

Abstract

We describe a useful advance in glycopeptide synthesis. We have developed a one-flask aspartylation/deprotection method, wherein long peptide fragments, bearing proximal pseudoproline functionality are merged with complex glycan domains. Following aspartylation, acidmediated global deprotection reveals the elaborated glycopeptide. The temporary pseudoproline functionality serves to suppress formation of aspartimide side products during solid phase peptide synthesis and aspartylation.
Keywords: glycopeptide, aspartylation, aspartimide, pseudoproline dipeptid
Our laboratory has a longstanding interest in the development of improved methods for the chemical synthesis of structurally complex proteins and glycoproteins.[1] Our efforts started with the study of new departures in oligosaccharide synthesis,[2] as well as in polypeptide synthesis.[1a] Building from the foundations of these key constituents, we have recently turned our attention to merging such subunits to produce glycoproteins.[3] Toward these pursuits, the complex, multiply glycosylated protein, erythropoietin (EPO),[4] has served as a defining synthetic target.[5] Our focusing goal; i.e. a total synthesis of homogeneous EPO has served to prompt a variety of ventures directed to enabling advances in glycoprotein synthesis. We disclose herein a valuable new aspartylation technology, developed in the context of our EPO program, which allows for the highly convergent synthesis of complex glycopeptides from fully elaborated peptide and carbohydrate fragments.
Several general strategies are commonly employed for the assembly of N-linked glycoprotein and glycopeptide targets,[6] though each approach suffers from significant limitations in scope or efficiency. According to the stepwise glycosylamino acid approach (Scheme 1a),[7] the carbohydrate domain is first appended to an Fmoc- or Boc-protected Asp residue. The resultant glycosylamino acid is subsequently used directly in solid phase peptide synthesis (SPPS). When the carbohydrate component is complex, the overall efficiency of this linear strategy is significantly compromised by low reaction yields obtained during the glycosylamino acid coupling step and the subsequent elongation of the next amino acid. Moreover, any sialic acid motifs in the glycan must be protected during SPPS. Glycopeptides of up to 20 amino acids in length may be generated through this method.[8]
Scheme 1.
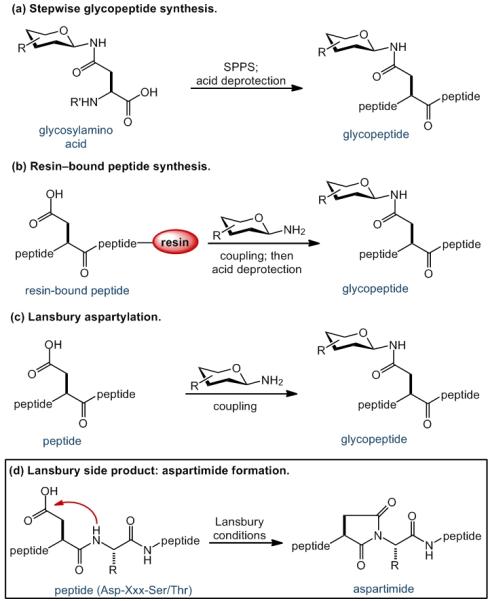
Strategies for Aspartylation.
A second SPPS-based approach offers enhanced convergence. According to the on-resin α-linked glycopeptide strategy[9] (Scheme 1b), the resin-bound peptide, assembled through SPPS, is selectively deprotected to reveal the Asp residue. Coupling of the glycan domain, followed by TFA-mediated cleavage from the resin, delivers the glycopeptide fragment. Two factors serve to mitigate the broad utility of this approach. In certain peptide sequences, particularly those incorporating aggregation-prone sectors, the SPPS-derived resin-bound peptide can suffer from significant impurities. Appendage of a “high-value” glycan domain to impure peptide substrate thus results in significant loss of precious material and formation of difficultly separable mixtures of glycopeptide product. Moreover, release from the resin delivers glycopeptide presenting a C-terminal carboxylic acid, which must then be converted to an activated thioester functionality prior to subsequent native chemical ligation[10] with a peptide or glycopeptide coupling partner.
Our laboratory has been employing[1] the convergent aspartylation approach pioneered by Lansbury[11] (Scheme 1c). In this protocol, a moderately sized, partially protected peptide, bearing the free aspartyl residue, is merged with the glycosyl amine to generate a glycopeptide fragment. While useful for producing short peptide fragments, this method is often compromised when long sequences are being joined with glycosylamine. Coupling yields may be badly undermined by peptide decomposition pathways, predominantly via aspartimide formation (Scheme 1d).[12] Accordingly, our general approach to glycoprotein synthesis has involved assembly of short glycopeptide fragments through Lansbury aspartylation, followed by ligation with a long peptide domain, which may itself contain a glycosylation site. Although not maximally convergent, this strategy is apt to offer ease of use and a high measure of control over glycopeptide purity.
In the context of our ongoing EPO synthesis, we sought to apply this less convergent strategy to the assembly of the EPO(29-78) glycopeptide fragment. Thus, glycopeptide 1, encompassing the EPO(29-40) peptide sector bearing hexasaccharide 4,[13] was prepared through Lansbury aspartylation. However, all efforts to couple 1 with peptide 2 [EPO(41-78)] delivered prohibitively low yields of 3 (Scheme 2). We attribute the failure of this attempted aspartylation to the low solubility of peptide 2, which contains a large hydrophobic domain. Even appendage of a C-terminal Pegylated thioester did not improve the solubility of the peptide to an acceptable level.[14]
Scheme 2.

Attempted synthesis of EPO fragment 3. Reagents and Conditions: 6 M GND•HCl, 0.1 M Na2HPO4, 50 mM TCEP•HCl, pH 6.8
An alternative approach, which would allow us to circumvent issues of peptide solubility and would provide maximum efficiency, envisions merger of the full peptide sequence with the glycan through aspartylation. The successful implementation of this strategy would require conditions under which the problematic aspartimide formation (see Scheme 1d) could be suppressed or even eliminated.
The consensus sequence for N-glycosylation is Asn–Xaa–Ser/Thr. With this generic structural pattern in mind, we evaluated a potential general solution to the challenge of aspartimide avoidance in the Lansbury coupling. Clearly, placement of a pseudoproline motif – derived from Ser or Thr – at the n+1 position necessarily blocks aspartimide formation at the n position.[15] Driven by curiosity, rather than by a convincing rationale, we wondered whether temporary installation of the pseudoproline motif at the n+2 position might also somehow serve to suppress the undesired aspartimide formation (Scheme 3). Because a Ser or Thr residue is universally located at the n+2 position (relative to Asp) in native protein sequences, success in this regard could bring with it major enabling progress in the chemical synthesis of homogeneous complex glycopolypeptides.
Scheme 3.
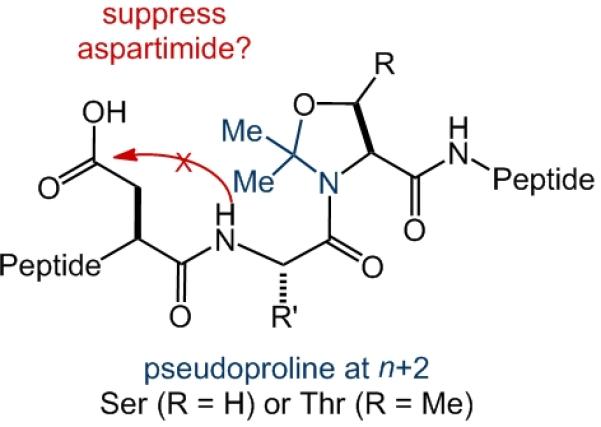
Pseudoproline dipeptide.
We examined this possibility in the context of the EPO(29-78) segment. Thus, through recourse to SPPS, we prepared partially protected peptide 5, incorporating several pseudoproline dipeptides[16] (highlighted in blue), including one at the n+2 position relative to Asp. In the crucial transformation, peptide 5 readily underwent coupling with chitobiose (7) under Lansbury conditions. Subsequent addition of TFA cocktail (TFA/phenol/water/TIPS:88/5/5/2) served to unmask the pseudoproline motifs and remove the peptide protecting groups, thereby delivering the target glycopeptide, 6a, with quantitative conversion and 53% isolated yield. Peptide 5 also underwent one-flask aspartylation/ deprotection with the more complex hexasaccharide, 4, to generate 6b with 75% conversion, albeit in somewhat lower isolated yield (38%).
In a further demonstration of the potential utility of this transformation, we accomplished the aspartylation of the modified EPO(30-78) peptide, 8, both with chitobiose and with the challenging dodecasaccharide, 9.[17] As outlined in Scheme 5, under our one-flask aspartylation/deprotection conditions, glycopeptide 10a was obtained in 54% yield and glycopeptide 10bwas isolated in 32% yield. Notably, the fucose and sialic acid motifs of the dodecasaccharide glycan survived under these conditions, despite the potential sensitivity of these functionalities to acid-mediated decomposition.
Scheme 5.
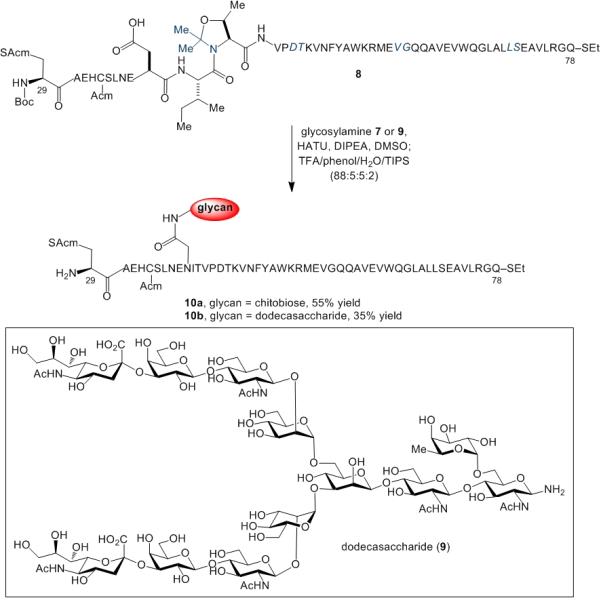
Synthesis of glycopeptides 10a and 10b. Pseudoproline dipeptides are depicted in blue. Acid-labile amino acid protecting groups are shown in bold. E = tBu, H = Trt, S = tBu, N = Trt, K = Boc, Y = tBu, W = Boc, R = Arg, Q = Trt.
Drawing encouragement from these early experiments, we sought to further establish the role of the n+2 pseudoproline in minimizing nonproductive peptide aspartimide formation. For these studies, we selected as a model peptide scaffold the 34-mer, 12, incorporating the aspartimide-prone Asp-Ala-Thr sequence. It is of note that the successful preparation of peptide 12, itself, through SPPS necessitated the installation of the Thr-based pseudoproline motif at the n+2 position (see 11). In the absence of pseudoproline protection, serious competition from aspartimide–containing peptide resulted, even at the SPPS stage (see Supporting Information for details). Apparently, the n+2 pseudoproline functionality effectively suppresses formation of aspartimide in SPPS, particularly at the stage of DBU/piperidine-mediated deprotection.
In our control experiment, we evaluated partially protected 12, wherein the amine and acid functionalities are masked by Allyl and Alloc groups and the n+2 residue is not protected as a pseudoproline (Scheme 6). As anticipated, exposure of peptide 12 to chitobiose under Lansbury conditions resulted in little or no glycopeptide formation. The only observed products were those incorporating aspartimide (13) and Asn (14) in place of the Asp residue. The latter product presumably arises from addition of trace ammonia[18] to the activated aspartate.
Scheme 6.
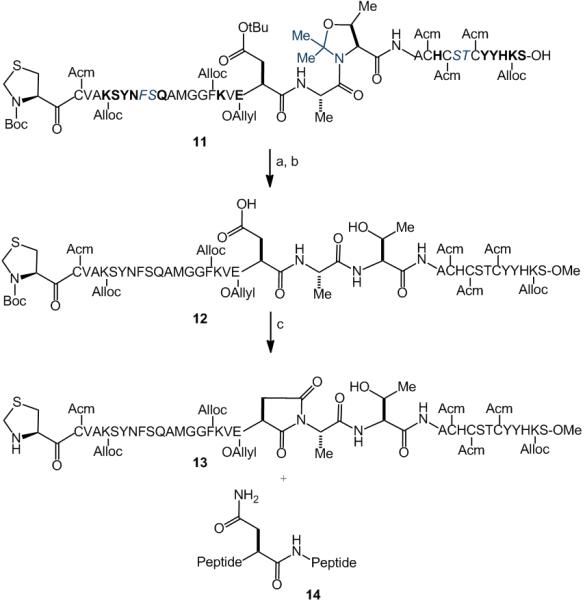
Attempted aspartylation of 12. Reagents and conditions: (a) TMS-Diazomethane CH2Cl2/MeOH; (b) TFA/PhOH/H2O/TIPS, 12% over 3 steps (c) glycosylamine 7, HATU, DIPEA, DMSO.
pseudoproline dipeptides are depicted in blue. Acid-labile amino acid protecting groups are shown in bold. E = Allyl, H = Trt, S = tBu, N = Dmcp, K = Alloc, Y = tBu, R = Pbf, Q = Dmcp.
In contrast, the fully protected peptide 15, incorporating the n+2 pseudoproline, was prepared through SPPS (Scheme 7). C-Terminal methyl ester formation, followed by Pd-mediated removal of the allyl group, yielded peptide 16. The latter readily underwent one-flask aspartylation with deprotection to deliver the desired chitobiose-containing glycopeptide 17 in 45% overall yield (starting from SPPS). The results of this comparison study (Scheme 6 vs. 7) clearly reveal the capacity for incorporation of a pseudoproline motif in the n+2 position to mitigate aspartimide formation at the n position.
Scheme 7.
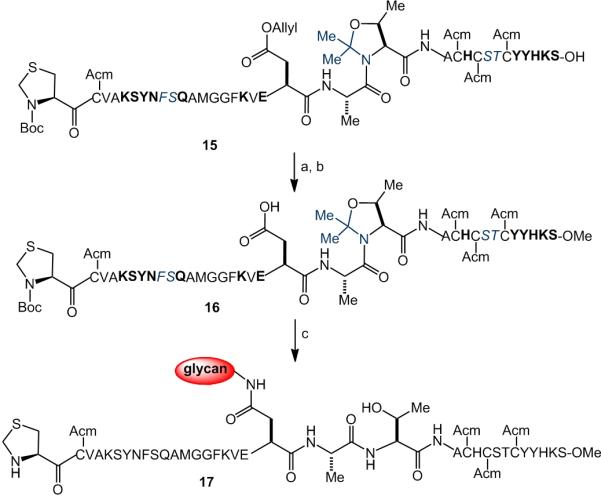
Synthesis of glycopeptide 17. (a) TMS-Diazomethane CH2Cl2/MeOH; (b) Pd(PPh3)4, CH2Cl2, PhSiH3, (c) glycosylamine 7, HATU, DIPEA, DMSO; then TFA/PhOH/H2O/TIPS, 45% over 4 steps. Pseudoproline dipeptides are depicted in blue. Acid-labile amino acid protecting groups are shown in bold. E = tBu, H = Trt, S = tBu, N = Dmcp, K = Boc, Y = tBu, R = Pbf, Q = Dmcp
In addition to the examples provided above, this protocol enabled the efficient and convergent syntheses of two key glycopeptide fragments en route to homogeneous EPO; namely, EPO(79-124) and EPO(1-28) (Scheme 8).19 Indeed, in the absence of the pseudoproline functionalities, the precursor peptide domains were not amenable to preparation through SPPS, presumably due to the propensity of the peptides to undergo aspartimide formation.
Scheme 8.
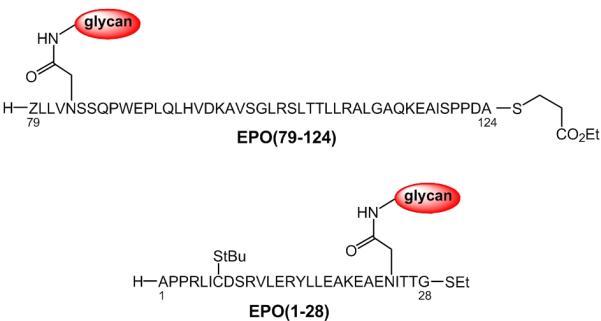
EPO(79-124) and EPO(1-28).
In summary, through incorporation of a pseudoproline motif at the n+2 Ser or Thr residue, it proved possible to suppress otherwise competitive aspartimide-based peptide decomposition pathways. This strategy is also effective for minimizing aspartimide formation during SPPS. Moreover, the aspartylation approach has been successfully applied to a convergent synthesis of the EPO(30-78) glycopeptide fragment. Though the reasons for this observed phenomenon are presently matters of conjecture, its consequences on this field of research are apt to be quite important. Indeed, if generalizable, it could well have solved one of the most serious problems in the assembly of complex glycopolypeptides by chemical means.[20]

Another line of conjecture focuses on the possible inhibition of the amidic NH deprotonation step necessary for aspartamide formation. Perhaps the hydrogen of the aspartamide NH group is transferred to the adjacent amide of the n + 1 amino acid via a hydrogen bond (to N or O). The tertiary amide character of the pseudoproline engagement, would impede such a transfer to either N or O. Clearly the question of the basis for the pseudoproline effect will be pursued in concerned laboratories, including our own.
Supplementary Material
Scheme 4.

Synthesis of glycopeptides 6a and 6b. Pseudoproline dipeptides are depicted in blue. Acid-labile amino acid protecting groups are shown in bold. E = tBu, H = Trt, S = tBu, N = Trt, K = Boc, Y = tBu, W = Boc, R = Arg, Q = Trt.
Footnotes
This research was supported by NIH grant CA103823 (S.J.D.).
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
Contributor Information
Ping Wang, Laboratory for Bioorganic Chemistry Sloan-Kettering Institute for Cancer Research 1275 York Avenue, New York, NY 10065 (USA).
Baptiste Aussedat, Laboratory for Bioorganic Chemistry Sloan-Kettering Institute for Cancer Research 1275 York Avenue, New York, NY 10065 (USA).
Samuel J. Danishefsky, Laboratory for Bioorganic Chemistry Sloan-Kettering Institute for Cancer Research 1275 York Avenue, New York, NY 10065 (USA) Department of Chemistry Columbia University 3000 Broadway, New York, NY 10027.
References
- 1.For recent reviews from our laboratory, see: Kan C, Danishefsky SJ. Tetrahedron. 2009;65:9047. doi: 10.1016/j.tet.2009.09.032.Wilson RM, Stockdill JL, Wu X, Li X, Vadola PA, Park PK, Wang P, Danishefsky SJ. Angew. Chem. 2012;124:2888. doi: 10.1002/anie.201106628.Angew. Chem. Int. Ed. 2012;51:2834.
- 2.Danishefsky SJ, Bilodeau MT. Angew. Chem. 1996;108:1482. [Google Scholar]; Angew. Chem. Int. Ed. 1996;35:1380. [Google Scholar]
- 3.a Shang S, Tan Z, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2011;108:4297. doi: 10.1073/pnas.1100195108. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Nagorny P, Sane N, Fasching B, Aussedat B, Danishefsky SJ. Angew. Chem. 2012;124:999. doi: 10.1002/anie.201107482. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:975. [Google Scholar]; c Aussedat B, Fasching B, Johnston E, Sane N, Nagorny P, Danishefsky SJ. J. Am. Chem. Soc. 2012;134:3532. doi: 10.1021/ja2111459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Sytkowski AJ. Erythropoietin. Wiley-VCH Verlag GmbH and Co; KGaG Weinheim: 2004. [Google Scholar]; b Lai PH, Everett R, Wang FF, Arakawa T, Goldwasser E. J. Biol. Chem. 1986;261:3116. [PubMed] [Google Scholar]
- 5.a Kan C, Trzupek JD, Wu B, Wan Q, Chen G, Tan Z, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5438. doi: 10.1021/ja808707w. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Yuan Y, Chen J, Wan Q, Tan Z, Chen G, Kan C, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5432. doi: 10.1021/ja808705v. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tan Z, Shang S, Halkina T, Yuan Y, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5424. doi: 10.1021/ja808704m. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Brailsford JA, Danishefsky SJ. Proc. Natl. Acad. Sci. USA. 2012;109:719. doi: 10.1073/pnas.1202762109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For recent reviews on glycoprotein synthesis, see: Davis BG. Chem. Rev. 2002;102:579. doi: 10.1021/cr0004310.Gamblin D, Scanlan EM, Davis BG. Chem. Rev. 2009;109:131. doi: 10.1021/cr078291i.Payne RJ, Wong CH. Chem. Commun. 2010:46. doi: 10.1039/b913845e.
- 7.a Kajihara Y, Yoshihara A, Hirano K, Yamamoto N. Carbohydrate Res. 2006;341:1333. doi: 10.1016/j.carres.2006.04.037. [DOI] [PubMed] [Google Scholar]; b Yamamoto N, Tanabe Y, Okamoto R, Dawson P, Kajihara Y. J. Am. Chem. Soc. 2008;130:501. doi: 10.1021/ja072543f. [DOI] [PubMed] [Google Scholar]; c Sakamoto I, Tezuka K, Fukae K, Ishii K, Taduru K, Maeda M, Ouchi M, Yoshida K, Nambu Y, Igarashi J, Hayashi N, Tsuji T, Kajihara Y. J. Am. Chem. Soc. 2012;134:5428. doi: 10.1021/ja2109079. [DOI] [PubMed] [Google Scholar]; d Takemura T, Hojo H, Nakahara Y, Ishimuzu T, Hase S. Org. Biomol. Chem. 2004;2:133. doi: 10.1039/b312413d. [DOI] [PubMed] [Google Scholar]; e Unverzagt C. Tetrahedron Lett. 1997;38:5627. [Google Scholar]; f Murakami M, Okamoto R, Izumi M, Kajihara Y. Angew. Chem. 2012;124:3627. doi: 10.1002/anie.201109034. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012;51:3567. [Google Scholar]; g Kunz H, Unverzagt C. Angew. Chem. 1988;100:1763. [Google Scholar]; Angew. Chem. Int. Ed. 1988;27:1697. [Google Scholar]; h Pratt MR, Bertozzi CR. J. Am. Chem. Soc. 2003;125:6149. doi: 10.1021/ja029346v. [DOI] [PubMed] [Google Scholar]; i Guo ZW, Shao N. Med. Res. Rev. 2005;25:655. doi: 10.1002/med.20033. [DOI] [PubMed] [Google Scholar]; j Piontek C, Varón Silva D, Heinlein C, Pöhner C, Mezzato S, Ring P, Martin A, Schmid FX, Unverzagt C. Angew. Chem. 2009;121:1974. doi: 10.1002/anie.200804735. [DOI] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2009;48:1941. [Google Scholar]
- 8.For recent improvements in the efficiency of the SPPS approach to glycopeptide synthesis, see: Heinlein C, Silva DV, Tröster A, Schmidt J, Gross A, Unverzagt C. Angew. Chem. 2011;123:6530–6534.Angew. Chem. Int. Ed. 2011;50:6406–6410. doi: 10.1002/anie.201101270.
- 9.a Chen R, Tolbert TJ. J. Am. Chem. Soc. 2010;132:3211. doi: 10.1021/ja9104073. [DOI] [PubMed] [Google Scholar]; b Conroy T, Jolliffe KA, Payne RJ. Org. Biomol. Chem. 2010;8:3723. doi: 10.1039/c003673k. [DOI] [PubMed] [Google Scholar]
- 10.Dawson PE, Muir TW, Clark-Lewis I, Kent SBH. Science. 1994;266:776. doi: 10.1126/science.7973629. [DOI] [PubMed] [Google Scholar]
- 11.a Anisfeld ST, Lansbury PT. J. Org. Chem. 1990;55:5560. [Google Scholar]; b Cohen-Anisfeld ST, Lansbury PT. J. Am. Chem. Soc. 1993;115:10531. [Google Scholar]
- 12.a Bodanszky M, Tolle JC, Deshmane SS, Bodanszky A. Int. J. Pept. Protein Res. 1978;12:57. [PubMed] [Google Scholar]; b Bodanszky M, Sigler GF, Bodanszky A. J. Am. Chem. Soc. 1973;95:2352. doi: 10.1021/ja00788a040. [DOI] [PubMed] [Google Scholar]
- 13.Wang P, Li X, Zhu J, Chen J, Yuan Y, Wu X, Danishefsky SJ. J. Am. Chem. Soc. 2011;133:1597. doi: 10.1021/ja110115a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Attempts to improve reactivity by increasing the solubility of peptide 2 through the use of dodecylphosphocholine were unsuccessful.
- 15.Haack T, Mutter M. Tetrahedron Lett. 1992;33:1589. [Google Scholar]
- 16.Installation of the Leu-Ser pseudoproline dipeptide at the C-terminus served to minimize peptide aggregation: Garcia-Martin F, White P, Steinauer R, Côté S, Tulla-Puche J, Albericio F. Biopolymers: Peptide Sci. 2006;84:566. doi: 10.1002/bip.20564.
- 17.Nagorny P, Fasching B, Li X, Chen G, Aussedat B, Danishefsky SJ. J. Am. Chem. Soc. 2009;131:5792. doi: 10.1021/ja809554x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Small amounts of ammonia are often present in the glycosylamine component, even following multiple lyophilizations.
- 19.Wang P, Dong S, Brailsford JA, Townsend SD, Zhang Q, Iyer K, Hendrickson RC, Shieh JH, Moore M, Danishefsky SJ. Angew. Chem. 2012 doi: 10.1002/anie.201206090. Submitted. [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. Int. Ed. 2012 Submitted. [Google Scholar]
- 20.The basis for the “n+2 pseudoproline” effect in suppressing aspartimide formation both in peptide synthesis as well as in asparagine glycosylation invites various interpretations. It has been argued, with computational support, that type II’ β–turn, stabilized by an intramolecular hydrogen bond (see structure i) of a type which cannot be present in the pseudoproline case (see structure ii). This effect could be operative at the kinetic level, leading to suppression of imide formation relative to biomolecular acylation. See: Capasso S, Mazzarella L, Zagari A. Chirality. 1995;7:605. doi: 10.1002/chir.530070808.Subiros-Funosas R, El-Faham A, Albericio F. Tetrahedron. 2011;67:8595.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.