

Abstract
Pulmonary hypertension (PH) is a progressive disease that leads to substantial morbidity and eventual death. Pulmonary multidetector CT angiography (MDCTA), pulmonary MR angiography (MRA) and MR-derived pulmonary perfusion (MRPP) imaging are non-invasive imaging techniques for the differential diagnosis of PH. MDCTA is considered the gold standard for the diagnosis of pulmonary embolism, one of the most common causes of PH. MRA and MRPP are promising techniques that do not require the use of ionising radiation or iodinated contrast material, and can be useful for patients for whom such material cannot be used. This review compares the imaging aspects of pulmonary MRA and 64-row MDCTA in patients with chronic thromboembolic or idiopathic PH.
Pulmonary hypertension (PH) is an insidious and progressive disease that leads to substantial morbidity and eventual death. PH results from a number of diseases with different physiopathologies, treatments and prognoses [1]. One of the most frequent causes of PH is chronic thromboembolic pulmonary hypertension (CTEPH).
The current classification of PH (Table 1), developed at the 2008 4th World Symposium on Pulmonary Hypertension in Dana Point, CA [2], resulted from a review of the previous classification developed at the 2003 3rd World Symposium in Venice, Italy. During the 4th World Symposium on PH, an international group of experts agreed to maintain the general philosophy and organisation of the Evian–Venice classifications. However, in response to a questionnaire regarding the previous classification, a majority (63%) of experts felt that modification of the Venice classification was required to accurately reflect information published in the past 5 years and to provide clarification in some areas [2].
Table 1. Classification of pulmonary hypertension according to the 4th World Symposium, Dana Point, CA, 2008 [2].
| 1. Pulmonary arterial hypertension (PAH) |
| 1.1. Idiopathic PAH |
| 1.2. Heritable PAH |
| 1.2.1. Bone morphogenetic protein receptor type 2 |
| 1.2.2. Activin receptor-like kinase type 1 (ALK1) |
| ALK1, endoglin (with or without hereditary haemorrhagic telangiectasia) |
| 1.2.3. Unknown |
| 1.3. Drug- and toxin-induced |
| 1.4. Associated with: |
| 1.4.1. Connective tissue diseases |
| 1.4.2. HIV infection |
| 1.4.3. Portal hypertension |
| 1.4.4. Congenital heart diseases |
| 1.4.5. Schistosomiasis |
| 1.4.6. Chronic haemolytic anaemia |
| 1.5. Persistent neonatal pulmonary hypertension |
| 1′. Pulmonary veno-occlusive disease and/or pulmonary capillary haemangiomatosis |
| 2. Pulmonary hypertension due to left heart disease |
| 2.1. Systolic dysfunction |
| 2.2. Diastolic dysfunction |
| 2.3. Valvular disease |
| 3. Pulmonary hypertension due to lung diseases and/or hypoxia |
| 3.1. Chronic obstructive pulmonary disease |
| 3.2. Interstitial lung disease |
| 3.3. Other pulmonary diseases with mixed restrictive and obstructive pattern |
| 3.4. Sleep-disordered breathing |
| 3.5. Alveolar hypoventilation disorders |
| 3.6. Chronic exposure to high altitude |
| 3.7. Developmental abnormalities |
| 4. Chronic thromboembolic pulmonary hypertension |
| 5. Pulmonary hypertension with unclear multifactorial mechanisms |
| 5.1. Haematological disorders: myeloproliferative disorders, splenectomy |
| 5.2. Systemic disorders: sarcoidosis, pulmonary Langerhans cell histiocytosis, lymphangioleiomyomatosis, neurofibromatosis, vasculitis |
| 5.3. Metabolic disorders: glycogen storage disease, Gaucher disease, thyroid disorders |
| 5.4. Other: tumoral obstruction, fibrosing mediastinitis, chronic renal failure on dialysis |
PH is a clinical and haemodynamic syndrome that results in increased vascular resistance in the pulmonary circulation, usually by a combination of mechanisms involving vasoconstriction and remodelling of the small vessels [3]. Haemodynamically, it is defined as a systolic pulmonary artery pressure of >35 mmHg, or a mean pulmonary artery pressure of >25 mmHg at rest or >30 mmHg with exertion [4,5]. An increase in pulmonary vascular resistance and subsequent compensatory right ventricular (RV) hypertrophy lead to elevated pulmonary pressure, which often results in increased RV afterload and failure. The disorder is progressive, leading to right heart failure and death within a median of 2.8 years after diagnosis [6,7].
The development of RV failure in patients with pulmonary arterial hypertension (PAH) is an ominous sign with major adverse prognostic implications. Patients with severe PAH or right heart failure die usually within 1 year without treatment. In the National Institutes of Health registry, approximately 50% of deaths in patients with PAH are attributed to RV failure [6]. Numerous factors may indicate a poor prognosis in patients with PAH and secondary RV failure, including age >45 years at presentation, New York Heart Association (NYHA) Class III or IV functional classification, failure to improve to a lower NYHA class during treatment, pericardial effusion, large right atrial size, elevated right atrial pressure, septal shift during diastole, decreased pulmonary arterial capacitance (stroke volume/pulmonary arterial pulse pressure), increased N-terminal brain natriuretic peptide level and hypocapnia [8,9].
Because patients with PH often present with non-specific symptoms, such as shortness of breath on minimal physical exertion, fatigue, chest pain and fainting, diagnosis often occurs late in the course of the disease, when the prognosis is poor and treatment options are limited [10]. A complete diagnostic evaluation includes a medical history, physical examination, pulmonary function tests, electrocardiogram, echocardiogram, cardiac catheterisation and advanced imaging. Invasive haemodynamic evaluation is mandatory, not only to confirm the diagnosis but also to address the prognosis and the patient's eligibility for the use of calcium channel blockers through an acute vasodilator challenge. Non-invasive surrogate response markers to the acute vasodilator test have been sought. In other studies, mean pulmonary artery distensibility (mPAD) has been evaluated using MRI to assess pulmonary haemodynamics and diagnose pulmonary vascular disease [11,12]. The mPAD may reflect the degree of vascular remodelling, making it a very interesting marker for the evaluation of patients with idiopathic PAH (IPAH) [13]. Jardim et al [14] found that the cardiac index, calculated after the determination of cardiac output using MRI and pulmonary artery catheterisation, showed excellent correlation, as did right atrial pressure and the RV ejection fraction. They also found that PAD was significantly higher in acute vasodilator test responders. A receiver operating characteristic curve analysis has shown that 10% distensibility can be used to differentiate responders from non-responders with 100% sensitivity and 56% specificity. This study suggested that MRI and PAD may be useful non-invasive tools for the evaluation of patients with PH. In some cases, definitive diagnosis requires a thoracoscopic lung biopsy [3]. Because CTEPH differs considerably from other forms of PH and may be treated surgically, an accurate diagnosis is essential [15].
The depiction of occluding thrombotic material and concomitant perfusion defects is a prerequisite for the correct and reliable diagnosis of CTEPH. Until recently, pulmonary perfusion could be assessed only by using radionuclide perfusion scintigraphy and conventional pulmonary angiography. The former technique has substantial limitations with respect to spatial and temporal resolution, and the latter requires invasive catheterisation of the right side of the heart and produces only two-dimensional projection images [16].
Pulmonary multidetector CT angiography (MDCTA), pulmonary MR angiography (MRA), and MR-derived pulmonary perfusion (MRPP) are non-invasive imaging techniques used to assess PH-related pulmonary vessel changes in the differential diagnosis [16]. MDCTA is considered the gold standard for the diagnosis of CTEPH because it depicts the occluding thrombotic material and concomitant lung changes [16]. However, the combined use of MRA and MRPP allows the evaluation of PH-related pulmonary vessel changes and concomitant perfusion defects without ionising radiation or iodinated contrast material, and can be useful for patients in whom such material cannot be used. Few studies to date have sought to determine the accuracy of MRA in distinguishing the various causes of PH [16-18].
MRI also contributes to the cardiac evaluation of patients with PH. Cardiac MRI is the gold standard technique for the assessment of ventricular function and the quantification of volumes and mass without geometric assumptions [19]. Recently, myocardial delayed enhancement after the intravenous administration of a gadolinium-based contrast agent has been shown at the insertion points of the RV free wall in the interventricular septum in patients with PAH and impaired ventricular function [20]. McCann et al [21] also suggested that the extent of hyperenhancement was not correlated with any clinical or haemodynamic variable, but was inversely correlated with RV dysfunction measured on cardiac MRI.
This review aims to compare the imaging aspects of pulmonary MRA and 64-row MDCTA in patients with CTEPH and IPAH, and to highlight the main differences between these techniques. Patients with other forms of PH are not considered here because CT is superior to MRI for the evaluation of lung parenchyma.
Normal anatomy of the pulmonary arteries
Normal pulmonary circulation is a low-pressure system with a high capacitance and approximately one-tenth of the flow resistance of the systemic circulation. The main pulmonary artery originates at the pulmonary valve and extends cranially and to the left before bifurcating into the shorter left and longer right arteries (Figure 1). The right pulmonary artery divides into three lobar branches at the right hilum. After arching over the left main bronchus, the left pulmonary artery also divides into two lobar branches in the left hilum [22]. The pulmonary arterial system is invariably related to the airways and divides with them. The segmental and subsegmental arteries have similar diameters to those of the adjacent airways.
Figure 1.
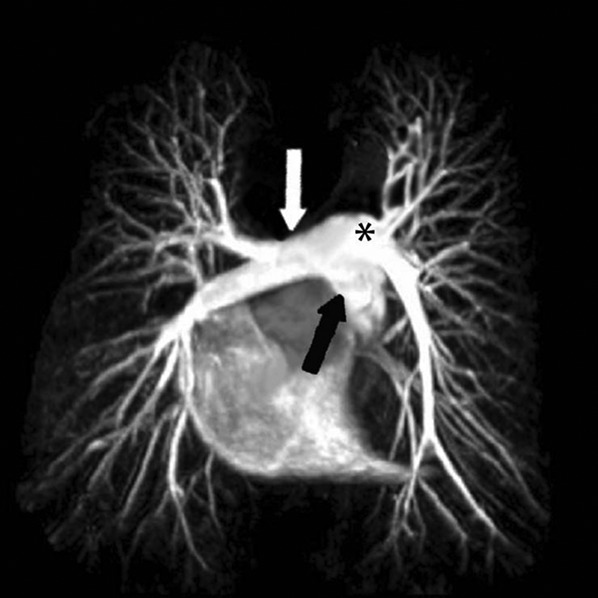
Normal 32-year-old female. MR angiography-derived maximum intensity projection reconstruction shows normal pulmonary artery anatomy: pulmonary artery main stem (black arrow), right pulmonary artery (white arrow) and left pulmonary artery (asterisk).
Bronchial circulation is a higher pressure system that also supplies the lung parenchyma. It arises from the thoracic aorta or intercostal arteries. The bronchial arteries are approximately 1.0–1.5 mm in diameter at the hilum. The pulmonary and bronchial vascular networks form rich anastomoses. Bronchial circulation is involved in gas exchange only in the presence of decreased pulmonary flow and ischaemia [22].
Comparison of idiopathic pulmonary hypertension and chronic thromboembolic pulmonary hypertension
Idiopathic and heritable pulmonary arterial hypertension
IPAH is sporadic and its aetiology and risk factors are unknown. Patients with IPAH usually lack family histories of PAH. The estimated incidence of IPAH is one or two cases per 1 million people. The condition is predominant in females, with a female-to-male ratio of 1.7–3.1:1. PAH is most prevalent between the ages of 20 and 40 years and shows no ethnic predilection. Most patients develop progressive dyspnoea and cor pulmonale, and die within a few years [1,3,23]. 6% of patients who participated in the North American National Registry had a family history of PAH [24]. Germline mutations in the bone morphogenetic protein receptor (BMPR2) gene are detected in 70% of familial PAH cases [25,26]. Because BMPR2 mutations have also been detected in 11–40% of apparently idiopathic cases with no family history [27,28], the distinction between idiopathic and familial BMPR2 mutations is artificial. In addition, no BMPR2 mutation has been identified in ≤30% of families with histories of PAH [29,30]. The category of “heritable PAH” does not mandate genetic testing in patients with IPAH or familial PAH. When called for, genetic testing should be performed as part of a comprehensive programme that includes genetic counselling and discussion of the risks, benefits and limitations of such testing [31].
The radiological findings of IPAH consist of dilated central pulmonary arteries, tortuous arteries, rapid tapering of the vessels (pruned tree sign—enlargement of the arterial roots with a paucity of peripheral arteries) and peripheral oligaemia (Figure 2) [16]. Additional findings include right heart enlargement and paradoxical septal wall motion, which are non-specific and can be found in all PH types [16]. Ventilation–perfusion scans can be normal or may show patchy non-segmental perfusion defects or diffuse reduction in perfusion, usually interpreted as having a low probability of representing thromboembolic disease [1,4,18,23].
Figure 2.

A 45-year-old female with idiopathic pulmonary arterial hypertension. Similar imaging aspects in multidetector CT angiography (MDCTA)-derived maximum intensity projection (MIP) (a), MR angiography (MRA)-derived MIP (b), MDCTA-derived multiplanar reconstruction (MPR) (c) and coloured MRA-derived MPR (d) show marked dilatation of the pulmonary artery main stem (white asterisk) and the right pulmonary artery (black asterisk), proximal calibre changes (pruned tree sign; white arrows on a, b), peripheral vessel reduction and vessel tortuosity (white circles on a, b). No occlusive criteria are visible in MRA or MDCTA images.
Although major therapeutic advances have been achieved for patients with PAH in the last decade, no currently approved therapy can cure this progressive disease. The search for such treatments continues, with promising new concepts arising from an improved understanding of the pathobiology of pulmonary vascular diseases. Patients and physicians should be encouraged to foster such research by participating in randomised clinical trials conducted at specialised PH centres [32].
Chronic thromboembolic pulmonary hypertension
CTEPH develops in approximately 1–5% of patients with acute pulmonary thromboembolism. Patients with malignant neoplasm, chronic cardiac or pulmonary disease, and/or clotting disorders are at increased risk of developing this condition. Patients may be asymptomatic for many years before developing symptoms similar to those of IPAH, such as progressive exertional dyspnoea, chest pain, cough, syncope and cor pulmonale. The management of patients with secondary PH is directed at the early recognition and treatment of the underlying disease (when it is still potentially reversible). For instance, left ventricular dysfunction should be treated with afterload-reducing agents, digoxin and diuretics. Surgery to correct structural cardiac and pulmonary anomalies can also be effective, and thromboendarterectomy in accessible chronic thromboemboli is potentially curative [18,23,33,34].
The choice of surgical treatment depends on the location of the obstruction (central vs more distal pulmonary arteries), the correlation between haemodynamic findings and angiographically assessed degree of mechanical obstruction, comorbidities, the patient's willingness and the surgeon's experience [35,36].
Patients who are not candidates for surgery may benefit from PAH-specific medical therapy [37,38], but the use of these medications in patients with CTEPH requires further evaluation in randomised controlled trials [39].
The bronchial circulation is increased in CTEPH owing to the development of systemic–pulmonary anastomoses, which help to maintain pulmonary blood flow. The radiological findings are more localised and consist of material adhering to the thrombotic wall, wall irregularities, vascular webs and stenosis (Figures 3–6), as well as abnormal proximal–distal tapering (abrupt change in vessel diameter) with perfusional defects and prominent bronchial arteries (Figure 7). Additional findings include right heart hypertrophy and enlargement with bowing of the interventricular septum, mosaic perfusion, air trapping, bronchiectasis and subpleural consolidation from prior pulmonary infarction [18,23,33].
Figure 3.

A 47-year-old female with secondary chronic thromboembolic pulmonary hypertension. Thin 10-mm maximum intensity projections derived from multidetector CT angiography (a, c) and MR angiography (b, d) images show thrombus in the left pulmonary artery that descends toward the basal trunk (white circles on a, b) and complete occlusion of the right pulmonary artery (black arrows on c, d).
Figure 6.

A 41-year-old female with secondary chronic thromboembolic pulmonary hypertension. Thin 10-mm maximum intensity projections derived from multidetector CT angiography (a) and MR angiography (b) images show a similar aspect of vascular bands (white circles).
Figure 7.

A 57-year-old male with secondary chronic thromboembolic pulmonary hypertension. Thin 10-mm maximum intensity projections derived from multidetector CT angiography (a) and MR angiography (b) images show a similar aspect of bronchial artery hypertrophy (white circles).
Figure 4.

A 53-year-old male with secondary chronic thromboembolic pulmonary hypertension. Multiplanar reconstructions derived from multidetector CT angiography (a, c) and MR angiography (b, d) images show the similar aspect of the thrombus in the right pulmonary artery bifurcation (white arrows on a, c; white circles on b, d).
Figure 5.

A 34-year-old female with secondary chronic thromboembolic pulmonary hypertension. Thin 10-mm maximum intensity projections derived from multidetector CT angiography (a) and MR angiography (b) images show a similar aspect of a small thrombus in the left basal trunk (white arrows).
Imaging in pulmonary hypertension using 64-multidetector CT angiography, MR angiography and MR-derived pulmonary perfusion
MR and CT acquisition protocols
According to the recent literature, MRA imaging for the diagnosis of PH can be performed using a high-resolution, breath-hold, contrast-enhanced, three-dimensional (3D), rapid, low-angle shot [repetition time (TR)/echo time (TE), 2.9/1.2 ms; flip angle, 25°] [16] obtained in the coronal orientation using an integrated parallel acquisition technique. This technique combines the generalised, autocalibrating, partially parallel acquisition (GRAPPA®; Siemens Medical Solutions, Erlangen, Germany) technique with sensitivity encoding (SENSE®; Siemens Medical Solutions, Erlangen, Germany) [31,32]. MRPP images can be obtained using high-field MR scanners (>1.5 T) with high-performance gradients (preferably 30–40 mT). Patients enter the scanner head-first in the supine position. A special receiver coil is necessary for parallel imaging.
For dynamic perfusion imaging (MRPP), Nikolaou et al [16] suggested the use of a turbo-speed, low-angle shot, gradient-echo sequence (TR/TE, 1.7/0.6 ms; flip angle, 25°) with an enhanced k-space-based reconstruction sequence (GRAPPA) and an acceleration factor of 2 [16]. The following further sequence parameters for perfusion MRI have been suggested: readout bandwidth, 1220 Hz per pixel; number of reference k-space lines for calibration, 24; section thickness, 4.0 mm; number of sections, 24; resulting slab thickness, 96 mm; acquisition matrix, 133×256 (52% phase resolution); field of view, 400 mm; and resulting spatial resolution, 2.9×1.5×4.0 mm. The k-space readout scheme is centrically reordered, and the acquisition time for one 3D data set of 24 sections is 1.1 s. A total of 24 slabs are acquired for each perfusion series, resulting in a total breath-hold time of 26 s. Image acquisition must be started 4 s before the estimated arrival time of the contrast material, so that unenhanced images can be acquired for subtraction purposes. All injections should be performed using a power injection system with 0.1 mmol gadobutrol or 0.2 mmol gadoterate meglumine per kilogram body weight, followed by a 25-ml saline flush. To ensure an optimised bolus profile, the use of a high injection rate (5 ml s−1) in combination with a 16-gauge intravenous catheter that can be inserted in an antecubital vein is recommended. Unenhanced images are subtracted from the contrast-enhanced images. For each phase (n=25), maximum intensity projections (MIPs) for all 24 sections are acquired for 3D display [16].
64-MDCT images can be obtained using standard protocols defined by the scanner manufacturers. The following parameters for a 64-row detector CT scanner were adapted from the recent literature [40]: caudocranial scanning direction, 120 kVp, 200 mAs, 0.5 mm collimation, 0.5 s rotation time (acquired during suspended breath after deep inspiration), 1.0 pitch, 1-mm reconstruction thickness and 0.5-mm reconstruction increment. A 125-ml injection of iodinated contrast is administered through an antecubital vein at a flow rate of 5 ml s−1. A bolus-tracking technique was used to determine the interval between the initiation of contrast material administration and that of scanning; measurements were obtained at the origin of the pulmonary artery, and an 8 s delay was added to the circulation time before scanning.
MRA and MDCTA images must be evaluated on dedicated workstations, depending on the scanner manufacturer, using multiplanar reconstruction, MIP and the volume-rendering technique.
Comparison of CT and MRI for the diagnosis in pulmonary hypertension
CT is a non-invasive imaging technique that allows evaluation of both the pulmonary circulation (by MDCTA) and the lung parenchyma. MDCTA is widely regarded as the technique of choice for the evaluation of acute and chronic pulmonary vascular disease. It also aids differential diagnoses that include conditions such as pulmonary artery sarcoma, extrinsic compression of pulmonary vessels and fibrosing mediastinitis. The success of thoracic MDCTA is the result, in part, of the high spatial resolution of CT, and advances in multisection technology have dramatically increased the speed and simplicity of this technique [41-43]. However, its main disadvantage is the use of ionising radiation and iodinated contrast material.
In the late 1970s and early 1980s, several investigators used MDCTA findings to measure the diameter of central pulmonary arteries and to predict the presence of PH [36-40]. The calibre of the pulmonary artery, the artery-to-bronchus ratio and the presence of pericardial effusion are associated with the severity of PH. Tan et al [44] also showed that CT-determined mean pulmonary artery diameter had excellent diagnostic value for the detection of PH in patients with advanced lung disease. However, the use of pulmonary artery diameter has limitations. Pulmonary artery dilatation can occur in the absence of PH in patients with pulmonary fibrosis, and is therefore an unreliable sign of PH in these patients [45]. Thus, standard chest CT scans may be used to screen for PH as a cause of exertional limitation in patients with parenchymal lung disease. Although MDCTA is currently the method of choice for the evaluation of patients with CTEPH, it provides poor functional information [16]. Kuriyama et al [46] found that a CT-determined mean pulmonary artery diameter of ≥29 mm predicted PH with 87% sensitivity and 89% specificity. This specificity reached 100% when this mean pulmonary artery diameter was accompanied by findings of a segmental artery-to-bronchus ratio greater than 1:1 in three of four pulmonary lobes [46].
MDCTA distinguishes CTEPH from IPAH by detecting thrombotic material, evaluates underlying lung disease and may help to identify rare causes of PH. MDCTA is quick, widely available and inexpensive. However, it is associated with a significant radiation burden and is unsuitable for serial examinations [47]. MDCTA will thus probably continue to have a pivotal role in the evaluation of patients with pulmonary vascular disease. However, renal impairment or contrast agent sensitivity prevent the use of this technique in a subset of patients. In other patients, the radiation burden of MDCTA may be considered undesirable owing to young age or the prospect of multiple follow-up examinations [48].
Dual-energy CT (DECT) pulmonary angiography seems to represent an important technique in pulmonary angiography. This technique not only enables imaging of emboli in the pulmonary vasculature but also provides an indication of their functional impact in terms of parenchymal perfusion. The DECT pulmonary angiography technique is similar in many respects to standard CT pulmonary angiography. A slightly longer delay time is used (around 7 s) to allow contrast material to perfuse to the lung periphery, and a saline flush helps minimise beam-hardening effects in the superior vena cava that can mimic perfusion defects in the upper lobes. However, DECT pulmonary angiography offers a functional aspect to CT pulmonary angiography and may improve its sensitivity for detecting subsegmental emboli [49]. DECT perfusion imaging is also able to display pulmonary perfusion defects with good agreement to scintigraphic findings. DECT can also provide a pulmonary CT angiogram, high-resolution morphology of the lung parenchyma and perfusion information in one single examination [50].
MRA and MRPP are non-invasive imaging methods that allow the morphological and functional study of PAH patients and the examination of cardiac anatomy and functional parameters without the use of ionising radiation or iodinated contrast material [51]. The combined use of these techniques can facilitate the differential diagnosis of IPAH and CTEPH [17,18,52,53]. MRPP can provide valuable information for the diagnosis of PH by demonstrating the exact extent of perfusion defects, and has increased diagnostic accuracy in the differentiation of various causes of PAH, depending on the specific patterns of perfusion defects [16,54]. MRI of lung perfusion is performed with rapid imaging of the first passage of contrast material through the lungs after bolus injection into a peripheral vein. Despite substantial recent improvements in the gradient technique, 3D imaging techniques have been limited with regard to spatial and temporal resolution [16]. Sufficient temporal resolution is critical for the visualisation of peak enhancement of the lungs because the transit time of the contrast material bolus through the lungs is usually 3–4 s. Unlike conventional MRI techniques, parallel imaging techniques acquire only a fraction of the phase-encoding lines and reconstruct the missing information to a full field-of-view image by using the inherent spatial encoding of the different receiver coils. A temporal resolution of 1.1 s can be obtained using these techniques [16]. Despite these improvements in temporal resolution, the breath-hold time for acquisition of a complete data set in perfusion MRI remains quite long (approximately 26 s) [16]. Long breath-hold times may be difficult for some patients with pulmonary disorders [16]. MRA allows the integration of morphological and functional information, which increases the diagnostic accuracy of the aetiological causes of PAH based on perfusion defect patterns [54].
MRA has become established as a powerful non-invasive tool for use in most vascular territories [55-57]. MRA was previously described as a highly sensitive and specific method for the diagnosis of acute pulmonary thromboembolism that does not require radiation exposure or the use of iodinated contrast media [52,53]. However, the sensitivity of MRA was lower than that of digital subtraction angiography or MDCTA for the detection of corkscrew phenomena or subsegmental webs and bands. A comparison of state-of-the-art MRA and MDCTA of the pulmonary vasculature found that MDCTA remains superior because of the ease of use, short acquisition time and high spatial resolution [17]. MRA imaging, however, offers the great advantage of combining morphological and functional information, which makes the use of MRI favourable in the comprehensive assessment of complex diseases such as PAH [17].
In both imaging techniques, the anatomy and presence of PH-related occlusive (CTEPH associated) and non-occlusive changes of the pulmonary arteries should be evaluated [16], as well as the dilatation of the pulmonary arterial main stem (diameter >3.0 cm), peripheral vessel reduction (peripheral oligaemia), proximal calibre changes (IPAH-associated pruned tree sign), focal vessel ectasia, vessel tortuosity, irregularity of the vessel walls, bronchial artery hypertrophy and the occlusive criteria, including complete vessel occlusion, free-floating or wall-adherent thrombus, webs and vascular bands.
In patients with CTEPH, currently available MRI techniques can be used to evaluate various aspects of the disease. With contrast-enhanced MRA, the pulmonary arterial vasculature can be sufficiently analysed to the subsegmental level so that characteristic angiographic findings are well delineated. Furthermore, this technique can demonstrate the proximal extent of the organised thromboembolic material with great certainty, a basic prerequisite for the identification of potential candidates for pulmonary endarterectomy [58].
Although MDCTA and MRA images can be very similar, we have found that thrombus detection is more difficult with MRA than with MDCTA because of the low signal intensity in MRI sequences. Careful evaluation and an experienced reader are required for accurate diagnosis. Figures 3–6 compare MDCTA and MRA images.
Perfusion data facilitate the visualisation of the characteristic loss of vascular perfusion. Advances in gradient systems and improved MR sequences have allowed the development of a 3D gradient-recalled echo sequence with an ultrashort TR/TE of <3.0/1.0 ms. With this sequence, qualitative regional perfusion differences in the entire lung can be evaluated with high temporal resolution [59,60]. Diagnostic criteria for perfusion imaging in the differential diagnosis of IPAH and CTEPH have been modified from earlier perfusion scintigraphy reports [61]. The perfusion patterns (Figure 8) are classified as normal (no perfusion defect), diffuse peripheral and/or patchy (IPAH), or segmental and/or focal defect (CTEPH; Figure 9).
Figure 8.

Evaluation criteria for pulmonary perfusion. Maximum intensity projection images of a single perfusion phase obtained with contrast-enhanced MR-derived pulmonary perfusion in three patients: (a) normal homogeneous perfusion (no perfusion defect), (b) bilateral diffuse non-segmental peripheral perfusion defects (typical of pulmonary arterial hypertension; white arrows) and (c) segmental or focal perfusion defect (typical of chronic thromboembolic pulmonary hypertension) in the upper right lobe (open arrow).
Figure 9.

Perfusional aspect in chronic thromboembolic pulmonary hypertension. Maximum intensity projection images of a single perfusion phase obtained with contrast-enhanced MR-derived pulmonary perfusion in three patients: (a) multiple perfusion defects (white arrows), (b) focal perfusion defect in the anterior segment of the right lung upper lobe (white arrow) and (c) complete perfusion defect in the right lung (white arrow).
As described in previous articles [62,63], there is good agreement between MRPP imaging and conventional radionuclide scintigraphy. However, MRPP imaging has various advantages over conventional scintigraphy, such as the avoidance of radiation exposure, higher spatial resolution and better anatomical information that allows data reconstruction in any desired imaging plane. In 2007, Ohno et al [64] showed that 3D dynamic contrast-enhanced MRPP has potential for the assessment of disease severity in patients with IPAH. Moreover, the quantitative assessment of regional pulmonary perfusion parameters from 3D dynamic contrast-enhanced MRPP images of patients with IPAH may offer the opportunity for non-invasive physiological and pathophysiological evaluation of the lungs with relatively high spatial resolution and without radiation exposure [64].
MDCTA and MRA are comprehensive and complementary methods of evaluating PH. MDCTA is superior to MRA in the visualisation of vascular abnormalities and the diagnosis of PH related to lung disease and bronchosystemic shunts [65]. However, the absence of ionising radiation and superior contrast profile favour MRA as a monitoring method. The use of MRA for other pathologies related to pulmonary circulation has not been investigated, and few studies have sought to determine its accuracy in the distinction of different forms of PH [16,47,58].
Conclusion
In this review, we compared the imaging findings of MRA and 64-row MDCTA in patients with known IPAH or CTEPH. We also detailed the MRPP imaging findings in these patients. MDCTA is currently considered the gold standard for the diagnosis of pulmonary embolism; however, we demonstrated that MRA can produce images similar to those of MDCTA in patients with IPAH or secondary CTEPH. Combined MRA and MRPP imaging allow the comprehensive evaluation of morphological and functional diagnostic information in patients with PAH. These can be helpful in the differential diagnosis and monitoring of the disease, and do not require the use of ionising radiation or iodinated contrast material.
Acknowledgments
The authors thank Dr Daniel Waetge and Dr Emerson Leandro Gasparetto for their help in the organisation of the manuscript; Professor Dudley J. Pennell for his help in the review of the manuscript; and Dr Fabio Magalhães Vargas and Dr Iugiro Roberto Kuroki for their help in MDCTA imaging analysis.
References
- 1.Barst RJ, McGoon M, Torbicki A, Sitbon O, Krowka MJ, Olschewski H, et al. Diagnosis and differential assessment of pulmonary arterial hypertension. J Am Coll Cardiol 2004;43Suppl. 12:40S–47S [DOI] [PubMed] [Google Scholar]
- 2.Simonneau G, Robbins IM, Beghetti M, Channick RN, Delcroix M, Denton CP, et al. Updated clinical classification of pulmonary hypertension. J Am Coll Cardiol 2009;54Suppl. 1:S43–54 [DOI] [PubMed] [Google Scholar]
- 3.Rabinovitch M. Pathobiology of pulmonary hypertension. Extracellular matrix. Clin Chest Med 2001;22:433–49 [DOI] [PubMed] [Google Scholar]
- 4.Rubin LJ. Primary pulmonary hypertension. N Engl J Med 1997;336:111–17 [DOI] [PubMed] [Google Scholar]
- 5.McLaughlin VV, Presberg KW, Doyle RL, Abman SH, McCrory DC, Fortin T, et al. Prognosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004;126 Suppl. 1:78S–92S [DOI] [PubMed] [Google Scholar]
- 6.D'Alonzo GE, Barst RJ, Ayres SM, Bergofsky EH, Brundage BH, Detre KM, et al. Survival in patients with primary pulmonary hypertension. Results from a national prospective registry. Ann Intern Med 1991;115:343–9 [DOI] [PubMed] [Google Scholar]
- 7.Fuster V, Steele PM, Edwards WD, Gersh BJ, McGoon MD, Frye RL. Primary pulmonary hypertension: natural history and the importance of thrombosis. Circulation 1984;70:580–7 [DOI] [PubMed] [Google Scholar]
- 8.Sitbon O, Humbert M, Nunes H, Parent F, Garcia G, Hervé P, et al. Long-term intravenous epoprostenol infusion in primary pulmonary hypertension: prognostic factors and survival. J Am Coll Cardiol 2002;40:780–8 [DOI] [PubMed] [Google Scholar]
- 9.Kuhn KP, Byrne DW, Arbogast PG, Doyle TP, Loyd JE, Robbins IM. Outcome in 91 consecutive patients with pulmonary arterial hypertension receiving epoprostenol. Am J Respir Crit Care Med 2003;167:580–6 [DOI] [PubMed] [Google Scholar]
- 10.Nauser TD, Stites SW. Diagnosis and treatment of pulmonary hypertension. Am Fam Physician 2001;63:1789–98 [PubMed] [Google Scholar]
- 11.Bogren HG, Klipstein RH, Mohiaddin RH, Firmin DN, Underwood SR, Rees RS, et al. Pulmonary artery distensibility and blood flow patterns: a magnetic resonance study of normal subjects and of patients with pulmonary arterial hypertension. Am Heart J 1989;118(5 Pt 1):990–9 [DOI] [PubMed] [Google Scholar]
- 12.Berger RM, Cromme-Dijkhuis AH, Hop WC, Kruit MN, Hess J. Pulmonary arterial wall distensibility assessed by intravascular ultrasound in children with congenital heart disease: an indicator for pulmonary vascular disease? Chest 2002;122:549–57 [DOI] [PubMed] [Google Scholar]
- 13.Casalino E, Laissy JP, Soyer P, Bouvet E, Vachon F. Assessment of right ventricle function and pulmonary artery circulation by cine-MRI in patients with AIDS. Chest 1996;110:1243–7 [DOI] [PubMed] [Google Scholar]
- 14.Jardim C, Rochitte CE, Humbert M, Rubenfeld G, Jasinowodolinski D, Carvalho CR, et al. Pulmonary artery distensibility in pulmonary arterial hypertension: an MRI pilot study. Eur Respir J 2007;29:476–81 [DOI] [PubMed] [Google Scholar]
- 15.Fedullo PF, Auger WR, Kerr KM, Rubin LJ. Chronic thromboembolic pulmonary hypertension. N Engl J Med 2001;345:1465–72 [DOI] [PubMed] [Google Scholar]
- 16.Nikolaou K, Schoenberg SO, Attenberger U, Scheidler J, Dietrich O, Kuehn B, et al. Pulmonary arterial hypertension: diagnosis with fast perfusion MR imaging and high-spatial-resolution MR angiography–preliminary experience. Radiology 2005;236:694–703 [DOI] [PubMed] [Google Scholar]
- 17.Kruger S, Haage P, Hoffmann R, Breuer C, Bücker A, Hanrath P, et al. Diagnosis of pulmonary arterial hypertension and pulmonary embolism with magnetic resonance angiography. Chest 2001;120:1556–61 [DOI] [PubMed] [Google Scholar]
- 18.Ley S, Fink C, Zaporozhan J, Borst MM, Meyer FJ, Puderbach M, et al. Value of high spatial and high temporal resolution magnetic resonance angiography for differentiation between idiopathic and thromboembolic pulmonary hypertension: initial results. Eur Radiol 2005;15:2256–63 [DOI] [PubMed] [Google Scholar]
- 19.Pennell DJ, Sechtem UP, Higgins CB, Manning WJ, Pohost GM, Rademakers FE, et al. Clinical indications for cardiovascular magnetic resonance (CMR): Consensus Panel report. Eur Heart J 2004;25:1940–65 [DOI] [PubMed] [Google Scholar]
- 20.Junqueira FP, Macedo R, Coutinho AC, Loureiro R, De Pontes PV, Domingues RC, et al. Myocardial delayed enhancement in patients with pulmonary hypertension and right ventricular failure: evaluation by cardiac MRI. Br J Radiol 2009;82:821–6 [DOI] [PubMed] [Google Scholar]
- 21.McCann GP, Gan CT, Beek AM, Niessen HW, Vonk Noordegraaf A, van Rossum AC. Extent of MRI delayed enhancement of myocardial mass is related to right ventricular dysfunction in pulmonary artery hypertension. AJR Am J Roentgenol 2007;188:349–55 [DOI] [PubMed] [Google Scholar]
- 22.Azevedo LF, Brum PC, Mattos KC, Junqueira CM, Rondon MU, Barretto AC, et al. Effects of losartan combined with exercise training in spontaneously hypertensive rats. Braz J Med Biol Res 2003;36:1595–603 [DOI] [PubMed] [Google Scholar]
- 23.Frazier AA, Galvin JR, Franks TJ, Rosado-De-Christenson ML. From the archives of the AFIP: pulmonary vasculature: hypertension and infarction. Radiographics 2000;20:491–524; quiz 530–1, 532 [DOI] [PubMed] [Google Scholar]
- 24.Gaine S. Pulmonary hypertension. JAMA 2000;284:3160–8 [DOI] [PubMed] [Google Scholar]
- 25.Aldred MA, Vijayakrishnan J, James V, Soubrier F, Gomez-Sanchez MA, Martensson G, et al. BMPR2 gene rearrangements account for a significant proportion of mutations in familial and idiopathic pulmonary arterial hypertension. Hum Mutat 2006;27:212–13 [DOI] [PubMed] [Google Scholar]
- 26.Cogan JD, Pauciulo MW, Batchman AP, Prince MA, Robbins IM, Hedges LK, et al. High frequency of BMPR2 exonic deletions/duplications in familial pulmonary arterial hypertension. Am J Respir Crit Care Med 2006;174:590–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Thomson JR, Machado RD, Pauciulo MW, Morgan NV, Humbert M, Elliott GC, et al. Sporadic primary pulmonary hypertension is associated with germline mutations of the gene encoding BMPR-II, a receptor member of the TGF-beta family. J Med Genet 2000;37:741–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Machado RD, Aldred MA, James V, Harrison RE, Patel B, Schwalbe EC, et al. Mutations of the TGF-beta type II receptor BMPR2 in pulmonary arterial hypertension. Hum Mutat 2006;27:121–32 [DOI] [PubMed] [Google Scholar]
- 29.Trembath RC, Thomson JR, Machado RD, Morgan NV, Atkinson C, Winship I, et al. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001;345:325–34 [DOI] [PubMed] [Google Scholar]
- 30.Chaouat A, Coulet F, Favre C, Simonneau G, Weitzenblum E, Soubrier F, et al. Endoglin germline mutation in a patient with hereditary haemorrhagic telangiectasia and dexfenfluramine associated pulmonary arterial hypertension. Thorax 2004;59:446–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McGoon M, Gutterman D, Steen V, Barst R, McCrory DC, Fortin TA, et al. Screening, early detection, and diagnosis of pulmonary arterial hypertension: ACCP evidence-based clinical practice guidelines. Chest 2004;126Suppl. 1:14S–34S [DOI] [PubMed] [Google Scholar]
- 32.Barst RJ, Gibbs JS, Ghofrani HA, Hoeper MM, McLaughlin VV, Rubin LJ, et al. Updated evidence-based treatment algorithm in pulmonary arterial hypertension. J Am Coll Cardiol 2009;54Suppl. 1:S78–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Reddy GP, Gotway MB, Araoz PA. Imaging of chronic thromboembolic pulmonary hypertension. Semin Roentgenol 2005;40:41–7 [DOI] [PubMed] [Google Scholar]
- 34.Moser KM, Auger WR, Fedullo PF. Chronic major-vessel thromboembolic pulmonary hypertension. Circulation 1990;81:1735–43 [DOI] [PubMed] [Google Scholar]
- 35.Jamieson SW, Kapelanski DP, Sakakibara N, Manecke GR, Thistlethwaite PA, Kerr KM, et al. Pulmonary endarterectomy: experience and lessons learned in 1,500 cases. Ann Thorac Surg 2003;76:1457–62; discussion 1462–4 [DOI] [PubMed] [Google Scholar]
- 36.Dartevelle P, Fadel E, Mussot S, Chapelier A, Hervé P, de Perrot M, et al. Chronic thromboembolic pulmonary hypertension. Eur Respir J 2004;23:637–48 [DOI] [PubMed] [Google Scholar]
- 37.Suntharalingam J, Treacy CM, Doughty NJ, Goldsmith K, Soon E, Toshner MR, et al. Long-term use of sildenafil in inoperable chronic thromboembolic pulmonary hypertension. Chest 2008;134:229–36 [DOI] [PubMed] [Google Scholar]
- 38.Jais X, D'Armini AM, Jansa P, Torbicki A, Delcroix M, Ghofrani HA, et al. Bosentan for treatment of inoperable chronic thromboembolic pulmonary hypertension: BENEFiT (Bosentan Effects in iNopErable Forms of chronIc Thromboembolic pulmonary hypertension), a randomized, placebo-controlled trial. J Am Coll Cardiol 2008;52:2127–34 [DOI] [PubMed] [Google Scholar]
- 39.Rubin LJ, Hoeper MM, Klepetko W, Galiè N, Lang IM, Simonneau G. Current and future management of chronic thromboembolic pulmonary hypertension: from diagnosis to treatment responses. Proc Am Thorac Soc 2006;3:601–7 [DOI] [PubMed] [Google Scholar]
- 40.Matsuoka S, Hunsaker AR, Gill RR, Oliva IB, Trotman-Dickenson B, Jacobson FL, et al. Vascular enhancement and image quality of MDCT pulmonary angiography in 400 cases: comparison of standard and low kilovoltage settings. AJR Am J Roentgenol 2009;192:1651–6 [DOI] [PubMed] [Google Scholar]
- 41.Alkadhi H, Wildermuth S, Desbiolles L, Schertler T, Crook D, Marincek B, et al. Vascular emergencies of the thorax after blunt and iatrogenic trauma: multi-detector row CT and three-dimensional imaging. Radiographics 2004;24:1239–55 [DOI] [PubMed] [Google Scholar]
- 42.Remy-Jardin M, Bouaziz N, Dumont P, Brillet PY, Bruzzi J, Remy J. Bronchial and nonbronchial systemic arteries at multi-detector row CT angiography: comparison with conventional angiography. Radiology 2004;233:741–9 [DOI] [PubMed] [Google Scholar]
- 43.Revel MP, Petrover D, Hernigou A, Lefort C, Meyer G, Frija G. Diagnosing pulmonary embolism with four-detector row helical CT: prospective evaluation of 216 outpatients and inpatients. Radiology 2005;234:265–73 [DOI] [PubMed] [Google Scholar]
- 44.Tan RT, Kuzo R, Goodman LR, Siegel R, Haasler GB, Presberg KW. Utility of CT scan evaluation for predicting pulmonary hypertension in patients with parenchymal lung disease. Medical College of Wisconsin Lung Transplant Group. Chest 1998;113:1250–6 [DOI] [PubMed] [Google Scholar]
- 45.Devaraj A, Wells AU, Meister MG, Corte TJ, Hansell DM. The effect of diffuse pulmonary fibrosis on the reliability of CT signs of pulmonary hypertension. Radiology 2008;249:1042–9 [DOI] [PubMed] [Google Scholar]
- 46.Kuriyama K, Gamsu G, Stern RG, Cann CE, Herfkens RJ, Brundage BH. CT-determined pulmonary artery diameters in predicting pulmonary hypertension. Invest Radiol 1984;19:16–22 [DOI] [PubMed] [Google Scholar]
- 47.Coulden R. State-of-the-art imaging techniques in chronic thromboembolic pulmonary hypertension. Proc Am Thorac Soc 2006;3:577–83 [DOI] [PubMed] [Google Scholar]
- 48.Nael K, Michaely HJ, Kramer U, Lee MH, Goldin J, Laub G, et al. Pulmonary circulation: contrast-enhanced 3.0-T MR angiography–initial results. Radiology 2006;240:858–68 [DOI] [PubMed] [Google Scholar]
- 49.Remy-Jardin M, Pistolesi M, Goodman LR, Gefter WB, Gottschalk A, Mayo JR, et al. Management of suspected acute pulmonary embolism in the era of CT angiography: a statement from the Fleischner Society. Radiology 2007;245:315–29 [DOI] [PubMed] [Google Scholar]
- 50.Thieme SF, Becker CR, Hacker M, Nikolaou K, Reiser MF, Johnson TR. Dual energy CT for the assessment of lung perfusion–correlation to scintigraphy. Eur J Radiol 2008;68:369–74 [DOI] [PubMed] [Google Scholar]
- 51.Pedersen MR, Fisher MT, van Beek EJ. MR imaging of the pulmonary vasculature–an update. Eur Radiol 2006;16:1374–86 [DOI] [PubMed] [Google Scholar]
- 52.Stein PD, Woodard PK, Hull RD, Kayali F, Weg JG, Olson RE, et al. Gadolinium-enhanced magnetic resonance angiography for detection of acute pulmonary embolism: an in-depth review. Chest 2003;124:2324–8 [DOI] [PubMed] [Google Scholar]
- 53.Oudkerk M, van Beek EJ, Wielopolski P, van Ooijen PM, Brouwers-Kuyper EM, Bongaerts AH, et al. Comparison of contrast-enhanced magnetic resonance angiography and conventional pulmonary angiography for the diagnosis of pulmonary embolism: a prospective study. Lancet 2002;359:1643–7 [DOI] [PubMed] [Google Scholar]
- 54.Fink C, Bock M, Puderbach M, Schmahl A, Delorme S. Partially parallel three-dimensional magnetic resonance imaging for the assessment of lung perfusion–initial results. Invest Radiol 2003;38:482–8 [DOI] [PubMed] [Google Scholar]
- 55.Ho VB, Corse WR. MR angiography of the abdominal aorta and peripheral vessels. Radiol Clin North Am 2003;41:115–44 [DOI] [PubMed] [Google Scholar]
- 56.Levy RA, Prince MR. Arterial-phase three-dimensional contrast-enhanced MR angiography of the carotid arteries. AJR Am J Roentgenol 1996;167:211–15 [DOI] [PubMed] [Google Scholar]
- 57.Prince MR, Narasimham DL, Stanley JC, Chenevert TL, Williams DM, Marx MV, et al. Breath-hold gadolinium-enhanced MR angiography of the abdominal aorta and its major branches. Radiology 1995;197:785–92 [DOI] [PubMed] [Google Scholar]
- 58.Kreitner KF, Kunz RP, Ley S, Oberholzer K, Neeb D, Gast KK, et al. Chronic thromboembolic pulmonary hypertension: assessment by magnetic resonance imaging. Eur Radiol 2007;17:11–21 [DOI] [PubMed] [Google Scholar]
- 59.Ohno Y, Hatabu H, Takenaka D, Adachi S, Hirota S, Sugimura K. Contrast-enhanced MR perfusion imaging and MR angiography: utility for management of pulmonary arteriovenous malformations for embolotherapy. Eur J Radiol 2002;41:136–46 [DOI] [PubMed] [Google Scholar]
- 60.Ohno Y, Hatabu H, Takenaka D, Adachi S, Kono M, Sugimura K. Solitary pulmonary nodules: potential role of dynamic MR imaging in management initial experience. Radiology 2002;224:503–11 [DOI] [PubMed] [Google Scholar]
- 61.Chapman PJ, Bateman ED, Benatar SR. Primary pulmonary hypertension and thromboembolic pulmonary hypertension–similarities and differences. Respir Med 1990;84:485–8 [DOI] [PubMed] [Google Scholar]
- 62.Berthezene Y, Croisille P, Wiart M, Howarth N, Houzard C, Faure O, et al. Prospective comparison of MR lung perfusion and lung scintigraphy. J Magn Reson Imaging 1999;9:61–8 [DOI] [PubMed] [Google Scholar]
- 63.Amundsen T, Torheim G, Kvistad KA, Waage A, Bjermer L, Nordlid KK, et al. Perfusion abnormalities in pulmonary embolism studied with perfusion MRI and ventilation-perfusion scintigraphy: an intra-modality and inter-modality agreement study. J Magn Reson Imaging 2002;15:386–94 [DOI] [PubMed] [Google Scholar]
- 64.Ohno Y, Hatabu H, Murase K, Higashino T, Nogami M, Yoshikawa T, et al. Primary pulmonary hypertension: 3D dynamic perfusion MRI for quantitative analysis of regional pulmonary perfusion. AJR Am J Roentgenol 2007;188:48–56 [DOI] [PubMed] [Google Scholar]
- 65.Ley S, Kreitner KF, Fink C, Heussel CP, Borst MM, Kauczor HU. Assessment of pulmonary hypertension by CT and MR imaging. Eur Radiol 2004;14:359–68 [DOI] [PubMed] [Google Scholar]