubMed] [Google Scholar]
- 198.Penacho N, Filipe A, Simoes S, Pedroso de Lima MC. Transferrin-associated lipoplexes as gene delivery systems: relevance of mode of preparation and biophysical properties. J Membr Biol. 2008;221:141–152. doi: 10.1007/s00232-008-9092-x. [DOI] [PubMed] [Google Scholar]
- 199.Xu L, Pirollo KF, Chang EH. Tumor-targeted p53-gene therapy enhances the efficacy of conventional chemo/radiotherapy. J Control Release. 2001;74:115–128. doi: 10.1016/s0168-3659(01)00324-8. [DOI] [PubMed] [Google Scholar]
- 200.Xu L, Pirollo KF, Tang WH, Rait A, Chang EH. Transferrin-liposome-mediated systemic p53 gene therapy in combination with radiation results in regression of human head and neck cancer xenografts. Hum Gene Ther. 1999;10:2941–2952. doi: 10.1089/10430349950016357. [DOI] [PubMed] [Google Scholar]
- 201.Xu L, Pirollo KF, Chang EH. Transferrin-liposome-mediated p53 sensitization of squamous cell carcinoma of the head and neck to radiation in vitro. Hum Gene Ther. 1997;8:467–475. doi: 10.1089/hum.1997.8.4-467. [DOI] [PubMed] [Google Scholar]
- 202.Xu L, Tang WH, Huang CC, Alexander W, Xiang LM, Pirollo KF, Rait A, Chang EH. Systemic p53 gene therapy of cancer with immunolipoplexes targeted by anti-transferrin receptor scFv. Mol Med. 2001;7:723–734. [PMC free article] [PubMed] [Google Scholar]
- 203.Xu L, Huang CC, Huang W, Tang WH, Rait A, Yin YZ, Cruz I, Xiang LM, Pirollo KF, Chang EH. Systemic tumor-targeted gene delivery by anti-transferrin receptor scFv-immunoliposomes. Mol Cancer Ther. 2002;1:337–346. [PubMed] [Google Scholar]
- 204.Yu W, Pirollo KF, Rait A, Yu B, Xiang LM, Huang WQ, Zhou Q, Ertem G, Chang EH. A sterically stabilized immunolipoplex for systemic administration of a therapeutic gene. Gene Ther. 2004;11:1434–1440. doi: 10.1038/sj.gt.3302304. [DOI] [PubMed] [Google Scholar]
- 205.Mendonca LS, Firmino F, Moreira JN, Pedroso de Lima MC, Simoes S. Transferrin receptor-targeted liposomes encapsulating anti-BCR-ABL siRNA or asODN for chronic myeloid leukemia treatment. Bioconjug Chem. 2010;21:157–168. doi: 10.1021/bc9004365. [DOI] [PubMed] [Google Scholar]
- 206.Loftsson T, Duchene D. Cyclodextrins and their pharmaceutical applications. Int J Pharm. 2007;329:1–11. doi: 10.1016/j.ijpharm.2006.10.044. [DOI] [PubMed] [Google Scholar]
- 207.Wenz G, Han BH, Muller A. Cyclodextrin rotaxanes and polyrotaxanes. Chem Rev. 2006;106:782–817. doi: 10.1021/cr970027+. [DOI] [PubMed] [Google Scholar]
- 208.Davis ME. The first targeted delivery of siRNA in humans via a self-assembling cyclodextrin polymer-based nanoparticle: from concept to clinic. Mol Pharm. 2009;6:659–668. doi: 10.1021/mp900015y. [DOI] [PubMed] [Google Scholar]
- 209.Bartlett DW, Davis ME. Physicochemical and biological characterization of targeted, nucleic acid-containing nanoparticles. Bioconjug Chem. 2007;18:456–468. doi: 10.1021/bc0603539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bellocq NC, Pun SH, Jensen GS, Davis ME. Transferrin-containing, cyclodextrin polymer-based particles for tumor-targeted gene delivery. Bioconjug Chem. 2003;14:1122–1132. doi: 10.1021/bc034125f. [DOI] [PubMed] [Google Scholar]
- 211.Pun SH, Tack F, Bellocq NC, Cheng J, Grubbs BH, Jensen GS, Davis ME, Brewster M, Janicot M, Janssens B, Floren W, Bakker A. Targeted delivery of RNA-cleaving DNA enzyme (DNAzyme) to tumor tissue by transferrin-modified, cyclodextrin-based particles. Cancer Biol Ther. 2004;3:641–650. doi: 10.4161/cbt.3.7.918. [DOI] [PubMed] [Google Scholar]
- 212.Hu-Lieskovan S, Heidel JD, Bartlett DW, Davis ME, Triche TJ. Sequence-specific knockdown of EWS-FLI1 by targeted nonviral delivery of small interfering RNA inhibits tumor growth in a murine model of metastatic Ewing’s sarcoma. Cancer Res. 2005;65:8984–8992. doi: 10.1158/0008-5472.CAN-05-0565. [DOI] [PubMed] [Google Scholar]
- 213.Heidel JD, Yu Z, Liu JY, Rele SM, Liang Y, Zeidan RK, Kornbrust DJ, Davis ME. Administration in non-human primates of escalating intravenous doses of targeted nanoparticles containing ribonucleotide reductase subunit M2 siRNA. Proc Natl Acad Sci U S A. 2007;104:5715–5721. doi: 10.1073/pnas.0701458104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Davis ME, Zuckerman JE, Choi CH, Seligson D, Tolcher A, Alabi CA, Yen Y, Heidel JD, Ribas A. Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature. 2010;464:1067–1070. doi: 10.1038/nature08956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Waehler R, Russell SJ, Curiel DT. Engineering targeted viral vectors for gene therapy. Nat Rev Genet. 2007;8:573–587. doi: 10.1038/nrg2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Wagner E, Zatloukal K, Cotten M, Kirlappos H, Mechtler K, Curiel DT, Birnstiel ML. Coupling of adenovirus to transferrin-polylysine/DNA complexes greatly enhances receptor-mediated gene delivery and expression of transfected genes. Proc Natl Acad Sci U S A. 1992;89:6099–6103. doi: 10.1073/pnas.89.13.6099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Kircheis R, Kupcu Z, Wallner G, Rossler V, Schweighoffer T, Wagner E. Interleukin-2 gene-modified allogeneic melanoma cell vaccines can induce cross-protection against syngeneic tumors in mice. Cancer Gene Ther. 2000;7:870–878. doi: 10.1038/sj.cgt.7700183. [DOI] [PubMed] [Google Scholar]
- 218.Schreiber S, Kampgen E, Wagner E, Pirkhammer D, Trcka J, Korschan H, Lindemann A, Dorffner R, Kittler H, Kasteliz F, Kupcu Z, Sinski A, Zatloukal K, Buschle M, Schmidt W, Birnstiel M, Kempe RE, Voigt T, Weber HA, Pehamberger H, Mertelsmann R, Brocker EB, Wolff K, Stingl G. Immunotherapy of metastatic malignant melanoma by a vaccine consisting of autologous interleukin 2-transfected cancer cells: outcome of a phase I study. Hum Gene Ther. 1999;10:983–993. doi: 10.1089/10430349950018382. [DOI] [PubMed] [Google Scholar]
- 219.Schwarzenberger P, Hunt JD, Robert E, Theodossiou C, Kolls JK. Receptor-targeted recombinant adenovirus conglomerates: a novel molecular conjugate vector with improved expression characteristics. J Virol. 1997;71:8563–8571. doi: 10.1128/jvi.71.11.8563-8571.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Schwarzenberger P, Huang W, Oliver P, Osidipe T, Theodossiou C, Kolls JK. Poly-L-lysine-based molecular conjugate vectors: a high efficiency gene transfer system for human progenitor and leukemia cells. Am J Med Sci. 2001;321:129–136. doi: 10.1097/00000441-200102000-00004. [DOI] [PubMed] [Google Scholar]
- 221.Kreppel F, Gackowski J, Schmidt E, Kochanek S. Combined genetic and chemical capsid modifications enable flexible and efficient de- and retargeting of adenovirus vectors. Mol Ther. 2005;12:107–117. doi: 10.1016/j.ymthe.2005.03.006. [DOI] [PubMed] [Google Scholar]
- 222.Kaikkonen MU, Lesch HP, Pikkarainen J, Raty JK, Vuorio T, Huhtala T, Taavitsainen M, Laitinen T, Tuunanen P, Grohn O, Narvanen A, Airenne KJ, Yla-Herttuala S. (Strept)avidin-displaying lentiviruses as versatile tools for targeting and dual imaging of gene delivery. Gene Ther. 2009;16:894–904. doi: 10.1038/gt.2009.47. [DOI] [PubMed] [Google Scholar]
- 223.Pariente N, Morizono K, Virk MS, Petrigliano FA, Reiter RE, Lieberman JR, Chen IS. A novel dual-targeted lentiviral vector leads to specific transduction of prostate cancer bone metastases in vivo after systemic administration. Mol Ther. 2007;15:1973–1981. doi: 10.1038/sj.mt.6300271. [DOI] [PubMed] [Google Scholar]
- 224.Morizono K, Xie Y, Helguera G, Daniels TR, Lane TF, Penichet ML, Chen IS. A versatile targeting system with lentiviral vectors bearing the biotin-adaptor peptide. J Gene Med. 2009;11:655–663. doi: 10.1002/jgm.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shimbo T, Kawachi M, Saga K, Fujita H, Yamazaki T, Tamai K, Kaneda Y. Development of a transferrin receptor-targeting HVJ-E vector. Biochem Biophys Res Commun. 2007;364:423–428. doi: 10.1016/j.bbrc.2007.09.135. [DOI] [PubMed] [Google Scholar]
- 226.Wu M, Sherwin T, Brown WL, Stockley PG. Delivery of antisense oligonucleotides to leukemia cells by RNA bacteriophage capsids. Nanomedicine. 2005;1:67–76. doi: 10.1016/j.nano.2004.11.011. [DOI] [PubMed] [Google Scholar]
- 227.Oh KS, Engler JA, Joung I. Enhancement of gene delivery to cancer cells by a retargeted adenovirus. J Microbiol. 2005;43:179–182. [PubMed] [Google Scholar]
- 228.Rojek JM, Kunz S. Cell entry by human pathogenic arenaviruses. Cell Microbiol. 2008;10:828–835. doi: 10.1111/j.1462-5822.2007.01113.x. [DOI] [PubMed] [Google Scholar]
- 229.Cotmore SF, Tattersall P. Parvoviral host range and cell entry mechanisms. Adv Virus Res. 2007;70:183–232. doi: 10.1016/S0065-3527(07)70005-2. [DOI] [PubMed] [Google Scholar]
- 230.Singh P, Destito G, Schneemann A, Manchester M. Canine parvovirus-like particles, a novel nanomaterial for tumor targeting. J Nanobiotechnology. 2006;4:2. doi: 10.1186/1477-3155-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]