

Abstract
Natural killer (NK) cells play a crucial role in early immune response against cytomegalovirus infection. A large and mounting body of data indicate that these cells are involved in the regulation of the adaptive immune response as well. By using mouse cytomegalovirus (MCMV) as a model, several groups provided novel insights into the role of NK cells in the development and kinetics of antiviral CD8+ T cell response. Depending on infection conditions, virus strain and the genetic background of mice used, NK cells are either positive or negative regulators of the CD8+ T cell response. At present, there is no unique explanation for the observed differences between various experimental systems used. In this review we discuss the mechanisms involved in the interplay between NK and CD8+ T cells in the early control of MCMV infection.
Keywords: Mouse cytomegalovirus, NK cells, CD8+ T cells, Ly49H
Introduction
Cytomegaloviruses (CMVs) are species-specific herpes viruses causing severe disease in immunocompromised or immunologically immature hosts. Murine cytomegalovirus (MCMV) is biologically similar to human cytomegalovirus (HCMV) and is therefore the most frequently used model for studying HCMV immunobiology and pathogenesis [1, 2]. Both innate and adaptive immunity are involved in the control of MCMV infection [1, 3, 4]. The innate immune system is induced rapidly after infection without the need for prior sensitization. The detection of virus infection is carried out by Toll-like receptors (TLRs), which have the ability to recognize the so-called pathogen-associated molecular patterns (PAMPs) [5]. After recognizing viral PAMPs, TLR3 and TLR9, expressed primarily by dendritic cells (DC), activate the NF-κB signaling pathway which triggers the innate and adaptive immune response by inducing the production of proinflammatory cytokines and the expression of costimulatory molecules during MCMV infection [6, 7]. These early events drive the activation of natural killer (NK) cells, the major effectors of innate immunity. NK cells express an array of germ line-encoded receptors that transmit either inhibitory or activating signals. The early activation of NK cells upon MCMV infection results in cytokine production and the release of cytotoxic granules containing perforins and granzymes [8].
Based on their ability to mount an NK cell response to MCMV, conventional mouse strains can be either MCMV-resistant, such as C57BL/6 mice, or MCMV-susceptible, such as BALB/c mice [1, 9]. In MCMV-infected C57BL/6 mice, NK cells are activated through Ly49H receptor specific to the MCMV protein m157 [10, 11] leading to an early control of viral replication [12, 13]. In contrast, BALB/c mice lack the Ly49H receptor and are unable to mount an efficient NK cell response to MCMV [14] due to the virus evading the NK-mediated control [15, 16]. For instance, MCMV encodes proteins that can engage inhibitory NK cell receptors, thus avoiding the recognition of infected cells by NK cells via the “missing self” axis [15]. In addition, in order to compromise NK cell activation, MCMV uses other strategies such as downmodulation of the ligands for activating receptors (e.g., NKG2D) [16, 17].
While NK cells restrict viral replication early upon infection, CD8+ T cells are important for the resolution of primary infection and maintenance of virus latency [3]. The number of virus-specific CD8+ T cells increases through intensive proliferation followed by a contraction phase and the generation of a stable pool of long-lived memory CD8+ T cells. Therefore, understanding the cellular and molecular mechanisms involved in the priming and maintenance of CD8+ T cell response is of uttermost importance for the development of CD8+ T cell–based immunotherapy and vaccines. In recent years, significant progress has been made in understanding the role of NK cells and other innate immune response mechanisms in the regulation of the magnitude and longevity of antigen-specific CD8+ T cell response (reviewed in [18]).
Innate regulation of the CD8+ T cell response
Activation of NK cells depends on an intricate balance of signals transmitted through their activating and inhibitory receptors [19]. In addition, NK cell activation is induced by proinflammatory cytokines such as type I interferons (IFNs) and interleukin-12 (IL-12) secreted by DCs upon MCMV infection [20]. Interferon gamma (IFN-γ) production by NK cell is induced by IL-12, whereas NK cell cytotoxicity is stimulated by type I IFNs [21]. It has been shown that in the early phase of MCMV infection, IFN-α and IL-12 production is dependent on MyD88/TLR9 signaling, whereas in the later phase of infection, IFN-a production is maintained in the plasmacytoid DCs (pDCs)- and MyD88-independent manner. In addition, in the latter phase of MCMV infection, IFN-c production by NK cells is decreased due to a reduced accessibility of IL-12 [22].
The adaptive immune response is linked to innate immunity by activation of the cells of the adaptive immune system: T and B lymphocytes. The mechanism relies on antigen presentation by antigen-presenting cells (APCs) and specific cytokines produced by various innate cells. DCs are professional APCs due to their powerful capacity to prime naive CD8+ T cells upon MCMV infection [23]. This process of antigen presentation, in the context of MHC class I molecules, can be achieved in two ways: The direct or endogenous presentation is mediated by proteins that are synthetized and processed by infected DCs and other APCs. In contrast, cross-presentation is performed by APCs that have endocytosed, processed and presented a foreign antigen on MHC class I molecules (e.g., from cells undergoing apoptosis) [24]. In addition to their essential contribution in priming of naive CD8+ T cells, DCs also cross talk with NK cells during MCMV infection [6, 25]. During this communication, DCs-derived cytokines induce NK cell activation. In addition, NK cells may enable the maintenance of DCs population during infection [26]. Thus, by influencing DCs function, NK cells can regulate development of specific immune response and eventually the outcome of a MCMV infection.
NK cells shape the CD8+ T cell response to MCMV
In addition to the well-established NK-DC cross talk during MCMV infection, a number of studies provide ample evidence for an interplay between NK cells and CD8+ T cells (reviewed in [15, 18]). Several studies have demonstrated that the effectiveness of the NK cell response determines the quality of the subsequent CD8+ T cell antiviral response [27–33]. Various murine models have been utilized in order to demonstrate the role of NK cells in shaping the antiviral CD8+ T cell response. While in some studies it has been shown that a strong NK cell response results in an impaired CD8+ T cell response [27, 29, 30,33], other studies suggest that a strong NK cell response can result in an enhanced CD8+ T cell response [28, 31,32]. These apparent discrepancies stem from differences in the genetic background of mouse strains used, the composition of innate immune receptors involved and the viral regulation of the immune response.
The study by Su and colleagues was first to report the immunoregulatory function of NK cells during early MCMV infection [27]. The absence of NK cells during MCMV infection resulted in an enhanced CD8+ T cell response characterized by IFN-γ production, BrdU incorporation and cell expansion. Andrews and colleagues have also shown that NK cell activation via the Ly49H receptor limits both the generation and long-term efficacy of specific T cells by altering the frequency and duration of infection of APCs [30]. In line with these data, we have also demonstrated that the requirement for the CD8+ T cells in the control of early MCMV infection of C57BL/6 mice inversely correlates with the capacity of NK cells to restrict viral replication via the Ly49H receptor (Fig. 1) [33]. The frequencies of epitope-specific CD8+ T cells and their activation status were higher in mice infected with virus lacking m157 (Δm157 MCMV), and therefore resistant to NK cell response, compared to mice infected with NK cell–sensitive virus (wild-type (WT) MCMV). Furthermore, we have shown that the infection of C57BL/6 mice with Δm157 MCMV resulted in a higher virus load during the first few days post-infection (p.i.) accompanied by a higher frequency of infected conventional DC (cDC). In addition, a higher virus load resulted in a dramatic increase in proinflammatory cytokines, which could contribute to an enhanced CD8+ T cell response [29, 33]. The immunoregulatory role for NK cells in limiting CD8+ T cell response and modulation of virus-induced disease was also demonstrated in lymphocytic choriomeningitis virus (LCMV) infection [34, 35]. These studies showed that, depending on the infection conditions and the virus dose used, NK cells can limit the CD8+ T cell response to LCMV by preventing virus clearance and promoting viral persistence. As demonstrated by Waggoner and colleagues, the impaired CD8+ T cell response to LCMV is a consequence of NK cells killing of the activated CD4+ T cells. Upon infection with a high virus dose, NK cells dampen immune pathology by supporting CD8+ T cell exhaustion and viral persistence, whereas during infection with a medium virus dose, the presence of NK cells leads to CD8+ T cell–mediated pathology and death [34]. The study by Lang and colleagues further supports the concept of negative regulation of the CD8+ T cell response to LCMV by NK cells. Although NK cells did not exert a direct antiviral effect on virus replication during LCMV infection, the activation through the NKG2D receptor led to the killing of CD8+ T cells in perforin-dependent manner, thus enabling viral persistence and immunopathology [35].
Fig. 1.

Early control of MCMV infection by NK cells negatively regulates the CD8+ T cell response. Infection of C57BL/6 mice with NK cell–sensitive virus results in limited CD8+ T cell response as a consequence of early restriction of viral replication by NK cells activated through Ly49H–m157 interaction, already on day 1.5 p.i. In contrast, infection of C57BL/6 mice with NK cell–resistant virus induces a strong CD8+ T cell response as early as 4 days p.i. and reaches the peak on day 7 p.i. This enhanced CD8+ T cell response is characterized by an increased proliferation measured by BrdU incorporation, a high frequency of IFN-γ (red dots) producing CD8+ T cells stimulated with MCMV peptides in vitro, and the upregulation of the activation marker granzyme B (blue dots) [33]
In a sharp contrast to the above-described inhibitory role of NK cells in governing the virus-specific CD8+ T cell response, several reports demonstrated the ability of NK cells to accelerate [28] or enhance the CD8+ T cell response [31, 32]. Robbins and colleagues have shown that NK cell activation via interaction between the Ly49H receptor and its viral ligand on infected cells may accelerate CD8+ T cell response in vivo [28]. According to the scenario proposed by the authors, the activation of NK cells via this axis limits IFN-α/β production by pDCs and consequently prevents the depletion of splenic cDCs causing a prompt induction of the CD8+ T cell response. Another study has demonstrated that the recognition of infected cells by licensed Ly49G2+ NK cells also results in a faster recovery of splenic cDCs and an enhanced antigen-specific CD8+ T cell response [32].
Data from our laboratory also indicate that the impact of NK cells on subsequent CD8+ T cell response cannot be explained only by the differential efficacy of virus control. The recombinant MCMV expressing RAE-1γ, a cellular ligand for the activating NK cell receptor NKG2D [36], has shown a dramatic NK cell–dependent early attenuation, but still the CD8+ T cell response to a variety of viral epitopes was equal or even stronger than in mice infected with WT MCMV [31]. Although there is no simple mechanistic explanation for the observed different outcomes in the above studies, it should be taken into account that the Ly49H receptor is exclusively expressed on NK cells [37], and the reduced CD8+ T cell response observed after WT MCMV infection could be a consequence of a reduced antigenic load. In contrast, NKG2D is also expressed as a costimulatory molecule on CD8+ T cells, suggesting that the engagement of this receptor by RAE-1γ expressed on infected DCs could contribute to an enhanced priming of CD8+ T cells regardless of the level of antigenic load [31].
To address the impact of NK cell activation other than via the Ly49H and NKG2D receptors, we have used MCMV which lacks m04, one of the three MCMV-derived glycoproteins that targets the MHC class I molecules [38]. Unlike m06 and m152, m04 does not downregulate MHC class I molecules, but instead, it rescues cell surface MHC class I molecules in order to engage the inhibitory Ly49 receptors [39, 40]. Therefore, the virus lacking m04 cannot prevent NK cells activation and virus control via the “missing self” mechanism [39]. We have now evidence that in spite of an early attenuation, Δm04 MCMV induces a potent CD8+ T cell response, which is essential for virus control in the spleen on day 7 p.i. (Fig. 2). Thus, unlike the Ly49H-dependent virus control in C57BL/6 mice, NK cell activation and virus control via the “missing self” axis upon infection of BALB/c mice with MCMV lacking m04 does not affect the development of the CD8+ T cell response in vivo. Yet, CD8+ T cells were less important for virus control in salivary glands (SG), indicating that different organs display a different need for CD8+ T cells in MCMV control (Fig. 2).
Fig. 2.

NK cell activation via the “missing self” mechanism does not compromise CD8+ T cell response to MCMV. BALB/c mice were intravenously (i.v.) injected with 2 × 105 plaque-forming units (PFU) of either WT or Δm04 MCMV. For in vivo depletion of CD8 T cells, mice were intraperitoneally (i.p.) injected with 300 μg of purified monoclonal antibody (mAb) to CD8 (YTS 169.4) on days 1 and 5 p.i. Control groups were treated with phosphate-buffered saline (PBS). On day 7 p.i., viral titers in indicated organs were determined by the plaque assay. Circles represent the titers of individual mice, and horizontal bars represent the median values. Dotted line, detection limit
Altogether, the above-presented scenarios of NK cell modulation of primary CD8+ T cell response to MCMV point to a complex set of host and viral interactions.
NK cells determine the dynamics of CD8+ T cell response to MCMV in different organs
It is well established that the dynamics of MCMV control in vivo is determined by various factors including the route of infection and the virus source. NK cell–mediated control of MCMV replication also varies between different organs [41]. Although both cytolytic and noncytolytic mechanisms contribute to NK cell–mediated antiviral control of MCMV infection, their contribution may vary in different organs [42]. The deletion of m157 resulted in a dramatic loss of MCMV control on day 3 p.i. in several organs of C57BL/6 mice but did not abolish virus control in liver [12], suggesting a different mechanism of virus control in this organ [41, 42].
The dynamics of virus control in SG is particularly informative and may depend on the virus source, route of infection as well as the timing after infection. We have shown previously that, unlike most of other organs, CD8+ T cells are unable to terminate productive infection in SG [43]. Moreover, MCMV control in SG requires CD4+ T cells [44, 45]. As reported recently, virus replication in different cellular compartments in SG can be explained by different immune response mechanisms involved in virus control [46].
A more recent work conducted in our laboratory also illustrates the differential requirements of immune response mechanisms in the control of MCMV in different tissues ([33] and Fig. 3). CD8+ T cells are not required for MCMV control in spleen on day 3 p.i., and their contribution at later time points is determined by the ability of NK cells to contain virus replication during the early days p.i. (Fig. 3). When C57BL/6 mice were infected with NK cell–resistant virus, the inability of NK cells to control virus replication was compensated by the CD8+ T cells which became essential for MCMV control on day 7 p.i. Yet, at 2 weeks p.i., neither CD8+ T cells nor NK cells are indispensable for MCMV control in the spleen, suggesting a robust physiological plasticity of the immune response.
Fig. 3.

The requirement for the CD8+ T cells in MCMV control is determined by the strength of the NK cell response. C57BL/6 mice were i.p. injected with 2 × 105 PFU of either WT or Δm157 MCMV. On days 1, 5 and 12 p.i., mice were i.p. injected with PBS or depleting anti-CD8 mAb. On days 3, 7 and 14 p.i., mice were killed and viral titers in indicated organs were determined by plaque assay. Data are presented as mean ± standard error of the mean (SEM). Dotted line, detection limit
MCMV control in SG is even more complex [45, 47,48]. Our study demonstrated that NK cell–mediated antiviral control in SG is dependent on the Ly49H–m157 interaction [33]. This might suggest that the absence of a strong NK cell control mediated through Ly49H–m157 interaction is providing the virus with the opportunity to reach the acinar glandular epithelial cells and establish persistent infection. Although our previous results suggest against the role of CD8+ T cells in control of persistent infection in SG [43], recent studies from our laboratory clearly demonstrated that CD8+ T cells play a role in the antiviral control in SG during the early days p.i. This is illustrated by a dramatic effect of the CD8+ T cell depletion on virus titer in SG on day 7 p.i. with NK cell–resistant MCMV (Fig. 3 and [33]). Notably, it appears that the route of infection may also influence the requirements for CD8+ T cells in virus control in SG. In contrast to dramatic effect of CD8+ T cell depletion on virus control in SG in mice infected intravenously [33], CD8+ T cell depletion after intraperitoneal infection had only minor effect on virus control in this organ (Fig. 3). These new data, showing that the virus replication in SG on day 7 p.i. is also mediated through CD8+ T cells, are—at a first glance—in a sharp contrast to the previously published work showing that MCMV control in SG is primarily CD4+ T cell dependent [43]. Our unpublished data indicate that antiviral control in SG is CD8+ T cell dependent only during the early p.i. period, before the virus enters the acinar glandular epithelial cells [44]. Therefore, it is likely that the cellular compartment in SG colonized by virus during the early days p.i. is under CD8+ T cell control, whereas CD4+ T cells become essential when the virus reaches the acinar glandular epithelial cells [49].
The role of TNFR type I and IFN type I signaling on the regulation of the CD8+ T cell response to MCMV
NK cells utilize several mechanisms to exert their antiviral functions. Direct antiviral activity can be achieved through cytotoxic mechanisms and the production of effector cytokines such as IFN-γ and tumor necrosis factor alpha (TNF-α) [50–52]. The immunoregulatory function of NK cells during MCMV infection, such as maintaining DCs population, and thus regulation of antigen presentation, can also be achieved through the production of effector cytokines. The members of TNF-receptor (TNFR) superfamily are expressed by different immune cells and, depending on the cell type, can trigger differentiation, proliferation, activation or cell death [53]. The TNFR type I (TNFRp55) signaling is rapid and highly specific and triggers activation of caspase, leading to the apoptotic cell death [54]. It has been shown that CMV infection inhibits signaling and decreases the expression of TNFRs [55, 56]. To assess the role of TNFRp55 in the development of CD8+ T cell response to early MCMV infection, we have infected mice lacking TNFRp55 (B6.TNFRp55−/− mice) with either NK cell–sensitive or NK cell–resistant MCMV and analyzed viral titers on day 7 p.i. Similar to normal C57BL/6 mice, B6.TNFRp55−/− mice efficiently controlled NK cell–sensitive virus in the spleen, whereas the control of NK cell–resistant virus required the activity of the CD8+ T cells (Fig. 4). Interestingly, B6.TNFRp55−/− mice were unable to completely clear NK cell–resistant virus from the SG on day 7 p.i., suggesting that NK cell–dependent virus control in SG is partially mediated through this receptor [57].
Fig. 4.
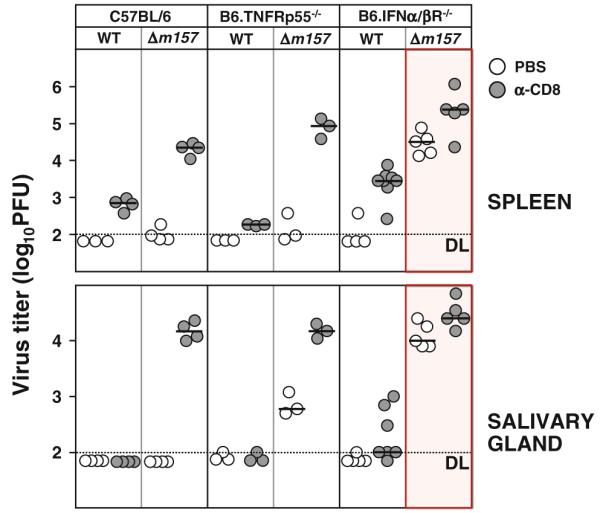
IFNα/βR but not TNFRp55 signaling is essential for the CD8+ T cell–dependent MCMV control under conditions of an insufficient NK cell response. C57BL/6 and B6.TNFRp55−/− mice were i.v. injected with 2 × 105 PFU, and B6.IFNα/βR−/− mice were i.v. injected with 5 × 104 PFU of either WT or Dm157 MCMV. On days 1 and 5 p.i., mice were i.p. injected with PBS or depleting anti-CD8 mAb. On day 7 p.i., viral titers in indicated organs were determined by plaque assay. Circles represent the virus titers in individual mice, and horizontal bars represent median values. Dotted line, detection limit
Type I IFNs are also important regulators of the immune response to different viral infections [20, 58]. Specifically, it has been shown that IFNα/β is potent inducer of NK cell cytotoxicity upon MCMV infection [59]. Previous studies demonstrated that mice lacking IFNα/β receptor (IFNα/βR−/−) exhibit a deficiency in homeostatic NK cell numbers, proliferation and killing capacity [60, 61]. In addition, a recent study by Geurs and colleagues showed that the engagement of Ly49H receptor after MCMV infection compensated to some extent the deficient NK cell proliferation observed in B6.IFNα/βR−/− mice, although the overall proliferation of NK cells in these mice was still reduced compared to C57BL/6 control mice [62]. Having in mind the above-mentioned NK cell deficiency of B6.IFNα/βR−/− mice, one might speculate that the immunoregulatory function of NK cells in shaping the subsequent CD8+ T cell response to MCMV is also compromised. To address this issue, we have infected B6.IFNα/βR−/− mice with either NK cell–sensitive or NK cell–resistant MCMV and analyzed viral titers on day 7 p.i. Similar to previously published work, the infection of B6.IFNα/βR−/− mice with NK cell–sensitive MCMV strain resulted in an efficient virus control on day 7 p.i., whereas the absence of Ly49H engagement upon infection with NK cell–resistant MCMV completely abolished any antiviral control ([62] and Fig. 4). Contrary to normal C57BL/6 mice, which were able to control NK cell–resistant virus on day 7 p.i. by CD8+ T cells, in B6.IFNα/βR−/− mice this antiviral control was abolished. Thus, the data suggest that type I IFNs play an important role in enhanced CD8+ T cell response under conditions of insufficient early NK cell antiviral control. The insufficient antiviral activity of CD8+ T cells in B6.IFNα/βR−/− mice was not unexpected, giving that the type I IFNs play important role in induction of CD8+ T cell response upon viral infection by upregulating the expression of MHC class I and costimulatory molecules on APCs, providing the “third signal” required for activation of naive CD8+ T cells and greatly augmenting their proliferation [63–66]. Altogether, we have shown that IFN type I signaling plays a major role in development of a strong CD8+ T cell response on day 7 p.i. with NK cell–resistant MCMV, whereas TNFRp55 signaling is dispensable for it.
Conclusion
The currently available body of data demonstrate that the impact of NK cells on specific CD8+ T cell response depends on numerous host and viral factors, including virus sensitivity to NK cell control, mode of NK cell activation, route of infection and host genetic factors. A better understanding of how NK cells and other innate immune response mechanisms regulate the generation and maintenance of virus-specific CD8+ T cell response is essential for designing new strategies in antiviral therapy and in the rational vaccine design.
Acknowledgments
This study was supported by NIH grants 1R01AI083201-01, and by the Impuls- und Vernetzungsfonds of the Helmholtz Association (grant “Viral strategies of immune evasion”; VH-VI-424-4) to S. Jonjic. J. Arapovic is supported by the Federal Ministry of Education and Science, Bosnia and Herzegovina (grant “Prevalence of congenital CMV infection in Herzegovina”).
Contributor Information
Maja Mitrović, Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, B. Branchetta 20, 51 000 Rijeka, Croatia.
Jurica Arapović, Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, B. Branchetta 20, 51 000 Rijeka, Croatia; Faculty of Medicine, University of Mostar, Mostar, Bosnia and Herzegovina.
Luka Traven, Department of Environmental Medicine, Faculty of Medicine, University of Rijeka, Rijeka, Croatia.
Astrid Krmpotić, Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, B. Branchetta 20, 51 000 Rijeka, Croatia.
Stipan Jonjić, Department of Histology and Embryology, Faculty of Medicine, University of Rijeka, B. Branchetta 20, 51 000 Rijeka, Croatia.
References
- 1.Krmpotic A, Bubic I, Polic B, Lucin P, Jonjic S. Pathogenesis of murine cytomegalovirus infection. Microbes Infect. 2003;5(13):1263–1277. doi: 10.1016/j.micinf.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 2.Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. Murine model of cytomegalovirus latency and reactivation. Curr Top Microbiol Immunol. 2008;325:315–331. doi: 10.1007/978-3-540-77349-8_18. [DOI] [PubMed] [Google Scholar]
- 3.Reddehase MJ. Antigens and immunoevasins: opponents in cytomegalovirus immune surveillance. Nat Rev Immunol. 2002;2(11):831–844. doi: 10.1038/nri932. doi:10.1038/nri932. [DOI] [PubMed] [Google Scholar]
- 4.Jonjic S, Bubic I, Krmpotic A. Innate immunity to cytomegaloviruses. In: Reddehase MJ, editor. Cytomegaloviruses: molecular biology and immunology. Caister Academic Press; Wymondham: 2006. pp. 285–319. [Google Scholar]
- 5.Takeda K, Akira S. Toll-like receptors. Curr Protoc Immunol. 2007;14:12. doi: 10.1002/0471142735.im1412s77. doi:10.1002/0471142735.im1412s77. [DOI] [PubMed] [Google Scholar]
- 6.Krug A, French AR, Barchet W, Fischer JA, Dzionek A, Pingel JT, Orihuela MM, Akira S, Yokoyama WM, Colonna M. TLR9-dependent recognition of MCMV by IPC and DC generates coordinated cytokine responses that activate antiviral NK cell function. Immunity. 2004;21(1):107–119. doi: 10.1016/j.immuni.2004.06.007. doi:10.1016/j.immuni.2004. 06.007. [DOI] [PubMed] [Google Scholar]
- 7.Tabeta K, Georgel P, Janssen E, Du X, Hoebe K, Crozat K, Mudd S, Shamel L, Sovath S, Goode J, Alexopoulou L, Flavell RA, Beutler B. Toll-like receptors 9 and 3 as essential components of innate immune defense against mouse cytomegalovirus infection. Proc Nat Acad Sci USA. 2004;101(10):3516–3521. doi: 10.1073/pnas.0400525101. doi:10.1073/pnas.0400525101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vidal SM, Lanier LL. NK cell recognition of mouse cytomegalovirus-infected cells. Curr Top Microbiol Immunol. 2006;298:183–206. doi: 10.1007/3-540-27743-9_10. [DOI] [PubMed] [Google Scholar]
- 9.Scalzo AA. Successful control of viruses by NK cells–a balance of opposing forces? Trends Microbiol. 2002;10(10):470–474. doi: 10.1016/s0966-842x(02)02441-1. [DOI] [PubMed] [Google Scholar]
- 10.Arase H, Mocarski ES, Campbell AE, Hill AB, Lanier LL. Direct recognition of cytomegalovirus by activating and inhibitory NK cell receptors. Science. 2002;296(5571):1323–1326. doi: 10.1126/science.1070884. doi:10.1126/science.1070884. [DOI] [PubMed] [Google Scholar]
- 11.Smith HR, Heusel JW, Mehta IK, Kim S, Dorner BG, Naidenko OV, Iizuka K, Furukawa H, Beckman DL, Pingel JT, Scalzo AA, Fremont DH, Yokoyama WM. Recognition of a virus-encoded ligand by a natural killer cell activation receptor. Proc Nat Acad Sci USA. 2002;99(13):8826–8831. doi: 10.1073/pnas.092258599. doi:10.1073/pnas.092258599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bubic I, Wagner M, Krmpotic A, Saulig T, Kim S, Yokoyama WM, Jonjic S, Koszinowski UH. Gain of virulence caused by loss of a gene in murine cytomegalovirus. J Virol. 2004;78(14):7536–7544. doi: 10.1128/JVI.78.14.7536-7544.2004. doi:10.1128/JVI.78.14.7536-7544.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fodil-Cornu N, Lee SH, Belanger S, Makrigiannis AP, Biron CA, Buller RM, Vidal SM. Ly49 h-deficient C57BL/6 mice: a new mouse cytomegalovirus-susceptible model remains resistant to unrelated pathogens controlled by the NK gene complex. J Immunol. 2008;181(9):6394–6405. doi: 10.4049/jimmunol.181.9.6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scalzo AA, Fitzgerald NA, Simmons A, La Vista AB, Shellam GR. Cmv-1, a genetic locus that controls murine cytomegalovirus replication in the spleen. J Exp Med. 1990;171(5):1469–1483. doi: 10.1084/jem.171.5.1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Babic M, Krmpotic A, Jonjic S. All is fair in virus-host interactions: NK cells and cytomegalovirus. Trends Mol Med. 2011;17(11):677–685. doi: 10.1016/j.molmed.2011.07.003. doi:10.1016/j.molmed.2011.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lisnic VJ, Krmpotic A, Jonjic S. Modulation of natural killer cell activity by viruses. Curr Opin Microbiol. 2010;13(4):530–539. doi: 10.1016/j.mib.2010.05.011. doi:10.1016/j.mib.2010.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lenac T, Arapovic J, Traven L, Krmpotic A, Jonjic S. Murine cytomegalovirus regulation of NKG2D ligands. Med Microbiol Immunol. 2008;197(2):159–166. doi: 10.1007/s00430-008-0080-7. doi:10.1007/s00430-008-0080-7. [DOI] [PubMed] [Google Scholar]
- 18.Biron CA. Yet another role for natural killer cells: cytotoxicity in immune regulation and viral persistence. Proc Nat Acad Sci USA. 2012;109(6):1814–1815. doi: 10.1073/pnas.1120528109. doi:10.1073/pnas.1120528109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Raulet DH. Interplay of natural killer cells and their receptors with the adaptive immune response. Nat Immunol. 2004;5(10):996–1002. doi: 10.1038/ni1114. doi:10.1038/ni1114. [DOI] [PubMed] [Google Scholar]
- 20.Dalod M, Salazar-Mather TP, Malmgaard L, Lewis C, Asselin-Paturel C, Briere F, Trinchieri G, Biron CA. Interferon alpha/beta and interleukin 12 responses to viral infections: pathways regulating dendritic cell cytokine expression in vivo. J Exp Med. 2002;195(4):517–528. doi: 10.1084/jem.20011672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Biron CA, Nguyen KB, Pien GC, Cousens LP, Salazar-Mather TP. Natural killer cells in antiviral defense: function and regulation by innate cytokines. Annu Rev Immunol. 1999;17:189–220. doi: 10.1146/annurev.immunol.17.1.189. doi:10.1146/annurev.immunol.17.1.189. [DOI] [PubMed] [Google Scholar]
- 22.Delale T, Paquin A, Asselin-Paturel C, Dalod M, Brizard G, Bates EE, Kastner P, Chan S, Akira S, Vicari A, Biron CA, Trinchieri G, Briere F. MyD88-dependent and -independent murine cytomegalovirus sensing for IFN-alpha release and initiation of immune responses in vivo. J Immunol. 2005;175(10):6723–6732. doi: 10.4049/jimmunol.175.10.6723. [DOI] [PubMed] [Google Scholar]
- 23.Andrews DM, Andoniou CE, Scalzo AA, van Dommelen SL, Wallace ME, Smyth MJ, Degli-Esposti MA. Cross-talk between dendritic cells and natural killer cells in viral infection. Mol Immunol. 2005;42(4):547–555. doi: 10.1016/j.molimm.2004.07.040. doi:10.1016/j.molimm.2004.07.040. [DOI] [PubMed] [Google Scholar]
- 24.Bevan MJ. Cross-priming. Nat Immunol. 2006;7(4):363–365. doi: 10.1038/ni0406-363. doi:10.1038/ni0406-363. [DOI] [PubMed] [Google Scholar]
- 25.Andoniou CE, van Dommelen SL, Voigt V, Andrews DM, Brizard G, Asselin-Paturel C, Delale T, Stacey KJ, Trinchieri G, Degli-Esposti MA. Interaction between conventional dendritic cells and natural killer cells is integral to the activation of effective antiviral immunity. Nat Immunol. 2005;6(10):1011–1019. doi: 10.1038/ni1244. doi:10.1038/ni1244. [DOI] [PubMed] [Google Scholar]
- 26.Andrews DM, Scalzo AA, Yokoyama WM, Smyth MJ, Degli-Esposti MA. Functional interactions between dendritic cells and NK cells during viral infection. Nat Immunol. 2003;4(2):175–181. doi: 10.1038/ni880. doi:10.1038/ni880. [DOI] [PubMed] [Google Scholar]
- 27.Su HC, Nguyen KB, Salazar-Mather TP, Ruzek MC, Dalod MY, Biron CA. NK cell functions restrain T cell responses during viral infections. Eur J Immunol. 2001;31(10):3048–3055. doi: 10.1002/1521-4141(2001010)31:10<3048::aid-immu3048>3.0.co;2-1. doi:10.1002/1521-4141(2001010)31:10<3048:AID-IMMU3048>3.0.CO;2-1. [DOI] [PubMed] [Google Scholar]
- 28.Robbins SH, Bessou G, Cornillon A, Zucchini N, Rupp B, Ruzsics Z, Sacher T, Tomasello E, Vivier E, Koszinowski UH, Dalod M. Natural killer cells promote early CD8 T cell responses against cytomegalovirus. PLoS Pathog. 2007;3(8):e123. doi: 10.1371/journal.ppat.0030123. doi:10.1371/journal.ppat.0030123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SH, Kim KS, Fodil-Cornu N, Vidal SM, Biron CA. Activating receptors promote NK cell expansion for maintenance, IL-10 production, and CD8 T cell regulation during viral infection. J Exp Med. 2009;206(10):2235–2251. doi: 10.1084/jem.20082387. doi:10.1084/jem.20082387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andrews DM, Estcourt MJ, Andoniou CE, Wikstrom ME, Khong A, Voigt V, Fleming P, Tabarias H, Hill GR, van der Most RG, Scalzo AA, Smyth MJ, Degli-Esposti MA. Innate immunity defines the capacity of antiviral T cells to limit persistent infection. J Exp Med. 2010;207(6):1333–1343. doi: 10.1084/jem.20091193. doi:10.1084/jem.20091193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slavuljica I, Busche A, Babic M, Mitrovic M, Gasparovic I, Cekinovic D, Markova Car E, Pernjak Pugel E, Cikovic A, Lisnic VJ, Britt WJ, Koszinowski U, Messerle M, Krmpotic A, Jonjic S. Recombinant mouse cytomegalovirus expressing a ligand for the NKG2D receptor is attenuated and has improved vaccine properties. J Clin Invest. 2010;120(12):4532–4545. doi: 10.1172/JCI43961. doi:10.1172/JCI43961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stadnisky MD, Xie X, Coats ER, Bullock TN, Brown MG. Self MHC class I-licensed NK cells enhance adaptive CD8 T-cell viral immunity. Blood. 2011;117(19):5133–5141. doi: 10.1182/blood-2010-12-324632. doi:10.1182/blood-2010-12-324632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mitrovic M, Arapovic J, Jordan S, Fodil-Cornu N, Ebert S, Vidal SM, Krmpotic A, Reddehase MJ, Jonjic S. The NK cell response to mouse cytomegalovirus infection affects the level and kinetics of the early CD8(+) T-cell response. J Virol. 2012;86(4):2165–2175. doi: 10.1128/JVI.06042-11. doi:10.1128/JVI.06042-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Waggoner SN, Cornberg M, Selin LK, Welsh RM. Natural killer cells act as rheostats modulating antiviral T cells. Nature. 2012;481(7381):394–398. doi: 10.1038/nature10624. doi:10.1038/nature10624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lang PA, Lang KS, Xu HC, Grusdat M, Parish IA, Recher M, Elford AR, Dhanji S, Shaabani N, Tran CW, Dissanayake D, Rahbar R, Ghazarian M, Brustle A, Fine J, Chen P, Weaver CT, Klose C, Diefenbach A, Haussinger D, Carlyle JR, Kaech SM, Mak TW, Ohashi PS. Natural killer cell activation enhances immune pathology and promotes chronic infection by limiting CD8+ T-cell immunity. Proc Nat Acad Sci USA. 2012;109(4):1210–1215. doi: 10.1073/pnas.1118834109. doi:10.1073/pnas.1118834109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerwenka A, Bakker AB, McClanahan T, Wagner J, Wu J, Phillips JH, Lanier LL. Retinoic acid early inducible genes define a ligand family for the activating NKG2D receptor in mice. Immunity. 2000;12(6):721–727. doi: 10.1016/s1074-7613(00)80222-8. [DOI] [PubMed] [Google Scholar]
- 37.Daniels KA, Devora G, Lai WC, O‘Donnell CL, Bennett M, Welsh RM. Murine cytomegalovirus is regulated by a discrete subset of natural killer cells reactive with monoclonal antibody to Ly49H. J Exp Med. 2001;194(1):29–44. doi: 10.1084/jem.194.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kielczewska A, Pyzik M, Sun T, Krmpotic A, Lodoen MB, Munks MW, Babic M, Hill AB, Koszinowski UH, Jonjic S, Lanier LL, Vidal SM. Ly49P recognition of cytomegalovirus-infected cells expressing H2-Dk and CMV-encoded m04 correlates with the NK cell antiviral response. J Exp Med. 2009;206(3):515–523. doi: 10.1084/jem.20080954. doi:10.1084/jem.20080954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Babic M, Pyzik M, Zafirova B, Mitrovic M, Butorac V, Lanier LL, Krmpotic A, Vidal SM, Jonjic S. Cytomegalovirus immunoevasin reveals the physiological role of “missing self” recognition in natural killer cell dependent virus control in vivo. J Exp Med. 2010;207(12):2663–2673. doi: 10.1084/jem.20100921. doi:10.1084/jem.20100921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kleijnen MF, Huppa JB, Lucin P, Mukherjee S, Farrell H, Campbell AE, Koszinowski UH, Hill AB, Ploegh HL. A mouse cytomegalovirus glycoprotein, gp34, forms a complex with folded class I MHC molecules in the ER which is not retained but is transported to the cell surface. EMBO J. 1997;16(4):685–694. doi: 10.1093/emboj/16.4.685. doi:10.1093/emboj/16.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Tay CH, Welsh RM. Distinct organ-dependent mechanisms for the control of murine cytomegalovirus infection by natural killer cells. J Virol. 1997;71(1):267–275. doi: 10.1128/jvi.71.1.267-275.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Loh J, Chu DT, O‘Guin AK, Yokoyama WM, Virgin HWt. Natural killer cells utilize both perforin and gamma interferon to regulate murine cytomegalovirus infection in the spleen and liver. J Virol. 2005;79(1):661–667. doi: 10.1128/JVI.79.1.661-667.2005. doi:10.1128/JVI.79.1.661-667.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jonjic S, Pavic I, Lucin P, Rukavina D, Koszinowski UH. Efficacious control of cytomegalovirus infection after long-term depletion of CD8+ T lymphocytes. J Virol. 1990;64(11):5457–5464. doi: 10.1128/jvi.64.11.5457-5464.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jonjic S, Mutter W, Weiland F, Reddehase MJ, Koszinowski UH. Site-restricted persistent cytomegalovirus infection after selective long-term depletion of CD4+ T lymphocytes. J Exp Med. 1989;169(4):1199–1212. doi: 10.1084/jem.169.4.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Campbell AE, Cavanaugh VJ, Slater JS. The salivary glands as a privileged site of cytomegalovirus immune evasion and persistence. Med Microbiol Immunol. 2008;197(2):205–213. doi: 10.1007/s00430-008-0077-2. doi:10.1007/s00430-008-0077-2. [DOI] [PubMed] [Google Scholar]
- 46.Walton SM, Mandaric S, Torti N, Zimmermann A, Hengel H, Oxenius A. Absence of cross-presenting cells in the salivary gland and viral immune evasion confine cytomegalovirus immune control to effector CD4 T cells. PLoS Pathog. 2011;7(8):e1002214. doi: 10.1371/journal.ppat.1002214. doi:10.1371/journal.ppat.1002214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Polic B, Hengel H, Krmpotic A, Trgovcich J, Pavic I, Luccaronin P, Jonjic S, Koszinowski UH. Hierarchical and redundant lymphocyte subset control precludes cytomegalovirus replication during latent infection. J Exp Med. 1998;188(6):1047–1054. doi: 10.1084/jem.188.6.1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pinto AK, Hill AB. Viral interference with antigen presentation to CD8+ T cells: lessons from cytomegalovirus. Viral Immunol. 2005;18(3):434–444. doi: 10.1089/vim.2005.18.434. doi:10.1089/vim.2005.18.434. [DOI] [PubMed] [Google Scholar]
- 49.Doom CM, Hill AB. MHC class I immune evasion in MCMV infection. Med Microbiol Immunol. 2008;197(2):191–204. doi: 10.1007/s00430-008-0089-y. doi:10.1007/s00430-008-0089-y. [DOI] [PubMed] [Google Scholar]
- 50.Orange JS. Formation and function of the lytic NK-cell immunological synapse. Nat Rev Immunol. 2008;8(9):713–725. doi: 10.1038/nri2381. doi:10.1038/nri2381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guidotti LG, Chisari FV. Noncytolytic control of viral infections by the innate and adaptive immune response. Annu Rev Immunol. 2001;19:65–91. doi: 10.1146/annurev.immunol.19.1.65. doi:10.1146/annurev.immunol.19.1.65. [DOI] [PubMed] [Google Scholar]
- 52.Voskoboinik I, Smyth MJ, Trapani JA. Perforin-mediated target-cell death and immune homeostasis. Nat Rev Immunol. 2006;6(12):940–952. doi: 10.1038/nri1983. doi:10.1038/nri1983. [DOI] [PubMed] [Google Scholar]
- 53.Hehlgans T, Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor super-family: players, rules and the games. Immunology. 2005;115(1):1–20. doi: 10.1111/j.1365-2567.2005.02143.x. doi:10.1111/j.1365-2567.2005.02143.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Strasser A, O‘Connor L, Dixit VM. Apoptosis signaling. Annu Rev Biochem. 2000;69:217–245. doi: 10.1146/annurev.biochem.69.1.217. doi:10.1146/annurev.biochem. 69.1.217. [DOI] [PubMed] [Google Scholar]
- 55.Baillie J, Sahlender DA, Sinclair JH. Human cytomegalovirus infection inhibits tumor necrosis factor alpha (TNF-alpha) signaling by targeting the 55-kilodalton TNF-alpha receptor. J Virol. 2003;77(12):7007–7016. doi: 10.1128/JVI.77.12.7007-7016.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Popkin DL, Virgin HWt. Murine cytomegalovirus infection inhibits tumor necrosis factor alpha responses in primary macrophages. J Virol. 2003;77(18):10125–10130. doi: 10.1128/JVI.77.18.10125-10130.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pavic I, Polic B, Crnkovic I, Lucin P, Jonjic S, Koszinowski UH. Participation of endogenous tumour necrosis factor alpha in host resistance to cytomegalovirus infection. J Gen Virol. 1993;74(Pt 10):2215–2223. doi: 10.1099/0022-1317-74-10-2215. [DOI] [PubMed] [Google Scholar]
- 58.Muller U, Steinhoff U, Reis LF, Hemmi S, Pavlovic J, Zinkernagel RM, Aguet M. Functional role of type I and type II interferons in antiviral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 59.Orange JS, Biron CA. Characterization of early IL-12, IFN-alphabeta, and TNF effects on antiviral state and NK cell responses during murine cytomegalovirus infection. J Immunol. 1996;156(12):4746–4756. [PubMed] [Google Scholar]
- 60.Dunn GP, Bruce AT, Sheehan KC, Shankaran V, Uppaluri R, Bui JD, Diamond MS, Koebel CM, Arthur C, White JM, Schreiber RD. A critical function for type I interferons in cancer immunoediting. Nat Immunol. 2005;6(7):722–729. doi: 10.1038/ni1213. doi:10.1038/ni1213. [DOI] [PubMed] [Google Scholar]
- 61.Nguyen KB, Salazar-Mather TP, Dalod MY, Van Deusen JB, Wei XQ, Liew FY, Caligiuri MA, Durbin JE, Biron CA. Coordinated and distinct roles for IFN-alpha beta, IL-12, and IL-15 regulation of NK cell responses to viral infection. J Immunol. 2002;169(8):4279–4287. doi: 10.4049/jimmunol.169.8.4279. [DOI] [PubMed] [Google Scholar]
- 62.Geurs TL, Zhao YM, Hill EB, French AR. Ly49H engagement compensates for the absence of type I interferon signaling in stimulating NK cell proliferation during murine cytomegalovirus infection. J Immunol. 2009;183(9):5830–5836. doi: 10.4049/jimmunol.0901520. doi:10.4049/jimmunol.0901520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huber JP, Farrar JD. Regulation of effector and memory T-cell functions by type I interferon. Immunology. 2011;132(4):466–474. doi: 10.1111/j.1365-2567.2011.03412.x. doi:10.1111/j.1365-2567.2011.03412.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Montoya M, Schiavoni G, Mattei F, Gresser I, Belardelli F, Borrow P, Tough DF. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 2002;99(9):3263–3271. doi: 10.1182/blood.v99.9.3263. [DOI] [PubMed] [Google Scholar]
- 65.Welsh RM, Bahl K, Marshall HD, Urban SL. Type 1 interferons and antiviral CD8 T-cell responses. PLoS Pathog. 2012;8(1):e1002352. doi: 10.1371/journal.ppat.1002352. doi:10.1371/journal.ppat.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Curtsinger JM, Valenzuela JO, Agarwal P, Lins D, Mescher MF. Type I IFNs provide a third signal to CD8 T cells to stimulate clonal expansion and differentiation. J Immunol. 2005;174(8):4465–4469. doi: 10.4049/jimmunol.174.8.4465. [DOI] [PubMed] [Google Scholar]