

Abstract
Plant viral expression vectors are advantageous for high-throughput functional characterization studies of genes due to their capability for rapid, high-level transient expression of proteins. We have constructed a series of tobacco mosaic virus (TMV) based vectors that are compatible with Gateway technology to enable rapid assembly of expression constructs and exploitation of ORFeome collections. In addition to the potential of producing recombinant protein at grams per kilogram FW of leaf tissue, these vectors facilitate either N- or C-terminal fusions to a broad series of epitope tag(s) and fluorescent proteins. We demonstrate the utility of these vectors in affinity purification, immunodetection and subcellular localisation studies. We also apply the vectors to characterize protein-protein interactions and demonstrate their utility in screening plant pathogen effectors. Given its broad utility in defining protein properties, this vector series will serve as a useful resource to expedite gene characterization efforts.
With the completion or near completion of genome sequencing of many plant species in the last decade, there is a growing need to functionally characterize the large number of predicted proteins and define their physical interaction networks. Consequently, the requirement for vectors that facilitate high-throughput gene cloning and expression of native proteins or derivatives fused with epitope tags and/or fluorescent proteins has become increasingly important. In plants, protein expression can be achieved either by a transient expression approach or by developing stably transformed lines. Transient expression has emerged as an appealing option compared to stable transgenic approaches due to its significant advantages in terms of short production time lines and low cost.
Virus-based expression systems in plants are particularly attractive versus alternative transient expression systems due to the high level of gene multiplication and concomitant elevated levels of expression achievable within a short period of time while minimizing impairment of host activities1,2. Tobacco mosaic virus (TMV) is one of the most extensively studied plant viruses and has thus become a natural choice for vector development. TMV-based vectors have led to recombinant protein yield as high as 80% of total soluble protein3,4. Agroinfection is inexpensive and reproducible, making it a preferred method of delivering viral expression vectors into plant tissues as part of the T-DNA of a binary vector carried by Agrobacterium tumefaciens5,6. First-generation TMV-derived vectors that utilized the entire virus genome were limited by low agroinfection efficiency and expression capability7. Significant improvements to agroinfection capability has resulted from extensive modification of the TMV genome, including removal of transcript splicing sites, altering codon usage of the RNA-dependent RNA polymerase (RdRP) and introduction of introns within RdRP and movement protein coding sequences8. Expression capability has been enhanced by co-expression of P19, a RNA silencing suppressor gene of Tomato Bushy Stunt Virus, leading to 10–25 fold increases in recombinant protein accumulation9. Additionally, expression capability has been increased multi-fold by positioning the open reading frame (ORF) of the target gene closer to the 3′ end of the TMV RNA10. This has been exploited recently to develop the TRBO (TMV RNA-based overexpression) vector that lacks the coat protein coding sequence, enabling placement of a foreign gene insert close to the 3′ end of the viral RNA11. TRBO vector can produce 100-fold more recombinant protein in plants than a P19-enhanced transient expression system9, and can be easily delivered into plants by agroinfection without the need for co-delivery of a P19 expression construct11. TRBO vector was capable of producing 3.3 to 5.5 g of recombinant GFP/kg of infiltrated Nicotiana benthamiana tissue11. Thus, TRBO vector has great potential for use in high-throughput plant proteomic studies. A major drawback, however, is that it relies on traditional restriction digestion and ligation based cloning for target gene insertion, hampering rapid gene cloning for high throughput functional analysis of proteins. Additionally, the curtailed polylinker region of TRBO vector with only three unique restriction enzyme sites (PacI, NotI, and AvrII) poses another hurdle in devising cloning strategies for large cDNAs, genomic DNA inserts or inserts carrying translational fusions. To overcome the limitations of restriction and ligation-based cloning, a modified version of the TRBO vector was developed12 to exploit ligation-independent cloning (LIC)13; however, an obvious downside of the LIC strategy is that sub-cloning or re-cloning is not efficient due to the inherent need for PCR re-amplification and, thus, sequencing of the inserts after each procedure14. A strict requirement for correct T4 DNA polymerase treatment of the insert and vector is another disadvantage of the LIC method.
The time requirements and limitations of traditional cloning procedures have been significantly reduced through development of the Gateway cloning system15 which employs site-specific recombination to enable inter-molecular transfer of DNA fragments between plasmid vectors. Gateway ‘entry’ clones of full-length ORFs for several organisms are currently available through public repositories and commercial suppliers. Alternatively, they can be assembled by topoisomerase-mediated cloning16 or conventional cloning procedures, both technically amenable due to the generally small size of entry vectors. To complement application of ORFs, the genomic coding regions of genes, encompassing the complete repertoire of potential splice variants thereby enabling broader analysis of gene function than possible using an ORF, can also be rapidly incorporated into the Gateway system using a bacteriophage-based homologous recombination system (recombineering)17. The Gateway vector system has been employed in large-scale plant genomic projects and compatible vectors have been developed for many applications, including ectopic overexpression, promoter-reporter fusions, RNA interference, complementation analysis, gene stacking, and affinity purification using epitope tags17,18,19,20,21. Epitope tagging has proved to be an indispensable way for analyzing protein functions22,23,24 by enabling specific detection of recombinant proteins using commercially available antibodies, thereby eliminating the need for generating antibodies for each protein to be studied. While several non-viral Gateway compatible binary expression vectors incorporating epitope tags have been described in the literature18,20, they are limited by lacking the autonomous replication and high transient expression capability of viral expression vectors. Examples of incorporating Gateway recombination sites into TMV, potato virus X and tobacco rattle virus based expression vectors have been reported25,26,27; however, with the single exception of an N-terminal GFP fusion variant26, the design of these vectors do not support fusion of target proteins with epitope tags or multiple fluorescent proteins, thus limiting their utility in comprehensive functional analysis studies.
Here we describe construction and functional validation of a unique set of viral expression vectors that combine the favourable attributes of the TRBO vector with Gateway technology, epitope-tagging and fluorescent protein-fusion systems to enable rapid and efficient cloning of gene inserts and production of native or recombinant proteins at very high yields within a short time frame. We further demonstrate the utility of these vectors for protein production, affinity purification, immunodetection, subcellular localisation and analysis of protein–protein interactions as well as plant pathogenic effector screening.
Results
Vector design and features
A major caveat with currently available viral expression vectors which depend on traditional in vitro cloning procedures is the inefficiency and laboriousness of cloning ORFs of gene candidates in multiple formats (e.g. epitope-tag variants). To enable rapid cloning of gene inserts and achievement of high level protein expression in a short time, we constructed a set of TMV-based Gateway compatible (TMV-Gate) expression vectors that facilitate production of native as well as epitope- or fluorescent protein-tagged variants (Figure 1). The backbone of these Destination vectors is derived from TRBO vector11 which includes a full-length cDNA clone of TMV U1 strain modified to lack the coat protein (CP) coding sequence and which is placed adjacent to the cauliflower mosaic virus (CaMV) 35S promoter in a T-DNA binary vector. All of the Destination vectors described in this study use the attR1/R2 RfA Gateway cassette which is linked in-frame to various epitope tags and/or fluorescent proteins and is cloned adjacent to the coat protein promoter of the TRBO vector. These assemblies were confirmed by restriction digests (data not shown) followed by DNA sequencing of the junctions between the TRBO vector, RfA cassette and epitope tag/fluorescent proteins.
Figure 1. Schematic illustration of the TMV-Gate vectors.
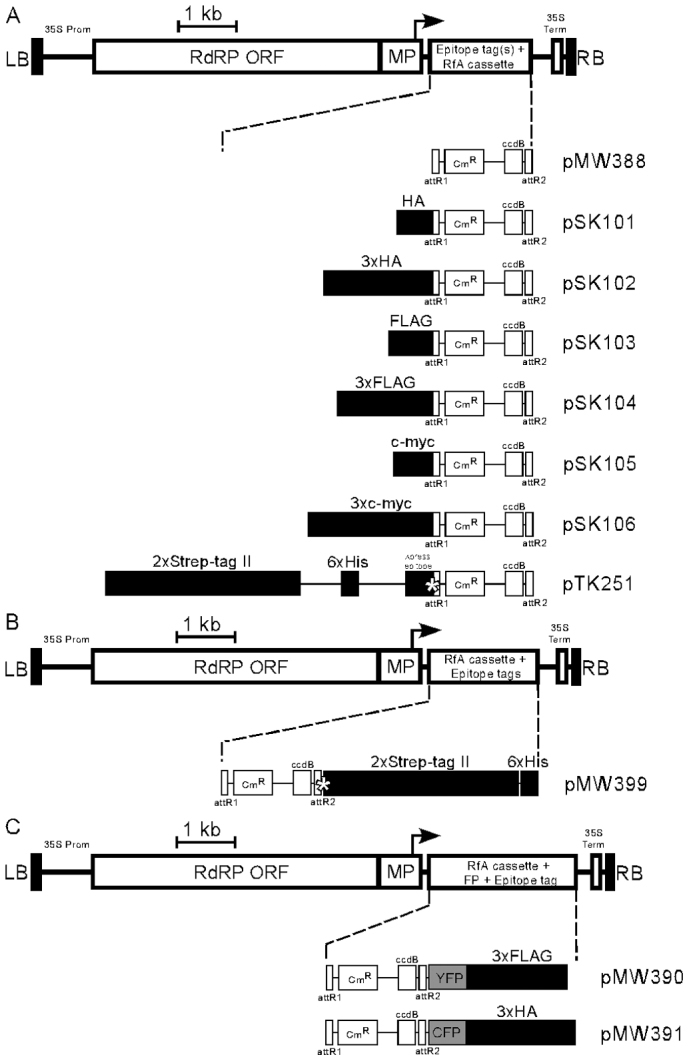
(A) Expression vectors facilitating N-terminal tagging of target protein with various epitope tags, including single- or triple-repeats of HA, FLAG or c-myc, as well as 2xStrep-tag II-6xHis-Xpress epitope. (B) Expression vector facilitating C-terminal tagging of target protein with 2xStrep-tag II-6xHis epitope tags. (C)Expression vectors facilitating C-terminal tagging of target protein with epitope tagged fluorescent proteins, including YFP-3xFLAG or CFP-3xHA. The illustrated vector features include: LB, T-DNA left border; RB, T-DNA right border; 35S Prom, CaMV 35S promoter; 35S term, CaMV 35S terminator; RdRP, RNA-dependent RNA polymerase; MP, movement protein; bent arrow, coat protein promoter; attR1 and attR2, Gateway recombination sites; CmR, chloramphenicol resistance marker; ccdB, E. coli DNA gyrase inhibitor; asterisk in pTK251, enterokinase protease recognition site; asterisk in pMW399, 3C protease recognition site; YFP, yellow fluorescent protein; CFP, cyan fluorescent protein. The epitope tag regions in the map are enlarged 1:20 for readability. DNA sequences for the vectors have been deposited at GenBank with accession numbers as follows: pMW388, JX971627; pSK101, JX971619; pSK102, JX971620; pSK103, JX971621; pSK104, JX971622; pSK105, JX971623; pSK106, JX971624; pTK251, JX971625; pMW399, JX971626; pMW390, JX971628; and pMW391, JX971629.
The structure of the T-DNA region of various TMV-Gate expression vectors is illustrated in Figure 1. In planta expression of the viral replicons in all eleven TMV-Gate vectors is driven by the CaMV 35S promoter (Figure 1). The TMV-Gate vector that allows expression of native proteins is designated as pMW388. TMV-Gate vectors that allow addition of N-terminal epitope tags, such as HA, 3xHA, FLAG, 3xFLAG, c-myc, 3xc-myc and 2xStrep-tag II-6xHis-Xpress epitope tag are designated as pSK101-pSK106 and pTK251, respectively. The TMV-Gate vector that allows addition of a C-terminal 2xStrep-tag II-6xHis epitope tag is designated as pMW399. pTK251 and pMW399 further encode an enterokinase cleavage site or 3C protease site, respectively, that permits proteolytic removal of the epitope tags from the recombinant protein. Another set of TMV-Gate vectors, designated as pMW390 and pMW391, allow generation of C-terminal fusions of target proteins to YFP with a 3xFLAG tag or CFP with 3xHA tag, respectively. All TMV-Gate vectors encode a kanamycin-resistance (KmR) bacterial selectable marker. Conventional pENTR Gateway vectors also utilise a KmR marker which may interfere in selection of recombinant clones following the ‘LR’ Gateway insert transfer reaction. To overcome this problem, the entry clone encoding the gene of interest needs to be linearized by a restriction enzyme that uniquely cleaves the backbone of the vector outside the attL sites prior to performing LR recombination reactions. Alternatively, a derivative of the Gateway Entry vector carrying a different bacterial selectable marker may be used. We have previously described a pENTR2B derivative vector, pJM1 (GenBank: FJ391469) and a pENTR1A derivative, pTK172 (GenBank: FJ410920) in which the KmR marker is inactivated by insertion of functional tetracycline and zeocin resistance genes, respectively17.
In planta evaluation and utility of TMV-Gate vectors for functional analysis of proteins
TRBO vector from which TMV-Gate vectors are derived has several favourable attributes, including high agroinfection rates, high protein expression levels, ease of scale-up and bio-containment11. TMV-Gate vectors should perform very similarly to the TRBO vector since none of the TRBO features have been modified during vector assembly. Functionality of TMV-Gate vectors was evaluated for transient expression of native and epitope-tagged proteins, affinity purification, immunodetection, analysis of protein-protein interaction, subcellular localisation and pathogen effector screening. Additionally, the agroinfectivity and expression capability of a subset of the TMV-Gate vectors were analyzed and compared with that of the TRBO vector. To enable these analyses, entry clones encoding ORFs of reporter genes such as mGFP528, GusPlus and NLS-mCherry as well as AtDMC1 (Arabidopsis thaliana disrupted meiotic cDNA 1), an orthologue of a RecA-like protein in yeast that facilitates meiotic homologous recombination29, and XopD (a type III effector protein from Xanthomonas campestris)30 were recombined into various TMV-Gate vectors as listed in Table 1.
Table 1. Recombinant expression constructs generated using TMV-Gate vectors to evaluate their functionality and utility.
| LR recombination | Recombinant clone | Recombinant protein | |
|---|---|---|---|
| TMV-Gate vector | Entry clone | ||
| For ectopic expression | |||
| pMW388 | pMW350 | pSK133 | mGFP5 |
| pTK251 | pSK6 | pSK154 | 2xStrep-tag II-6xHis-GusPlus |
| For ectopic expression, affinity purification and analysis of protein-protein interactions | |||
| pSK101 | pMW350 | pSK143 | HA-mGFP5 |
| pSK102 | pMW350 | pSK144 | 3xHA-mGFP5 |
| pSK103 | pMW350 | pSK145 | FLAG-mGFP5 |
| pSK104 | pMW350 | pSK146 | 3xFLAG-mGFP5 |
| pSK105 | pMW350 | pSK147 | c-myc-mGFP5 |
| pSK106 | pMW350 | pSK148 | 3xc-myc-mGFP5 |
| pTK251 | pMW350 | pSK149 | 2xStrep-tag II-6xHis-mGFP5 |
| pSK104 | pSW45 | pSK84 | 3xFLAG-GusPlus |
| pSK101 | pTK135 | pSK109 | HA-AtDMC1G138D |
| pSK102 | pTK135 | pSK110 | 3xHA-AtDMC1G138D |
| pSK103 | pTK135 | pSK111 | FLAG-AtDMC1G138D |
| pSK104 | pTK135 | pSK112 | 3xFLAG-AtDMC1G138D |
| pSK105 | pTK135 | pSK113 | c-myc-AtDMC1G138D |
| pSK106 | pTK135 | pSK114 | 3xc-myc-AtDMC1G138D |
| pTK251 | pTK135 | pTK255 | 2xStrep-tag II-6xHis-AtDMC1G138D |
| pMW399 | pMW389 | pMW403 | AtDMC1G138D -2xStrep-tag II-6xHis |
| For analysis of subcellular localisation | |||
| pMW388 | pMK6 | pMW412 | NLS-mCherry |
| pMW390 | pSK212 | pSK215 | NLS-mCherry-YFP-3xFLAG |
| pMW391 | pSK212 | pSK216 | NLS-mCherry-CFP-3xHA |
| For pathogen effector screening | |||
| pSK103 | pSU73 | pSU101 | FLAG-XopD |
Agroinfectivity of TMV-Gate vectors
To test the agroinfectivity of TMV-Gate vectors A. tumefaciens cells carrying a subset of expression constructs encoding native or epitope-tagged mGFP5 (pSK133 and pSK143-149; Table I) or 2xStrep-tag II-6xHis-GusPlus (pSK154; Table I) were infiltrated into N. benthamiana leaves and expression of mGFP5 and GusPlus was monitored for 6 days post infiltration (DPI). Expression of these reporter genes was detected at 3 DPI and increased gradually over the next 2–3 days reaching a maximum at 5–6 DPI. At 5 DPI, most of the cells in the infiltrated leaf area were found to be expressing mGFP5 (Figure 2A) as well as GusPlus (Figure 2B), thus confirming the high agroinfection capability and expression potential of TMV-Gate vectors.
Figure 2. Transient expression of mGFP5 and GusPlus in N. benthamiana leaves by using TMV-Gate vectors.
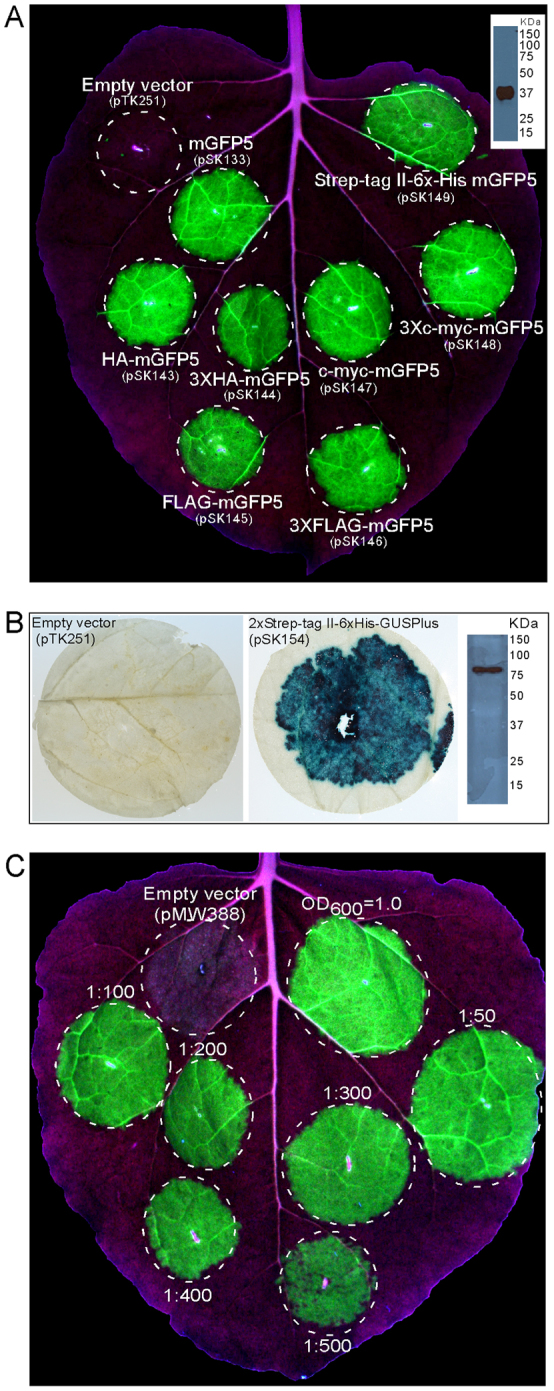
(A) N. benthamiana leaf infiltrated with A. tumefaciens carrying empty vector or viral expression vector encoding mGFP5 with or without translational fusion to either HA, 3xHA, FLAG, 3xFLAG, c-myc, 3xc-myc or 2xStrep-tag II-6xHis-Xpress epitope tag(s) was illuminated under UV light at 5 DPI and photographed. Inset shows immunoblot detection of 2xStrep-tag II-6xHis-Xpress epitope-tagged mGFP5 (in silico predicted size of 37.8 kDa) in crude extract using anti-6xHis antibody. (B) N. benthamiana leaf discs from the area infiltrated with A. tumefaciens carrying empty vector (pTK251) or viral expression vector encoding 2xStrep-tag II-6xHis-GusPlus (pSK154) were excised 5 DPI and stained for GUS activity. Inset in right panel shows immunoblot detection of 2xStrep-tag II-6xHis-Xpress epitope-tagged GusPlus (in silico predicted size of 81.5 kDa) in crude leaf extract using anti-6xHis antibody. (C) N. benthamiana leaf infiltrated with various dilutions of a cell suspension of A. tumefaciens at OD600 of 1.0 carrying pSK133 (pMW388 encoding mGFP5) and photographed with UV illumination at 5 DPI.
The CP gene is required for systemic movement of virus particles31,32. Thus, TMV CP deletion vectors, such as TRBO11 and those developed by ICON Genetics3, cannot form virions and are unable to move systemically in plants, desirable attributes for bio-containment. TMV-Gate replicons share the feature of non-systemic movement as evident from the infiltration zone-restricted expression of mGFP5 and GusPlus (Figure 2). This feature of TMV-Gate vectors thus possesses the desirable attribute of bio-containment by reducing plant to plant transmission and persistence in the environment.
Lindbo11 demonstrated that N. benthamiana leaves can be efficiently agroinoculated with TRBO-GFP replicon over a wide range of A. tumefaciens cell densities (upto 1:300 dilution from a starting optical density at 600 nm (OD600) of 1.0). To determine whether dilute suspensions of TMV-Gate vectors also can be used to efficiently inoculate N. benthamiana leaves, we evaluated various dilute suspensions (upto 1:500 dilution from a starting OD600 of 1.0; Figure 1C) of A. tumefaciens carrying pSK133 (pMW388 encoding mGFP5; Table I) and the expression of mGFP5 was monitored over the next five days. Expression of mGFP5 in all treatments began to be visible at 3 DPI and nearly all cells in the leaf zone infiltrated with A. tumefaciens cell suspension diluted upto 1:400 expressed mGFP5 at 5 DPI, suggesting that, similar to the TRBO vector, the agroinfectivity of TMV-Gate vectors remains high at a wide range of cell densities of inoculum. A low density of A. tumefaciens cells [dilution of 1:500 (Figure 2C) and beyond (data not shown)] resulted in reduced mGFP5 expression, while higher A. tumefaciens cell density (OD600>1.0) occasionally resulted in yellowing and necrotic spots in the infiltration zone (data not shown), probably due to host cell death triggered as a defence mechanism in response to the high titre of inoculum. These results illustrate the inoculum cell density and the length of infection by A. tumefaciens cells carrying TMV-Gate-based expression constructs may be varied to achieve optimal levels of recombinant protein production for different applications.
Expression of native and fusion proteins using TMV-Gate vectors
To determine the protein production efficiency of TMV-Gate vectors, mGFP5 accumulation in N. benthamiana leaves infiltrated with A. tumefaciens cells carrying empty vector (pMW388), TRBO-GFP11, pSK133 (pMW388 encoding mGFP5) and pSK143 (pSK101 encoding HA-tagged mGFP5) was quantified using an ExperionTM automated electrophoresis system. Functionality of the constructs was first confirmed by assessing GFP fluorescence in agroinfiltrated N. benthamiana leaves (Figure 3A). Protein extracts from infiltrated leaf sectors were prepared at 5 DPI and were resolved using an Experion Pro260 protein chip. A virtual gel image of the proteins detectable in each extract is shown in Figure 3B. As expected, the sample from the empty viral replicon extract did not possess a protein representing GFP, whereas distinct bands for GFP (~27 kDa) and HA-GFP (~30 kDa) were seen in protein extracts from N. benthamiana leaves agroinfected with TRBO-GFP or pSK133 and pSK143, respectively. Quantification of resolved proteins indicated that TRBO-GFP, pSK133 and pSK143 produced 4.7, 2.5 and 2.8 mg of GFP g−1 FW, respectively (Figure 3C). This GFP production by TRBO-GFP is in agreement with values (3.3 to 5.5 mg GFP g−1 FW) reported earlier11. It has been previously demonstrated that the level of recombinant protein production increases proportionately with the proximity of the corresponding gene insert to the viral 3′ terminus10,11. Thus, the relatively lower levels of protein produced by TMV-Gate replicons (pSK133 and pSK143) may be due to the incorporation of ~70 bp attR sequence between the GFP coding region and the 3′ terminus of the TMV RNA genome. Nevertheless, the levels of protein produced by the TMV-Gate replicons is much higher than the amount of GFP reported11 to be produced by conventional non-TMV based agroinfiltration vectors (0.03–0.04 mg GFP g−1 FW), full-length TMV genome based vectors (1.5–1.7 mg GFP g−1 FW) or pEAQ series of Cowpea mosaic virus-derived binary vectors (0.7–1.25 mg GFP g−1 FW)33.
Figure 3. Quantitative analysis of transient GFP production in N. benthamiana using TRBO or TMV-Gate vectors.

(A) Fluorescence emitted from GFP transiently produced in a N. benthamiana leaf by infiltration with A. tumefaciens carrying TRBO-GFP (Lindbo, 2007b) or TMV-Gate constructs pSK133 encoding mGFP5, pSK143 encoding HA-mGFP5 or pMW388 (empty vector) and photographed with UV illumination at 5 DPI. (B) Assessment of GFP accumulation by automated capillary electrophoresis analysis. N. benthamiana leaf discs from the area outlined in (A) were excised at 5 DPI and corresponding protein extracts were resolved and GFP identified using the Experion capillary electrophoresis system. Position of the GFP band in each sample is indicated by downward pointing triangles in the simulated gel view produced by the Experion software. The shift in the molecular weight of GFP in pSK143 is due to the presence of an HA tag at the N-terminus. (C) Quantification of GFP resolved by automated capillary electrophoresis analysis using the Experion system. Error bars represent standard deviation. FW, fresh weight.
Affinity purification and immunodetection of epitope-tagged proteins expressed using TMV-Gate vectors
TMV-Gate vectors designed to express epitope-tagged proteins allow addition of single or triple-repeats of several commonly used epitope tags, such as HA, FLAG, c-myc, Strep-tag II, 6xHis, and Xpress epitope, to either the N- or C-terminus of target proteins (Figure 1). To demonstrate the utility of these vectors in expression, detection and purification of recombinant proteins, the ORF of an AtDMC1 variant with a defect in its ATPase motif (AtDMC1G138D; see below) was recombined into TMV-Gate vectors, including pSK101-106, pTK251 and pMW399, resulting in expression constructs pSK109-114, pTK255 and pMW403, respectively (Table I). Leaf tissues encompassing zones agroinfiltrated with these constructs were collected at 5 DPI, total protein extracted and the epitope tagged proteins affinity purified by using either agarose resins possessing epitope tag-specific antibody or Strep-Tactin matrix columns. Immunodetection of various epitope-tagged versions of AtDMC1G138D expressed from TMV-Gate vectors is shown in Figure 4. The anti-HA, -FLAG, -c-myc, -Strep-tag II and -6xHis antibodies effectively recognised both the single- (molecular mass of ~42 kDa) and triple-repeat (molecular mass of ~44 kDa) epitope tagged AtDMC1G138D fusion proteins from the input as well as affinity purified fractions. These results demonstrate the functionality of the epitope and affinity purification modules of the TMV-Gate vectors.
Figure 4. Affinity purification and detection of epitope-tagged AtDMC1G138D transiently expressed in N. benthamiana leaves by using TMV-Gate vectors.

Epitope-tagged recombinant AtDMC1G138D immobilized on anti-HA, anti-FLAG or anti-c-myc antibody-conjugated agarose beads were eluted either by boiling the beads in SDS-PAGE sample buffer (SDS eluate) or by incubating the beads with 3xFLAG peptide (peptide eluate). The Strep-tag II-tagged AtDMC1G138D immobilized on Strep-Tactin matrix was eluted using desthiobiotin. Equal volume aliquots of the input protein extract and eluted AtDMC1G138D proteins were subjected to SDS–PAGE, transferred onto PVDF membrane then subjected to immunoblot analysis. WB, western blot. (A) Immunoblot detection of HA- or 3xHA-tagged AtDMC1G138D protein affinity captured using anti-HA agarose affinity gel. (B) Immunoblot detection of FLAG- or 3xFLAG-tagged AtDMC1G138D protein affinity captured using anti-FLAG agarose affinity gel. (C) Immunoblot detection of c-myc- or 3xmyc-tagged AtDMC1G138D protein affinity captured using anti-c-myc conjugated agarose beads. (D) Immunoblot detection of 2xStrep-tag II-6xHis-Xpress epitope-tagged AtDMC1G138D in crude extract using anti-Strep-tag II antibody. (E) Immunoblot detection of 2xStrep-tag II-6xHis-Xpress epitope-tagged AtDMC1G138D in crude extract using anti-6xHis antibody. (F) Capillary electrophoresis analysis using the Experion system of 2xStrep-tag II-6xHis-Xpress epitope-tagged AtDMC1G138D protein affinity purified by using Strep-Tactin matrix.
Epitope tagged proteins are most commonly eluted from affinity matrices using SDS-PAGE sample buffer. This elution method typically results in denaturation and loss of activity of the purified proteins. Proteins can be eluted under native conditions by competitive elution using epitope tag-specific synthetic peptides or counter ligands. To test this possibility, the single or triple-repeat FLAG-tagged AtDMC1G138D expressed using TMV-Gate vectors and immobilized on anti-FLAG agarose affinity resin was eluted using 3xFLAG peptide. As shown in Figure 4B, the 3xFLAG peptide efficiently eluted the triple-repeat FLAG-tagged AtDMC1G138D; however, as suggested by the peptide manufacturer, it failed to elute the single-repeat FLAG-tagged AtDMC1G138D. Similarly, the 2xStrep-tag II-6xHis–Xpress epitope-tagged AtDMC1G138D immobilized on Strep-Tactin matrix was eluted using desthiobiotin as a counter ligand. Experion-based analysis of the purified AtDMC1G138D revealed the presence of a single band at ~50 KDa (Figure 4F), the predicted size of the fusion protein, indicating high purity of the product. Subsequently, the epitope tags from the purified fusion protein were cleaved off by treatment with enterokinase and the resultant AtDMC1G138D was successfully used for raising antibody in a rabbit (data not shown). Taken together, the results of these experiments establish the suitability of TMV-Gate vectors for expression of recombinant proteins, their immunodetection and affinity purification to obtain native proteins of high purity.
Utility of TMV-Gate vectors for analysis of protein-protein interaction
Protein-protein interactions play a central role in the maintenance and control of cellular and organismal processes. The study of protein-protein interactions has been greatly enhanced by epitope-tagging technology facilitating ‘pull-down’ experiments that can be used to detect and validate protein interactions. In order to establish the utility of epitope-tagging TMV-Gate vectors for analysing protein-protein interactions we employed AtDMC1. The yeast and human DMC1 orthologues have been shown to physically interact homotypically by yeast two-hybrid (Y2H) analysis and, in the case of human DMC1, by immunoprecipitation34. DMC1 DNA strand exchange activity in vitro is dependent upon ATP binding and hydrolysis mediated by an ATPase motif highly conserved between prokaryote and eukaryote RecA-like proteins35, including AtDMC1. Notably, mutating the ATP-binding motif in yeast DMC1 (yDMC1G126D) results in defective meiotic homologous recombination; however, homotypic interactions of yDMC1G126D have not been demonstrated because of instability of the Y2H plasmid expressing this protein34. In characterising AtDMC1 we created a variant (AtDMC1G138D) analogous to yDMC1G126D and sought to assess if it could interact homotypically. Self-interaction of AtDMC1G138D was initially analysed by Y2H assay wherein interaction of the bait and prey fusion proteins results in expression of a HIS3 reporter gene enabling growth of his3 auxotrophic yeast cells on a selective medium lacking histidine. When the AtDMC1G138D bait was expressed with the non-interacting empty prey vector no growth of yeast cells was detected in histidine-deficient plates containing 15 mM 3-amino-1,2,4-triazole (3-AT) which was added to suppress autoactivation of the reporter gene by the AtDMC1G138D bait. In contrast, co-expression of AtDMC1G138D as bait and prey enabled growth of yeast cells, indicating that AtDMC1G138D can interact homotypically (Figure 5A) and, as proposed for yDMC1G126D 34, given the substitution of glycine by the relatively bulky and negatively charged aspartate likely does so independently of ATP binding.
Figure 5. Protein-protein interaction of AtDMC1G138D expressed using TMV-Gate vectors.
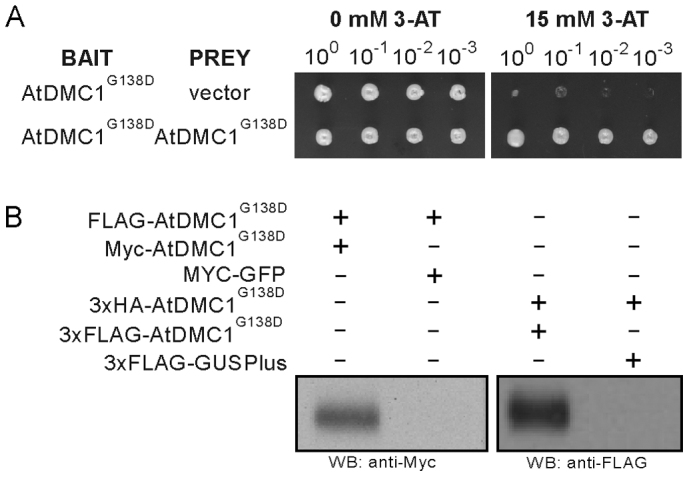
(A) Yeast two-hybrid analysis demonstrating self-interaction of AtDMC1G138D. (B) Confirmation of AtDMC1G138D self-interaction by pull-down experiment. The FLAG-tagged AtDMC1G138D and 3xHA-tagged AtDMC1G138D immobilized on anti-FLAG and anti-HA agarose beads, respectively, were incubated with total protein extract of N. benthamiana leaf discs transiently expressing c-myc-tagged AtDMC1G138D or c-myc-tagged mGFP5 (negative control) and 3xFLAG-tagged AtDMC1G138D or 3xFLAG-tagged GusPlus (negative control), respectively; corresponding TMV-Gate vectors and recombinant plasmids are listed in Table I. After washing the affinity matrices, proteins were eluted using SDS-PAGE sample buffer, resolved by SDS-PAGE and visualized by immunoblotting with antibodies as indicated. WB, western blot.
To validate the results of the Y2H assay, we performed pull-down experiments with various versions of epitope tagged-AtDMC1G138D expressed using TMV-Gate vectors. FLAG-tagged AtDMC1G138D was first immobilized on anti-FLAG agarose beads and then incubated with crude protein extracts containing c-myc-tagged AtDMC1G138D or c-myc-tagged mGFP5 to allow protein complex formation. Western blot analysis of the proteins eluted off the anti-FLAG beads after thorough washing revealed the presence of c-myc-tagged AtDMC1G138D, with expected molecular mass of 42 kDa, but not the c-myc-tagged mGFP5 (Figure 5B, left panel), demonstrating that FLAG-tagged AtDMC1G138D interacted with c-myc-tagged AtDMC1G138D but not with c-myc-tagged mGFP5. In a similar manner, 3xHA-tagged AtDMC1G138D was immobilised on anti-HA antibody conjugated beads and incubated with crude protein extract of 3xFLAG-tagged AtDMC1G138D or 3xFLAG-tagged GusPlus then eluted proteins were probed with anti-FLAG antibody. As predicted, 3xHA-tagged AtDMC1G138D interacted with 3xFLAG-tagged AtDMC1G138D but not with 3xFLAG-tagged GusPlus (negative control; Figure 5B, right panel). These results, apart from confirming the ability of AtDMC1G138D to interact homotypically and validating the results of the Y2H assay, demonstrate the applicability of TMV-Gate vectors for the analysis of protein-protein interactions.
Utility of TMV-Gate vectors for analysis of subcellular localisation of proteins
In recent years, translational fusion of candidate proteins to a fluorescent protein combined with microscopy has become a powerful tool for defining protein function through determining its subcellular localisation. TMV-Gate vectors designed to express C-terminal YFP or CFP fusion proteins include an in-frame 3xFLAG or 3xHA epitope tag, respectively (Figure 1), and thus enable simultaneous analyses, including (i) in planta evaluation of subcellular localisation of fluorescent protein fusions and (ii) immunodetection, affinity purification and analysis of protein-protein interaction by virtue of the epitope tags attached at the C-terminal ends of the fusion proteins. To test the suitability of these vectors for studying subcellular localisation of proteins, the mCherry red fluorescent protein linked to a nuclear localisation signal derived from the SV40 large T-antigen (NLS-mCherry) was recombined with pMW388; additionally, a variant lacking the stop codon (NLS-mCherryΔSTOP) was recombined with the TMV-Gate vectors pMW390 or pMW391 to generate in-frame fusions with YFP-3xFLAG or CFP-3xHA, respectively (Figure 6A). The resultant constructs, designated as pMW412, pSK215 and pSK216, respectively (Table I), were agroinfiltrated into N. benthamiana leaves and subcellular localisation of the fusion proteins assessed by fluorescent microscopy. The subcellular fluorescence signal emitted by NLS-mCherry overlapped with the DAPI (4′,6-diamidino-2-phenylindole; a nuclear stain) signal (Figure 6B), demonstrating that NLS-mCherry localizes to the nucleus and can be utilized as a positive control in nuclear localisation experiments. As expected, both NLS-mCherry-YFP-3xFLAG and NLS-mCherry-CFP-3xHA fusion proteins localized to the nucleus, as deduced by comparing the fluorescence signal with the DAPI signal (Figure 6B). These results demonstrate pMW390 and pMW391 TMV-Gate vectors can be utilized for detection of subcellular localisation of proteins in planta.
Figure 6. Nuclear localizsation of NLS-mCherry expressed using TMV-Gate vectors.
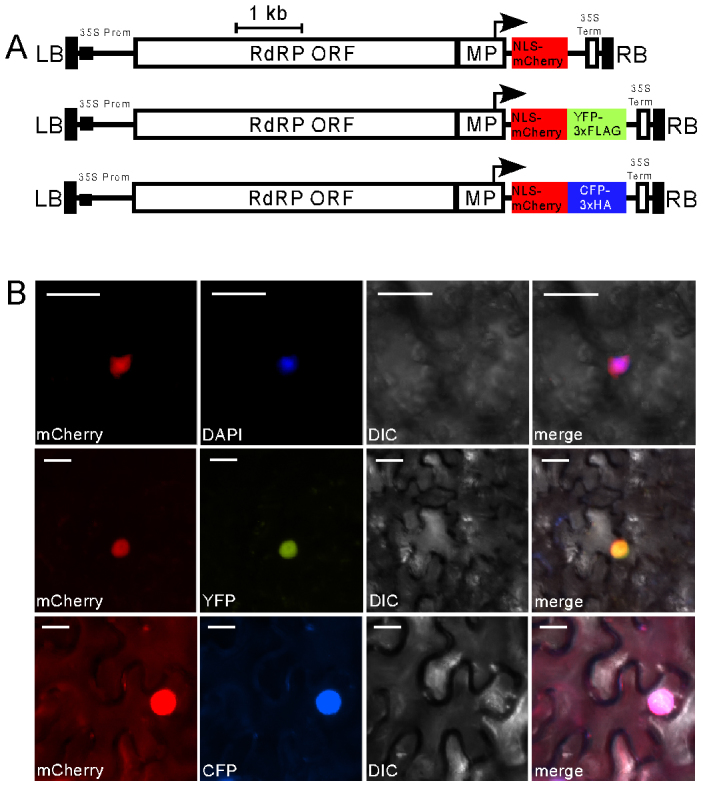
(A) Schematic diagram showing constructs used for analysis of subcellular localisation of NLS-mCherry. mCherry carrying a nuclear localization signal (NLS) was introduced unmodified into pMW388. A NLS-mCherry variant without translational stop codon was linked in-frame with the coding sequence of YFP-3xFLAG or CFP-3xHA in pMW390 and pMW391, respectively. Annotated features as per Figure 1. (B) Epifluorescent microscopy images showing nuclear localisation of NLS-mCherry (upper panel), NLS-mCherry linked to YFP-3xFLAG (middle panel) or NLS-mCherry linked to CFP-3xHA in cells of N. benthamiana leaves infiltrated with A. tumefaciens carrying the constructs described in (A) and imaged 3 DPI. The nucleus was detected by staining cells with DAPI. Images were taken using YFP, CFP, mCherry or DAPI filter sets as indicated. Scale bar, 20 µM. DIC, differential interference contrast.
Utility of TMV-Gate vectors in pathogen effector protein screening
Effectors are pathogen-derived molecules that manipulate host cell structure and function, thereby facilitating infection and/or triggering disease resistance, culminating in a hypersensitive response (HR)-based programmed cell death36,37. In recent years, the combination of genome sequencing of plant pathogens with computational data mining has dramatically accelerated the identification of effector candidates. Developing high-throughput vector platforms for functional screening of candidate effectors thus would provide an important complement to the bioinformatics approach of identifying effectors. Ectopic expression of effector proteins in plant cells often leads to phenotypic effects, such as induction of HR-like necrosis38. To test the suitability of TMV-Gate vectors for the expression and functional screening of effector proteins we selected XopD which has been shown to elicit HR-like tissue chlorosis and necrosis symptoms in N. benthamiana39. The ORF of XopD was recombined in-frame with the FLAG epitope tag of pSK103, resulting in pSU101. This and pSK145 encoding mGFP were agroinfiltrated into N. benthamiana leaves at 1:5 dilution from the initial OD600 of 1.0 (optimised to overcome the necrosis potentially induced by A. tumefaciens). Transient expression of FLAG-XopD resulted in localized tissue chlorosis by 5–6 DPI and necrosis by 9–10 DPI (Figure 7). In contrast, transient expression of FLAG-mGFP5 did not elicit such symptoms. These data, in agreement with a previous report by Kim et al.39, illustrate that XopD is capable of eliciting an HR response in N. benthamiana and also confirm the suitability of TMV-Gate vectors for effector screening.
Figure 7. TMV-Gate vector based expression of XopD effector protein in N. benthamiana leaves.

N. benthamiana leaves were infiltrated with A. tumefaciens strains expressing FLAG-tagged XopD (pSU101) or mGFP5 (negative control; pSK145) and the phenotypes were photographed at 2, 4, 6, 8, 10 and 12 DPI.
Discussion
In this study, we describe construction and validation of a viral vector system for expedited production and characterisation of recombinant proteins via transient expression in plant leaves. The vectors are derived from the TRBO variant of TMV previously reported11 as enabling high-level production of recombinant proteins but which is severely constrained in practical applications by requiring reliance on traditional DNA cloning procedures. The vectors described in this study are unique from other plant virus vectors integrating Gateway technology25,26,27 by further incorporating a very broad series of epitope, affinity purification and fluorescent protein tags to more readily exploit the attributes of TMV for rapid production and analysis of protein attributes regarding their biochemical and cell biology properties. The compatibility of this expression system with Gateway technology enhances its suitability for high-throughput functional analysis studies. Gateway technology has been widely adapted by the research community to facilitate high-throughput proteomic research. We anticipate that this vector system will complement the on-going and future efforts to clone and functionally characterize ORFeomes using Gateway pENTR vectors40.
We demonstrate the utility of the vector system through examples of protein-protein interaction analysis and protein sub-cellular localisation. Effective protein expression using the system was demonstrated by immunoblot analysis of AtDMC1 and GFP with eight different tags or GusPlus with 2xStrep-tag II-6xHis tag revealing no truncated recombinant proteins; furthermore, in all of these cases the proteins were extracted without a denaturation step, collectively supporting their accumulation as intact soluble cellular constituents and not as truncated proteins or insoluble aggregates. Because of the capability to generate grams of recombinant protein per kg of fresh weight biomass within a short time frame, this vector set has great potential to be utilized in large scale protein production applications. Additionally, the mid- to high-level of protein production inherent to TMV-Gate vectors combined with the flexibility of modulating protein accumulation through altering A. tumefaciens cell density and length of infection has utility in experiments involving toxic proteins which when expressed at very high levels may confer detrimental effects on host cells. Recombinant proteins produced using this system can be easily separated from cellular material using anti-epitope tag affinity matrices and be readily detected by either using anti-epitope tag antibodies or by fluorescence microscopy. Such capabilities can be useful for functional genomic studies by defining protein properties, including analysis of protein-protein interactions and subcellular localisation. Functionality of the system for subcellular localisation studies was illustrated using NLS-mCherry which maintained its ability to localise to the nucleus when expressed in fusion with fluorescent proteins. Similarly, AtSAP18 (At2g45640), an established corepressor involved in chromatin remodelling and previously demonstrated to localize to the nucleus41, when expressed in fusion with CFP using pMW391 reproducibly localised to the nucleus (Supplementary Figure S1), thereby further validating the utility of the TMV-Gate vectors in cell biology applications. It is evident from these results that TMV-Gate driven high-level expression of proteins does not affect their integrity, solubility or localisation. To-date, this system has been used to successfully express 45 native or fusion proteins, including: fluorescent protein, GusPlus, AtDMC1 and XopD variants as listed in Table I (with insert sizes ranging from 741–2358 bp); four transcriptional regulators (654–939 bp; data not shown); a corepressor involved in chromatin remodelling (1422 bp; data not shown); and 17 pathogen effectors (234–1335 bps; data not shown) from Albugo candida42. In the past, several viral-based expression vector systems have been utilized for functional screening of effector proteins43,44,45; however, TMV-Gate vectors, by virtue of their ability to produce native as well as various epitope- or fluorescent protein-tagged recombinant proteins, provide a more complete and integrated platform for functional evaluation of effector candidates in N. benthamiana. Collectively, this illustrates the robustness and broad utility of the system.
In summary, the vector system described here allows rapid and versatile assembly of expression constructs for production of intact, soluble and functionally active recombinant proteins at a very high yield within a short time-frame and overcomes some limitations of prokaryotic expression systems, thereby providing an invaluable tool to expedite plant functional genomics and proteomics research. While this study employs plant proteins as examples, it is expected the vector system will be attractive for applications with proteins from animal or other species given the combination of tags incorporated and the potential for rapid, high-level production of heterologous proteins.
Methods
Plant material
N. benthamiana plants were grown for 4–6 weeks in a greenhouse at ~25°C on a 16 h light and 8 h dark cycle before infiltration with A. tumefaciens cultures carrying various expression constructs.
Construction of Destination vectors
Cloning techniques were performed using standard protocols46. Nucleotide sequences of the primers (Integrated DNA Technologies) used in this study are listed in Supplementary Table S1. Resynthesised DNA cassettes were obtained from the National Research Council (Saskatoon, SK). Gateway reagents and vectors were obtained from Invitrogen. PCR amplifications were performed using PicoMaxx polymerase (Stratagene). All destination vector assemblies as described below were transformed into One Shot ccdB Survival 2 T1 Phage-Resistant Escherichia coli cells (Invitrogen), and positive transformants were selected in the presence of kanamycin (50 mg/L) and chloramphenicol (30 mg/L).
Vectors encoding RfA-cassette with or without N-terminal epitope tag(s)
The RfA-cassette encoding a chloramphenicol resistance marker and ccdB flanked by attR1 and attR2 was amplified by PCR using primers Dest-5′-SnaMfePac and Dest-3′-SphPspAvr. Coding sequences for the RfA-cassette linked to the HA (YPYDVPDYA) or 3xHA (YPYDVPDYAGYPYDVPDYAGSYPYDVPDYA) epitope tags were amplified by PCR from pEarleyGate20120 using sense primers pEG201-5′-BspPac or pEG201-5′-BspPac+2xHA, respectively, and a common antisense primer DEST-3′AvrPsm. Similarly, the FLAG (DYKDDDDK) or 3xFLAG (DYKDDDDKGDYKDDDDKGDYKDDDDK) tags linked to the RfA-cassette were amplified from pEarleyGate20220 using the primers pEG202-5′-BspPac or pEG202-5′-BspPac+2xFLAG, respectively, as sense primers and the common antisense primer DEST-3′AvrPsm. To amplify c-myc (EQKLISEEDLN) or 3xc-myc (EQKLISEEDLNGEQKLISEEDLNGEQKLISEEDL) tags linked to the RfA-cassette, pEarleyGate20320 was used as template and the primers pEG203-5′-BspPac or pEG203-5′-BspPac+2xMyc, respectively, were used as sense primers along with the common antisense primer DEST-3′AvrPsm. The 2xStrep-tag II-6xHis-Xpress epitope-linked RfA-cassette was amplified using as template pTK249, a pEXP1-Dest (Invtirogen) derivative encoding a 2xStrep-tag II-6xHis-Xpress epitope-linked RfA-cassette (see Supplementary Methods), and primers Strep-5′-PciNdePac and Exp1-Dest-3′-PspAvr. The PCR amplicons (~1.8 kb) encoding the RfA-cassettes with or without epitope tags from the above PCR reactions were digested with PacI and AvrII then ligated into the TRBO vector digested with the same enzymes resulting in the respective TMV-Gate vectors pMW388, pSK101, pSK102, pSK103, pSK104, pSK105, pSK106 and pTK251, as depicted in Figure 1.
Vector encoding RfA-cassette with C-terminal epitope tag
The 2xStrep-tag II-6xHis epitope assembly at the 3′ end of the RfA cassette in pMW394, a pEXP2-Dest (Invitrogen) derivative (see Supplementary Methods), was amplified by PCR using the primers EXP2-Dest-5′-XhoSnaPac and EXP2-Dest-3′-SacPspAvr. The 1.95 kb amplicon was digested with PacI and AvrII then ligated into the TRBO vector digested with the same enzymes, resulting in pMW399 (Figure 1).
Vectors encoding RfA-cassette with C-terminal epitope-tagged fluorescent protein
Plasmids pMW397 and pMW396 encoding assemblies of the RfA-cassette linked to C-terminal YFP or CFP and 3x concatamers of the FLAG or HA tag, respectively, were derived from pEarleyGate101 and pEarleyGate10220 (see Supplementary Methods). The RfA-YFP-3xFLAG and RfA-CFP-3xHA cassettes were isolated by digestion of pMW397 and pMW396 (Supplementary Methods), respectively, with PacI and AvrII then ligated into the TRBO vector digested with the same enzymes, resulting in pMW390 and pMW391 (Figure 1).
Gateway Entry clones
The Gateway Entry clones pMK6, pMW350, pSK6, pSW45, pSU73 and pTK135 (Table I) were assembled as described in the Supporting Information (see Supplementary Methods). These plasmids were digested with PvuI, uniquely cutting the KmR gene, gel-purified and recombined with Destination vectors pSK101-106, pTK251, pMW388, pMW390-391, or pMW399 (Table I) using LR clonase II Invitrogen.
Agroinfection
Expression constructs (Table I) encoding mGFP5, GusPlus, AtDMC1G138D, NLS-mCherry and XopD were introduced into A. tumefaciens GV3101::pMP9047 by electroporation. Agroinfection of N. benthamiana leaves was carried out as described previously11 and summarised in the Supporting Information (see Supplementary Methods).
Visualization of mGFP5 and GusPlus activity
Five days after infiltration of N. benthamiana leaves with A. tumefaciens carrying various TMV-Gate expression vectors encoding GFP and GusPlus reporter genes, leaf material encompassing the infiltrated area was excised. Leaves expressing GFP were photographed under UV illumination generated by a transilluminator (VWR Scientific). GusPlus activity was visualized by histochemical staining48 with overnight incubation at 37°C.
Yeast two-hybrid assay
The GAL4-based ProQuest (Invitrogen) Y2H system was used. To assemble the bait and prey constructs, the ORF encoding AtDMC1G138D was transferred from pTK135 to pDEST32 (bait vector) and pDEST22 (prey vector) using the LR clonase reaction, resulting in pTK149 and pTK145, respectively. Yeast strain MaV203 (Invitrogen) was transformed with plasmid combinations listed in Figure 5A according to the method of Gietz and Woods49. Assays were conducted on plates containing minimal medium lacking leucine, tryptophan and histidine supplemented with 15 mM 3-AT (Sigma-Aldrich).
Extraction of leaf proteins
Proteins from agroinfected N. benthamiana leaves were extracted by grinding tissue in the presence of liquid nitrogen then suspending in 4–5 volumes of extraction buffer [50 mM Tris HCl, pH 8.0, 150 mM NaCl, 1 mM EDTA, 1% Triton-X-100, 1 mM PMSF and 1x plant protease inhibitor mix (Sigma-Aldrich)]. The homogenate was centrifuged twice at 14,000 rpm for 15 min at 4 °C. Protein concentration was determined by the Bradford method50 (BioRad).
Experion automated electrophoresis analysis
Samples for Experion Pro260 (BioRad) analysis were prepared by mixing 2 µl of sample buffer with 4 µl samples of protein extracts then loaded onto chips as recommended by the manufacturer. Electrophoresis runs were analyzed using the Experion software (BioRad) as summarised in Supporting Information (Supplementary Methods).
Affinity purification and pull-down experiment
Protein extracts prepared as described above were either incubated with 50 µL (bed volume) of pre-washed EZview red agarose beads (Sigma-Aldrich) conjugated with anti-HA, anti-FLAG or anti-c-myc antibodies, or passed through Strep-Tactin matrix (IBA GmbH). Bound protein was then washed three times with extraction buffer. Tagged proteins affinity captured on the beads were eluted either by boiling the beads in SDS-PAGE sample buffer or, in the case of FLAG-based purification, by mixing with antibody-specific peptide (3xFLAG peptide; Sigma-Aldrich) or for the Strep-Tactin matrix eluted by addition of 2.5 mM desthiobiotin. Eluted proteins were analysed by SDS-PAGE followed by immunoblotting using antibodies specific to each of the epitope tags (see Supplementary Methods).
For pull-down experiments, FLAG or 3xHA-tagged AtDMC1G138D fusions immobilized on anti-FLAG or anti-HA-antibody-conjugated agarose beads, respectively, were washed three times with 500 µL of extraction buffer, then incubated for ~90 min with total protein extract prepared from N. benthamiana leaves expressing c-myc- or 3xFLAG-tagged AtDMC1G138D, mGFP5 (negative control) or GusPlus (negative control). The beads were then washed three times with 500 µL of extraction buffer. Proteins retained on the beads were eluted using SDS-PAGE sample buffer and analyzed by SDS–PAGE followed by immunoblotting with anti-c-myc or anti-FLAG antibodies (see Supplementary Methods).
Epifluorescence microscopy
Epifluorescent images of leaf discs expressing NLS-mCherry, NLS-mCherry-YFP-3xFLAG and NLS-mCherry-CFP-3xHA were obtained by using a Zeiss Axio Imager.Z1 microscope equipped with an EC Plan-Neofluar 20X/0.50 M27 objective and an AxioCam HRm camera and using AxioVision software (Carl Zeiss Canada Ltd.). The light source was a broad spectrum X-cite 120 series mercury bulb (Lumen Dynamics Group Inc.). Nuclei were detected by staining with 4′,6-diamidino-2-phenylindole (DAPI). Filter sets (Carl Zeiss Canada Ltd.) include HE DsRed (excitation: 538–562; emission: 570–640), YFP (excitation: 490-510; emission: 520–550), HE Cyan (excitation: 424–448; emission: 460–500) and DAPI (excitation: 335–383; emission: 420–470). Exposure times were between 0.2 to 0.9 seconds.
Author Contributions
KR conceived the study. KR and SK designed and coordinated the study, and wrote the manuscript. SK developed and established protocols, and assembled constructs in concert with MW and TB. SK performed transient expression, purification and characterization of recombinant protein experiments. WY performed Y2H experiments. SU and MHB designed and performed pathogen effector screening experiments. All authors read and approved the final manuscript.
Supplementary Material
SUPPLEMNTRAY INFORMATION
Acknowledgments
We thank J. Lindbo for providing the TRBO vector and TRBO-GFP construct and R. Tsien for providing mCherry. We also thank Hyun-hye Kim for technical assistance. Milosz Kaczmarek and Song Wang contributed to assembly of NLS-mCherry and the GusPlus ORF, respectively. GusPlus was provided by CAMBIA. pEarleyGate vectors were obtained from the Arabidopsis Biological Resource Centre. This work was supported by funding from the Canadian Crop Genomics Initiative of Agriculture and Agri-Food Canada provided to K.R.
References
- Scholthof H. B., Scholthof K. B. & Jackson A. O. Plant virus gene vectors for transient expression of foreign proteins in plants. Annu Rev Phytopathol 34, 299–323 (1996). [DOI] [PubMed] [Google Scholar]
- Turpen T. H. Tobacco mosaic virus and the virescence of biotechnology. Philos Trans R Soc Lond B Biol Sci 354, 665–673 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marillonnet S. et al. In planta engineering of viral RNA replicons: efficient assembly by recombination of DNA modules delivered by Agrobacterium. Proc Natl Acad Sci USA 101, 6852–6857 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusibov V., Shivprasad S., Turpen T. H., Dawson W. & Koprowski H. Plant viral vectors based on tobamoviruses. Curr Top Microbiol Immunol 240, 81–94 (1999). [DOI] [PubMed] [Google Scholar]
- Grimsley N., Hohn B., Hohn T. & Walden R. “Agroinfection,” an alternative route for viral infection of plants by using the Ti plasmid. Proc Natl Acad Sci USA 83, 3282–3286 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiser R. M. et al. Agroinfection as an alternative to insects for infecting plants with beet western yellows luteovirus. Proc Natl Acad Sci USA 89, 9136–9140 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gleba Y., Klimyuk V. & Marillonnet S. Viral vectors for the expression of proteins in plants. Curr. Opin. Biotechnol. 18, 134–141 (2007). [DOI] [PubMed] [Google Scholar]
- Marillonnet S., Thoeringer C., Kandzia R., Klimyuk V. & Gleba Y. Systemic Agrobacterium tumefaciens-mediated transfection of viral replicons for efficient transient expression in plants. Nat Biotechnol 23, 718–723 (2005). [DOI] [PubMed] [Google Scholar]
- Lindbo J. A. High-efficiency protein expression in plants from agroinfection-compatible Tobacco mosaic virus expression vectors. BMC Biotechnol 7, 52 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Culver J. N., Lehto K., Close S. M., Hilf M. E. & Dawson W. O. Genomic position affects the expression of tobacco mosaic virus movement and coat protein genes. Proc Natl Acad Sci USA 90, 2055–2059 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindbo J. A. TRBO: a high-efficiency tobacco mosaic virus RNA-based overexpression vector. Plant Physiol 145, 1232–1240 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh S. K., Kim S. B., Yeom S. I., Lee H. A. & Choi D. Positive-selection and ligation-independent cloning vectors for large scale in planta expression for plant functional genomics. Mol Cells 30, 557–562 (2010). [DOI] [PubMed] [Google Scholar]
- Aslanidis C. & de Jong P. J. Ligation-independent cloning of PCR products (LIC-PCR). Nucleic Acids Res 18, 6069–6074 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Rybel B. et al. A versatile set of ligation-independent cloning vectors for functional studies in plants. Plant Physiol 156, 1292–1299 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartley J. L., Temple G. F. & Brasch M. A. DNA cloning using in vitro site-specific recombination. Genome Res 10, 1788–1795 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuman S. Novel approach to molecular cloning and polynucleotide synthesis using vaccinia DNA topoisomerase. J Biol Chem 269, 32678–32684 (1994). [PubMed] [Google Scholar]
- Rozwadowski K. L., Yang W. & Kagale S. Homologous recombination-mediated cloning and manipulation of genomic DNA regions using Gateway and recombineering systems. BMC Biotechnol 8, 88 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis M. D. & Grossniklaus U. A gateway cloning vector set for high-throughput functional analysis of genes in planta. Plant Physiol 133, 462–469 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin M. J., Bowler C. & Benvenuto G. A modified Gateway cloning strategy for overexpressing tagged proteins in plants. Plant Methods 4, 3 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Earley K. W. et al. Gateway-compatible vectors for plant functional genomics and proteomics. Plant J 45, 616–629 (2006). [DOI] [PubMed] [Google Scholar]
- Karimi M., Depicker A. & Hilson P. Recombinational cloning with plant gateway vectors. Plant Physiol 145, 1144–1154 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brizzard B. Epitope tagging. Biotechniques 44, 693–695 (2008). [DOI] [PubMed] [Google Scholar]
- Jarvik J. W. & Telmer C. A. Epitope tagging. Annu Rev Genet 32, 601–618 (1998). [DOI] [PubMed] [Google Scholar]
- Terpe K. Overview of tag protein fusions: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol 60, 523–533 (2003). [DOI] [PubMed] [Google Scholar]
- Liu Y., Schiff M. & Dinesh-Kumar S. P. Virus-induced gene silencing in tomato. Plant J 31, 777–786 (2002). [DOI] [PubMed] [Google Scholar]
- Lacorte C., Ribeiro S. G., Lohuis D., Goldbach R. & Prins M. Potato virus X and Tobacco mosaic virus-based vectors compatible with the Gateway™ cloning system. J Virol Methods 164, 7–13 (2010). [DOI] [PubMed] [Google Scholar]
- Avesani L. et al. Stability of Potato virus X expression vectors is related to insert size: implications for replication models and risk assessment. Transgenic Res 16, 587–597 (2007). [DOI] [PubMed] [Google Scholar]
- Siemering K. R., Golbik R., Sever R. & Haseloff J. Mutations that suppress the thermosensitivity of green fluorescent protein. Curr Biol 6, 1653–1663 (1996). [DOI] [PubMed] [Google Scholar]
- Bishop D. K., Park D., Xu L. & Kleckner N. DMC1: a meiosis-specific yeast homolog of E. coli recA required for recombination, synaptonemal complex formation, and cell cycle progression. Cell 69, 439–456 (1992). [DOI] [PubMed] [Google Scholar]
- Noel L., Thieme F., Nennstiel D. & Bonas U. Two novel type III-secreted proteins of Xanthomonas campestris pv. vesicatoria are encoded within the hrp pathogenicity island. J Bacteriol 184, 1340–1348 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donson J., Kearney C. M., Hilf M. E. & Dawson W. O. Systemic expression of a bacterial gene by a tobacco mosaic virus-based vector. Proc Natl Acad Sci USA 88, 7204–7208 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson W. O., Bubrick P. & Grantham G. L. Modifications of the tobacco mosaic virus coat protein gene affecting replication, movement, and symptomatology. Phytopathology 78, 783–789 (1988). [Google Scholar]
- Sainsbury F., Thuenemann E. C. & Lomonossoff G. P. pEAQ: versatile expression vectors for easy and quick transient expression of heterologous proteins in plants. Plant Biotechnol J 7, 682–693 (2009). [DOI] [PubMed] [Google Scholar]
- Dresser M. E. et al. DMC1 functions in a Saccharomyces cerevisiae meiotic pathway that is largely independent of the RAD51 pathway. Genetics 147, 533–544 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyman C. & Kanaar R. Homologous recombination: down to the wire. Curr Biol 14, R629–631 (2004). [DOI] [PubMed] [Google Scholar]
- Dangl J. L. & Jones J. D. Plant pathogens and integrated defence responses to infection. Nature 411, 826–833 (2001). [DOI] [PubMed] [Google Scholar]
- Kamoun S. A catalogue of the effector secretome of plant pathogenic oomycetes. Annu Rev Phytopathol 44, 41–60 (2006). [DOI] [PubMed] [Google Scholar]
- Kjemtrup S., Nimchuk Z. & Dangl J. L. Effector proteins of phytopathogenic bacteria: bifunctional signals in virulence and host recognition. Curr Opin Microbiol 3, 73–78 (2000). [DOI] [PubMed] [Google Scholar]
- Kim J. G. et al. XopD SUMO protease affects host transcription, promotes pathogen growth, and delays symptom development in xanthomonas-infected tomato leaves. Plant Cell 20, 1915–1929 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong W. et al. Genome-wide ORFeome cloning and analysis of Arabidopsis transcription factor genes. Plant Physiol 135, 773–782 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pendle A. F. et al. Proteomic analysis of the Arabidopsis nucleolus suggests novel nucleolar functions. Mol Biol Cell 16, 260–269 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Links M. G. et al. De novo sequence assembly of Albugo candida reveals a small genome relative to other biotrophic oomycetes. BMC Genomics 12, 503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones L. et al. RNA-DNA interactions and DNA methylation in post-transcriptional gene silencing. Plant Cell 11, 2291–2301 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vleeshouwers V. G. et al. Agroinfection-based high-throughput screening reveals specific recognition of INF elicitins in Solanum. Mol Plant Pathol 7, 499–510 (2006). [DOI] [PubMed] [Google Scholar]
- Ratcliff F., Martin-Hernandez A. M. & Baulcombe D. C. Tobacco rattle virus as a vector for analysis of gene function by silencing. Plant J 25, 237–245 (2001). [DOI] [PubMed] [Google Scholar]
- Sambrook J. & Russell D. W. Molecular Cloning: A Laboratory Manual. (Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press) (2001). [Google Scholar]
- Koncz C. & Schell J. The promoter of TL-DNA gene 5 controls the tissue-specific expression of chimaeric genes carried by a novel type of Agrobacterium binary vector. Mol Gen Genet 204, 383–396 (1986). [Google Scholar]
- Jefferson R. A., Kavanagh T. A. & Bevan M. W. GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz R. D. & Woods R. A. Yeast transformation by the LiAc/SS Carrier DNA/PEG method. Methods Mol Biol 313, 107–120 (2006). [DOI] [PubMed] [Google Scholar]
- Bradford M. M. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72, 248–254 (1976). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPLEMNTRAY INFORMATION