

Abstract
The nervous system develops through a program that first produces neurons in excess and then eliminates as many as half in a specific period of early postnatal life. Neurotrophins are widely thought to regulate neuronal survival, but this role has not been clearly defined in the CNS. Here we show that neurotrophins promote survival of young neurons by promoting spontaneous activity. Survival of hippocampal neurons in neonatal rat requires spontaneous activity that depends on the excitatory action of GABA. Neurotrophins facilitate recruitment of cultured neurons into active networks, and it is this activity, combined with integrin receptor signaling, that controls neuronal survival. In vivo, neurotrophins require integrin signaling to control neuron number. These data are the first to link the early excitatory action of GABA to the developmental death period and to assign an essential role for activity in neurotrophin-mediated survival that establishes appropriate networks.
Introduction
The importance of cell death in the development of the nervous system was revealed by pioneering work in the periphery showing that the size of the target (muscle) regulates the number of motor neurons. Nerve growth factor (NGF) was shown to control neuronal differentiation and survival in the PNS (Levi-Montalcini, 1987). These findings led to the influential idea that neurons are produced in excess, and that an activity-dependent competition for a limited supply of factors regulates neuron numbers. NGF forms a gene family with BDNF, neurotrophin-3 (NT3), and neurotrophin-4/5 (Barde, 1994; Bibel and Barde, 2000). However, in contrast to dramatic effects observed in the PNS, deleting the genes that encode neurotrophins or their receptors produces only minor effects on neuron number in the CNS (Jones et al., 1994; Minichiello and Klein, 1996; Alcántara et al., 1997; Rauskolb et al., 2010). As a consequence, no clear model has been developed to explain the survival of CNS neurons.
The hippocampus is widely studied because it plays an important role in memory formation (Cooke and Bliss, 2006). As in the other regions of CNS, death of hippocampal neurons occurs during the first postnatal week (Gould et al., 1991; Ferrer et al., 1994). The neonatal hippocampus displays spontaneous synchronous activity mediated by depolarizing GABA, and this activity is thought to be important for the maturation of functional networks (Ben-Ari et al., 1997). However, whether or how such neonatal activity is linked to neuronal survival has not been tested.
Neurotrophins activate signaling pathways through the tropomyosin receptor kinase (Trk), and retrograde transport of the signaling endosome containing internalized Trk with the ligand is thought to control neuronal survival (Zweifel et al., 2005). However, neurons receive multiple extracellular survival signals. Integrins, which consist of heterodimers (α and β) and interact with extracellular matrix, regulate various cell functions including survival (Hynes, 2002). Integrins play critical roles in the survival of developing and adult hippocampal neurons (Gary et al., 2003; Wakselman et al., 2008), as well as in the migration of neurons (Stanco et al., 2009) and maturation of synapses (Chavis and Westbrook, 2001). It remains unclear whether and how integrin signals are integrated with other extracellular cues and activity to control the number of neurons during the period of developmental neuronal death.
Here we show that GABA-mediated spontaneous activity regulates neuronal death in the neonatal hippocampus. In vitro, neuron number is reduced during a brief period when GABA-dependent calcium responses develop in the neurons that preferentially survive. Specifically during this period, neurotrophins regulate neuron numbers indirectly by promoting this GABA-mediated spontaneous activity. Neurotrophins transiently activate Trk receptors to promote survival through the sustained activation of the serine-threonine kinase Akt. This sustained activation of Akt requires both neuronal activity and integrin signaling. Acute pharmacological manipulations in vivo support a central role for integrins in neurotrophin-mediated survival of neonatal hippocampal neurons. These data define neuronal activity and integrin signaling as the direct mediators of neurotrophin-activated survival mechanisms. Our findings have implications for understanding the development of hippocampal networks.
Materials and Methods
Reagents.
dl-2-Amino-5-phosphonovaleric acid (APV), bicuculline, tetrodotoxin (TTX), fluorouridine (FUDR), apotransferrin, putrescin, sodium selenite, progesterone, corticosterone, triiodothyronine, insulin, and echistatin were purchased from Sigma-Aldrich. LY294002 [2-(4-morpholinyl)-8-phenyl-4H-1-benzopyran-4-one] and bromodeoxyuridine (BrdU) were purchased from Roche. Recombinant human (rh) BDNF and rhNT3 were purchased from R&D Systems. Modified MEM, GlutaMAX, and penicillin/streptomycin were purchased from Invitrogen.
Antibodies.
Antibodies were used with the following dilutions: monoclonal mouse anti-MAP2 (Sigma-Aldrich), 1:500; polyclonal rabbit anti-GABA, anti-c-cas3 (anti-cleaved caspase 3) (Cell Signaling Technology), 1:500; polyclonal goat anti-TrkB, anti-TrkC (R&D Systems), 1:40 (concentration for function blocking recommended by the manufacturer); monoclonal rat anti-BrdU (Accurate Chemical and Scientific), 1:200; monoclonal mouse anti-NeuN (Millipore); polyclonal rabbit anti-phospho Thr-308 Akt, anti-phospho Ser-473 Akt for Western blot, and immunohistochemistry, anti-phospho Tyr-490 TrkA/Tyr-516 TrkB (Cell Signaling Technology), 1:200; function-blocking hamster anti-integrin β1 and mouse anti-integrin β3 (BD Biosciences), 1:20; Alexa 488-conjugated goat anti-mouse IgG and Alexa 546-conjugated goat anti-rabbit IgG, 1:100; HRP-conjugated goat anti-rabbit IgG (Invitrogen), 1:2000.
Cell cultures.
Culture was prepared as described previously (Murase and McKay, 2006). Hippocampi from embryonic day 18 Sprague Dawley rat embryos were used for both astrocyte (plated at a density of 80,000 cells/ml) and neuron (density, 200,000 cells/ml) cultures. Neurons were cultured in neuron medium (modified MEM containing 2 mm GlutaMAX, penicillin/streptomycin, 200 mg/L apotransferrin, 0.5 mm putrescin, 30 nm sodium selenite, 40 nm progesterone, 40 ng/ml corticosterone, 20 ng/ml triiodothyronine, 25 μg/ml insulin and FUDR). One-third of the medium was exchanged every 3–4 d.
Bcl-XL expression.
A nontoxic derivative of anthrax toxin was used to deliver Bcl-XL peptide into the cytosolic compartment of cultured neurons (Liu et al., 2001). The first 254 residues of anthrax lethal factor (LFn) fused with Bcl-XL and protective antigen (PA) were a gift from Dr. Richard J. Youle (National Institute of Neurological Disorders and Stroke, National Institutes of Health, Bethesda, MD). Treatment was performed between the fifth day in vitro (DIV5) and DIV7 with either LFn-Bcl-XL (50 μg/ml) and PA (40 μg/ml), or BSA (50 μg/ml) and PA (40 μg/ml) as a control.
Transfection.
Using Lipofectamine 2000 (Invitrogen), cotransfection with 1.6 μg/ml pEGFPC1 vector (Clontech) and/or 8 μg/ml activated Akt1/pUSE vector (Millipore) per culture was performed for 1.5 h, 1 d before the experiments.
Immunocytochemistry.
Cultures were fixed with 4% paraformaldehyde, permeabilized in 0.5% Triton X-100, and blocked with 5% normal goat serum (NGS) (Vector Laboratories).
Rat neonatal brains were fixed with 4% paraformaldehyde followed by 20% sucrose at 4°C. Coronal cryostat sections, 16 μm thick, were blocked with 4% NGS in PBS with 0.1% Triton X-100. Antibodies were diluted in 4% NGS. Every fourth sequential section was used for immunohistochemistry.
Cell quantification.
The fluorescent images were obtained with a Zeiss 510 confocal microscope running laser scanning microscopy using a 25× objective lens. Because the densities of neurons on the edge of coverslips were higher than in other regions, images were taken from the following 5 fields: one from the center of the coverslip, two vertically, and two horizontally 400–3000 μm from the center. Numbers represent mean ± SEM.
Each coverslip is defined as an individual culture. All the results were obtained from multiple cultures. All the analyses were performed blind.
Analyses of hippocampal sections were performed using NIH ImageJ. Every fourth sequential section was stained for c-cas3 analyses. Identification of individual cells was confirmed by DAPI (4′,6′-diamidino-2-phenylindole dihydrochloride) staining (to visualize nuclei), and cell type was determined by NeuN staining. Partly due to dimensional changes in the location of the hippocampus during development, the total numbers of sections from rostral to caudal ends of hippocampus were ∼160 throughout development [postnatal day 1 (P1)–P10] despite the increase in total volume of hippocampus. Although the c-cas3 immunostaining produced a relatively high background, distinctive positive signals from apoptotic cells could be detected reliably using the threshold function in ImageJ.
BrdU.
For experiments using BrdU, cultures were treated with BDNF and NT3 (10 ng/ml) or a vehicle control (0.1% BSA in PBS) at DIV4, pulsed with BrdU 1 μm at DIV5, and fixed at DIV6. Cells were fixed in 4N HCl in distilled H2O for 10 min at room temperature, and blocked with 10% NGS and 0.3% Triton X-100 in PBS for 1 h at room temperature before immunostaining.
Western blot.
Western blot was performed as described previously (Murase and McKay, 2006).
In vivo injection.
In vivo injection to CA1 was described previously (Cunningham and McKay, 1993). Briefly, Sprague Dawley rat pups (P2) of either sex were anesthetized by hypothermia (in ice for 5 min) before the surgery. The anesthetized animal was placed on ice in a stereotaxic instrument. The stereotaxic coordinates from bregma are as follows: anteroposterior, +1.5; mediolateral, ±1.8; ventrodorsal, −1.8 mm. Reagents (0.3 μl) were delivered at a rate of 0.1 μl/min using a Hamilton syringe with a LASI needle attached to a pump. Concentrations of reagents were as follows: anti-β1 antibody, 0.5 mg/ml; BDNF, 50 μg/ml; TTX, 30 μm; bicuculline, 1 mm. PBS was used as control. The incision was closed using a 5–0 suture. Animals were allowed to recover at 37°C for 1–2 h.
Calcium imaging.
The cultures were incubated with 2 μm Fluo-4 AM (Invitrogen) for 15 min. The images were obtained with a Zeiss Axiovert 135TV microscope equipped with a 25× objective lens. The cultures were imaged in HEPES-buffered saline (HBS) (containing, in mm: 110 NaCl, 5.4 KCl, 1.8 CaCl2, 0.8 MgCl2, 10 d-glucose, 10 HEPES-NaOH, pH 7.4, 290 mOsm) at 37°C using a CCD camera (Hamamatsu) at 3 Hz for 30 s.
Electrophysiological recording.
Whole-cell recording was performed with pipettes containing the following internal solution (in mm): 136 K-gluconate, 10 KCl, 5 NaCl, 0.1 EGTA, 0.3 Na-GTP, 1 Mg-ATP, 5 phosphocreatine, 10 HEPES, pH 7.2. The resistance of the pipettes was in the range of 1–10 MΩ. Signals were amplified with a DC amplifier (model 440, Brownlee Precision). Electrophysiological recordings were synchronized with calcium images (20 Hz) by custom software written in Labview (National Instruments). During recording sessions, neuron cultures were perfused with HBS at a flow rate of 2.5 ml/min; 2 ml of 10 ng/ml BDNF was applied followed by HBS for 5 min.
Detection of apoptotic neurons with annexin V.
The cultures were imaged in Neurobasal medium supplemented with B27 containing 5 μl/ml PE annexin V (BD Biosciences) with a Zeiss Axiovert 135TV microscope equipped with a 10× objective lens. The cultures were incubated at 37°C with 10% CO2 using a live cell chamber (Zeiss). Phase and fluorescent images were taken every hour for 3 d.
Statistical analyses.
Statistical significance between two groups was determined with a two-tailed paired Student's t test. For multiple groups, statistical comparisons were made by ANOVA followed by individual group tests corrected for multiple comparisons.
Results
Spontaneous neuronal activity and death
As have others (Ferrer et al., 1994), we observed apoptosis during the first postnatal week in the rat hippocampus. The numbers of NeuN+ neurons in CA1 and CA3 cell layers decreased by 30–50% between P1 and P10 (Fig. 1A). Thus, in the hippocampus, developmental death controls number of neurons.
Figure 1.

Spontaneous activity is involved in survival. A, In vivo, numbers of hippocampal neurons decrease during the postnatal period. Mean numbers of NeuN+ cells in CA1 and CA3 cell layer are compared between P1 and P10 (n = 6). B, Apoptotic neurons were analyzed by immunostaining of c-cas3 and a neuronal marker, NeuN (blue, DAPI; green, NeuN; red, c-cas3). Inhibiting spontaneous activity increased death. Either 0.3 μl of 30 μm TTX or 1 mm bicuculline (Bic) was injected at P2. cnt, Control. c-cas3+ neurons were analyzed at P3 (n = 8). *p < 0.05 (one-way ANOVA). C, The neuronal number declined between DIV4 and DIV9 (5–16 cultures). MAP2+ cells were counted (n = 8). D, Proportion of spontaneously active neurons. Calcium imaging with Fluo-4 AM was used (n = 5). E, Spontaneous activity was blocked by 30 μm bicuculline. F, GABAergic neurons became spontaneously active earlier than glutamatergic neurons at DIV5 (n = 4). G, From left: Phase image of DIV6 culture, location of active neurons, location of inactive neurons (red dots, annexin+ within 24 h), proportion of neurons that became annexin− to annexin+ within 24 h period after calcium imaging (n = 5). *p < 0.05.
GABA-dependent network activity in a specific period of neonatal hippocampus development has been well defined (Garaschuk et al., 1998; Wakselman et al., 2008), but it is not known whether this activity is linked to neuronal death. To directly test the effect of neuronal activity on survival, we injected antagonists of voltage-dependent sodium channels (TTX) and GABAA receptors (bicuculline) to the neonatal hippocampi. By monitoring apoptotic neurons with immunostaining against c-cas3, we found that bilateral injection of TTX or bicuculline to P2 hippocampus promoted neuron death (Fig. 1B), showing that GABA-dependent neuronal activity regulates neuronal survival in vivo.
We further investigated the relationship between neuronal activity and survival with dissociated hippocampal neurons. Interactions with astrocytes are known to be important for the maturation and survival of neurons (Blondel et al., 2000; Ullian et al., 2004; Elmariah et al., 2005; Nagai et al., 2007). To study neuronal death, we used hippocampal neurons plated onto astrocytes and showed that, in the central region of 3-mm-diameter coverslips, the density of neurons was reproducible and insensitive to changing the medium or adding the solvent DMSO (1% DMSO; 102.5 ± 4.1% and 95.3 ± 4.1% of control, n = 6). Under these conditions, the number of surviving neurons was stable for the first 4 d but reproducibly decreased between DIV4 and DIV9 before becoming stable once more (Fig. 1C). Incubation for 2 d with the antimitotic drug, FUDR, did not affect the neuron number, showing that neurons were not generated from dividing precursors (80 μm FUDR; 108.2 ± 5.3% of control, n = 15 at DIV4). Including a caspase inhibitor (20 μm) or transfecting a plasmid encoding the anti-apoptotic protein Bcl-XL effectively reduced death (119.0 ± 11.3% and 135.5 ± 9.4% of control, n = 4). These results define a specific period when postmitotic neurons undergo spontaneous apoptosis that eliminates ∼40% of total neurons.
To monitor activity in these developing neurons, we used time-lapse imaging following incubation with Fluo-4 AM, a nonfluorescent acetoxymethyl ester that is cleaved inside cells to the green-fluorescent calcium indicator, Fluo-4. These data show that spontaneous calcium activity was first evident in a small proportion of neurons on DIV4–DIV5, but by DIV11–DIV12, almost all neurons exhibited activity (Fig. 1D). This activity was synchronous throughout the population of neurons (data not shown) and was completely blocked by the GABAA antagonist, bicuculline (Fig. 1E). When this activity first emerged, a larger proportion of the active neurons expressed high levels of the neurotransmitter GABA (Fig. 1F). These data suggest that synchronous calcium activity develops during the period when some neurons die, and that GABA plays an early role in this process.
Next, we performed live-imaging experiments to determine the correlation between activity and death. Annexin V binds to negatively charged phospholipids that are normally present on the inner surface of the plasma membrane but exposed on the surface of dying cells (Koopman et al., 1994). Neuronal activity was monitored optically, and then the cell surface expression of annexin V was monitored for the next 24 h. At DIV6, one-third of the neurons showed spontaneous calcium transients. Only 1% of these active cells became annexin+, whereas >10% of the inactive neurons became annexin+ in the next 24 h (Fig. 1G). These results show that inactive neurons were 10 times more likely to die than active ones.
BDNF facilitates activity while promoting survival
Simultaneous whole-cell recording and calcium imaging have shown that calcium transients [also known as early network oscillations (ENOs)] are associated with action potentials (APs) in hippocampal slices (Garaschuk et al., 1998). In dissociated hippocampal neurons, we found that ENOs were also associated with APs when they become spontaneously active (Fig. 2A). The frequency of these GABA-dependent ENOs increased following application of BDNF (Fig. 2A). This effect of BDNF was observed only during the death period (DIV7, 188 ± 17.7%, n = 6; DIV11, 108 ± 17.5% of control, n = 4).
Figure 2.
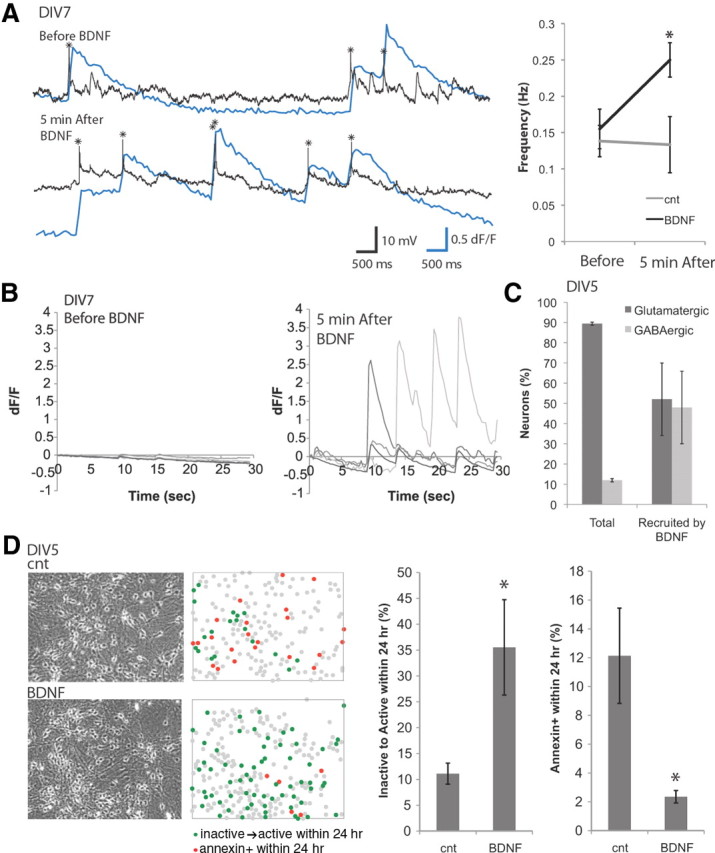
BDNF promotes spontaneous activity and survival. A, Left, Calcium signal (blue line) and intracellular recording (black line, action potentials marked with asterisks) show increased activity 5 min after application of 10 ng/ml BDNF. Right, Frequency of calcium activity is enhanced by 10 ng/ml BDNF in 5 min (n = 6). B, Example of inactive neurons (DIV7) becoming active 5 min after application of 10 ng/ml BDNF. C, GABAergic neurons were preferentially recruited by BDNF at DIV5 (n = 7). D, BDNF promoted spontaneous activity while suppressing apoptosis. Calcium imaging with Fluo-4 AM was performed at DIV5, and then the cultures were incubated with or without 10 ng/ml BDNF for 24 h, before the second calcium imaging was performed at DIV6 (n = 5). cnt, Control. *p < 0.05.
At DIV7, BDNF acutely recruited inactive cells into the active population (Fig. 2B). Treatment with BDNF increased the number of spontaneously active neurons by 20.4 ± 6.4% (20% of inactive neurons are recruited). At DIV5, GABAergic neurons were preferentially activated by BDNF (Fig. 2C). The calcium transients in both previously active neurons and BDNF-recruited neurons were blocked entirely by bicuculline (data not shown). These observations show that BDNF acutely recruits inactive cells into the active population and that GABAergic cells are initially more likely to be activated.
To determine whether exposure to BDNF leads to a long-term change in the proportion of active neurons, we measured neuronal activity by Fluo-4 AM imaging 24 h after treatment with BDNF. BDNF treatment increased the proportion of active neurons more than threefold, and this treatment reduced the number of annexin V+ neurons by fivefold (Fig. 2D). These results suggest that BDNF has a long-term effect on neuronal activity and survival.
BDNF fails to promote survival of neurons in the absence of spontaneous activity
Our results show (1) that inactive neurons are likely to become annexin+ and (2) that BDNF rapidly promotes activity and reduces the number of annexin+ neurons. Different models could explain these observations. One possibility is that BDNF supported neuronal survival by promoting activity. Alternatively, there may be two distinct populations of neurons; one that dies and another that shows altered activity in response to BDNF. Therefore, we tested whether BDNF required activity to exert its survival effect. Treatment with BDNF from DIV5 to DIV7 resulted in a significant increase in neuron number (Fig. 3). This effect was abolished in the presence of activity blockade by bicuculline (Fig. 3). This result shows directly that exogenous BDNF requires activity to support neuronal survival.
Figure 3.

BDNF fails to rescue neurons without spontaneous activity. Left, MAP2 immunostaining. Right, Numbers of neurons (n = 7). Neurons were treated with 10 ng/ml BDNF and/or 30 μm bicuculline (Bic) from DIV5 to DIV7. *p < 0.05 (one-way ANOVA); ns, not significant; cnt, control.
L-type channels are critical for calcium activity, and activate Akt
Our calcium-imaging data suggested that the survival effects of BDNF may be mediated through a calcium-dependent system. Nifedipine, the L-type calcium channel antagonist, completely blocked spontaneous ENOs (Fig. 4A). Blocking L-type channels also reduced neuron numbers (Fig. 4B), suggesting that L-type calcium channels are involved in survival. BDNF did not increase the number of surviving neurons in the presence of nifedipine (Fig. 4B). These results show that BDNF requires both GABAA receptors and the depolarization mediated by L-type calcium channels to support neuronal survival.
Figure 4.

L-type channels are required for spontaneous activity and survival. A, Calcium events were attenuated by nifedipine (Nif). B, Neuronal death was induced by nifedipine (n = 5). C, Activation of L-type channels by depolarization (100 μm GABA for 5 min) induced Akt phosphorylation. Concentrations of 30 μm bicuculline, 5 mm EGTA, 100 μm APV, 10 μm nifedipine, and 10 μm LY294002 were used. cnt, Control. *p < 0.05 (one-way ANOVA).
Cell survival responses in many systems are controlled by a rapid activation of the phosphoinositol kinase (PI3K) and recruitment of the serine-threonine kinase Akt to the cell surface (Toker and Newton, 2000; Jacinto et al., 2006). Treating hippocampal neurons with GABA induced the phosphorylation of Ser-473 Akt, which can be blocked by LY294002, an inhibitor for PI3K (Fig. 4C). The phosphorylation of Akt was impaired by bicuculline, and when extracellular calcium was depleted with EGTA, or L-type channels were inhibited by nifedipine, GABA did not cause Akt phosphorylation (Fig. 4C). In contrast, the NMDA receptor antagonist APV did not block Akt phosphorylation (Fig. 4C). These results show that depolarization drives L-type channels to regulate the survival of hippocampal neurons, and suggest that this effect may be mediated by the activation of Akt.
Neurotrophins initiate Akt activation that is maintained by L-type channels
To directly determine how activity is involved in neurotrophin-induced cell signaling, we first monitored activation of the tyrosine kinase receptor Trk in response to BDNF and NT3. Trk phosphorylation endured for <1 h in the continued presence of these ligands (Fig. 5A). Preincubation with antibodies against TrkB and TrkC, which block receptor–ligand interaction, blocked tyrosine phosphorylation of the receptors (Fig. 5A). EGTA and bicuculline treatment did not block tyrosine phosphorylation of Trk receptors (Fig. 5A).
Figure 5.

Neurotrophins initiate Akt activation that is maintained by L-type channels. A, BDNF and NT3 (10 ng/ml) induced phosphorylation of Trk, and sustained phosphorylation of Ser-473 Akt; 50 μg/ml anti-TrkB and anti-TrkC, 30 μm bicuculline (Bic), 5 mm EGTA, and 10 μm nifedipine were used. B, Neurons were treated from DIV5 to DIV7 (n = 7); 10 ng/ml BDNF and NT3, 50 μg/ml anti-TrkB and anti-TrkC, and 10 μm LY294002 (LY) were used. C, Overexpression of constitutively active Akt by transfection of a plasmid encoding Akt cDNA with a myristoylation signal. Transfection was performed at DIV4, and neurons were treated with 10 μm nifedipine from DIV5 to DIV7 (n = 5). cnt, Control. *p < 0.05 (one-way ANOVA).
In contrast to the short-lasting activation of Trk, treatment with neurotrophins caused activation of Akt for 4 h (Fig. 5A). Preincubation with anti-TrkB and anti-TrkC blocked Akt activation (Fig. 5A). Buffering external calcium with EGTA, blocking GABAA receptors or L-type channels, permitted Akt phosphorylation for 1 h but caused premature downregulation of Akt phosphorylation (Fig. 5A). These data suggest that Akt activation is initiated by Trk activation, but is sustained by activity-driven calcium influx that requires GABAA and L-type channels.
Akt is necessary and sufficient for survival
To determine whether Akt activation is required for neurotrophin survival signaling, neurons were treated with neurotrophins in the presence of LY294002, a drug that is widely used as a selective inhibitor of PI3K. Incubation with BDNF promoted survival, and the addition of NT3 did not enhance this effect (Fig. 5B). Incubation with anti-TrkB and anti-TrkC, on the other hand, reduced the number of surviving neurons below control levels, suggesting a role for endogenous neurotrophins (Fig. 5B). In the presence of LY294002, the survival effect of neurotrophins was blocked, leading to numbers of surviving neurons similar to those observed when neurotrophin signaling was blocked (Fig. 5B). This result suggests that activation of Akt is necessary for neurotrophin-dependent survival.
Akt activation promotes its localization to the membrane, and when Akt is tagged with a myristoylation site, it becomes constitutively active (Kohn et al., 1996). To test whether Akt activation is sufficient for survival, we overexpressed myristoylated Akt in neurons. When neurons were transfected with a control plasmid expressing only green fluorescent protein (GFP), the decline in numbers of transfected neurons from DIV5 to DIV7 was exacerbated by nifedipine (Fig. 5B). However, when neurons were cotransfected with myristoylated Akt, the numbers of neurons did not change over time, and nifedipine had no effect on the number (Fig. 5C). These results suggest that constitutively active Akt is sufficient to suppress neuronal death.
Integrin β1 signaling is necessary for neurotrophin-dependent survival
Akt has two phosphorylation targets for PI3K at Thr-308 and Ser-473 that mediate distinct downstream effects in survival and growth signaling (Jacinto et al., 2006). We found that depolarizing hippocampal neurons by applying elevated K+ induced the phosphorylation of both sites, and these phosphorylation events were both attenuated by nifedipine (Fig. 6A). Because the extracellular matrix is known to play an important role in cell survival, and laminin acting through integrin receptors is known to influence the survival of hippocampal neurons (Gary et al., 2003), we explored the role of integrin in this survival pathway.
Figure 6.
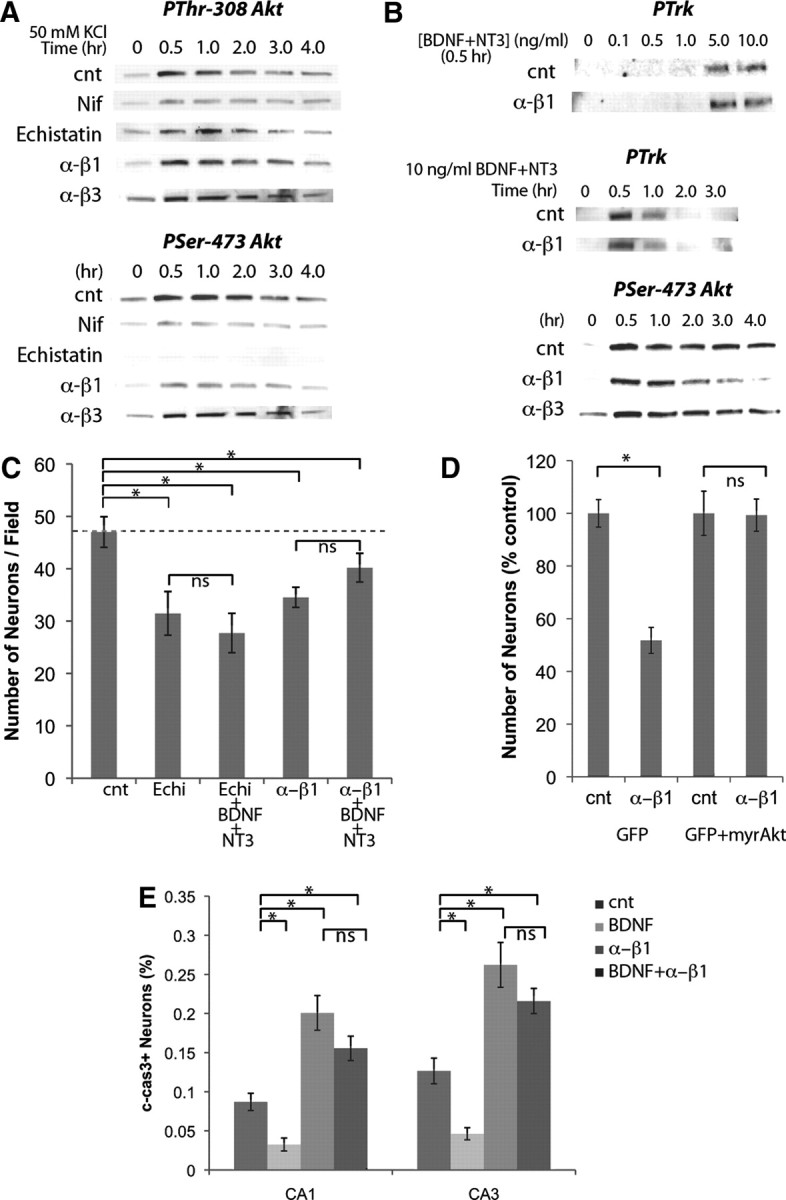
L-type channels cooperate with integrin β1 to activate Akt and promote survival. A, Activation of Akt by depolarization with 50 mm KCl for 5 min required L-type channels. Phosphorylation of Ser-473 but not Thr-308 was impaired by echistatin or anti-integrin β1 antibody. B, Neurotrophin-induced phosphorylation of Trk receptors was not affected by anti-integrin β1 antibody, but the sustained Akt activation was impaired. C, Incubation with echistatin or anti-integrin β1 antibody reduced the number of surviving neurons and attenuated the survival effect of neurotrophins. Treatment was from DIV5 to DIV7 (n = 6). D, Cotransfection of GFP and myristoylated Akt blocked the effect of anti-integrin β1 antibody on neuronal survival. Transfection was at DIV4 and treatment from DIV5 to DIV7 (n = 6); 10 μm nifedipine (Nif), 0.1 μm echistatin, 50 μg/ml anti-integrin β1 and anti-integrin β3 antibodies, and 10 ng/ml BDNF and NT3 were used. E, In vivo delivery of BDNF reduced apoptosis while anti-β1 antibody increased. BDNF failed to reduce apoptosis when integrin β1 was inhibited; 0.3 μl of 0.5 mg/ml anti-β1, 50 μg/ml BDNF were injected at P2, and c-cas3+ neurons were analyzed at P4 (n = 6). cnt, Control. *p < 0.05 (one-way ANOVA).
Echistatin, a disintegrin derived from snake venom that blocks specifically β1/β3-containing integrins (Pfaff et al., 1994), and a function-blocking antibody against the β1 subunit of integrin (Mendrick and Kelly, 1993), specifically impaired phosphorylation of Ser-473 (Fig. 6A). On the other hand, a function-blocking antibody against integrin β3 (Helfrich et al., 1992) had no effect on either phosphorylation site (Fig. 6A). These results suggest that L-type channel-induced phosphorylation of Akt at Ser-473 requires integrin β1 signaling.
If integrin β1 is required for L-type channel-induced Akt activation, the sustained Akt activation by neurotrophins should also be integrin β1 dependent. Neither the sensitivity of Trk activation nor the duration of Trk activation was affected by anti-integrin β1 antibody (Fig. 6B). While initial transient activation of Akt was intact in the presence of integrin β1 blockade, the sustained Akt activation was impaired (Fig. 6B). Inhibition of integrin β3, on the other hand, had no effect on Akt activation (Fig. 6B). Although it has been reported that neurotrophins directly bind to integrin α9 β1 (Staniszewska et al., 2008), the function blocking anti-integrin β1 antibody did not block the transient Akt activation by neurotrophins, which was completely attenuated by anti-TrkB and anti-TrkC antibodies (Fig. 6B). Therefore, it is likely that the transient Akt activation was triggered by Trk receptor activation. These results show that, independent of the initial activation mechanism, integrin β1 is required for sustained Akt activation on Ser-473 that is triggered by neurotrophins.
If integrin β1 is required for neurotrophin-dependent survival of neurons, inhibition of integrin β1 should attenuate the survival effect of neurotrophins. Indeed, we found that in the presence of echistatin or anti-integrin β1 antibody, the number of surviving neurons was decreased, and neurotrophins failed to promote neuronal survival (Fig. 6C). Anti-integrin β3 antibody did not affect the number of surviving neurons (105.7 ± 4.2% of control, n = 6). When neurons were cotransfected with GFP and myristoylated Akt, the number of transfected neurons did not change by the anti-integrin β1 antibody (Fig. 6D), confirming that sustained Akt activation is sufficient to rescue neurons from integrin β1 blockade. These results suggest that the survival effects induced by neurotrophins require integrin β1 signaling.
Manipulations in vivo confirm the survival pathway is conserved
Molecules involved in the survival of hippocampal neurons during the death period play important roles in earlier stages of development, and mice lacking neurotrophin or integrin receptors have been shown not to survive through the period when hippocampal neurons die (Stephens et al., 1995; Silos-Santiago et al., 1997). To achieve precisely timed manipulations of survival signals, we made microinjections into the neonatal hippocampus. When BDNF was injected into P2 hippocampus, the number of c-cas3+ neurons in both CA1 and CA3 regions was reduced (Fig. 6E). In contrast, injection of the anti-integrin β1 antibody led to increased numbers of c-cas3+ neurons (Fig. 6E). As with in vitro experiments, the survival effect of BDNF was not observed in the presence of anti-integrin β1 antibody (Fig. 6E). These results suggest that neurotrophins promote survival through an integrin-dependent mechanism in vivo.
Discussion
The goal of this study was to understand whether neurotrophins control neuronal number during the specific stage of postnatal development when spontaneous neuron loss occurs. As summarized in Figure 7, we first found that neuronal survival in the postnatal hippocampus required spontaneous GABA-dependent activity. We then used in vitro systems to show that BDNF promoted GABA-dependent network activity while suppressing apoptosis (Fig. 7A). Transient Trk receptor activation by neurotrophins triggered Akt activation that was maintained by calcium influx through L-type channels and also required signals from integrin receptors (Fig. 7B). Finally, experiments performed in vivo demonstrated that neurotrophins regulated neuron death through an integrin-dependent mechanism. These results support a new view of the survival mechanisms activated by neurotrophins.
Figure 7.
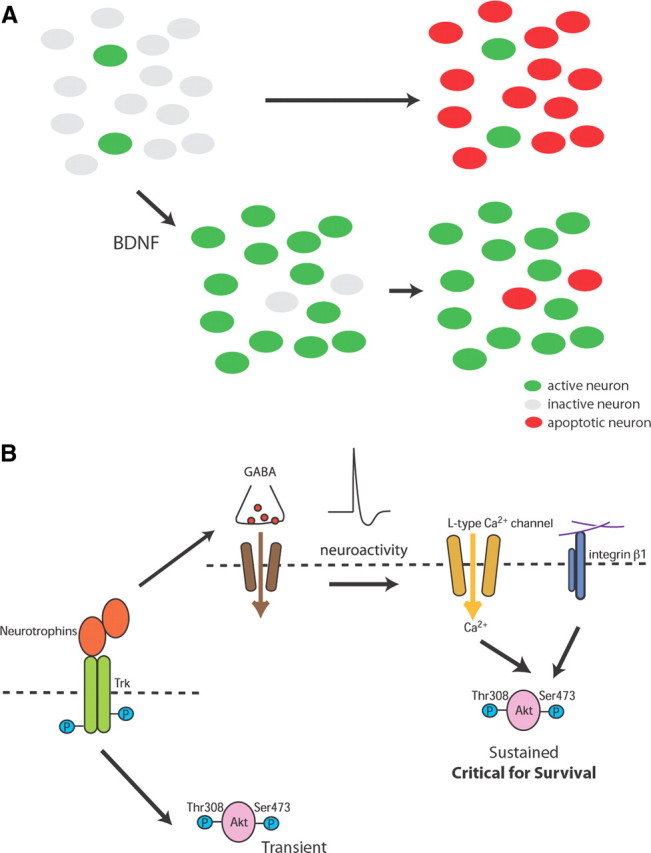
Schematic drawings of how neurotrophins promote survival of neurons in neonate. A, During the neonatal period when spontaneous network activity is developing, BDNF promotes spontaneous activity while suppressing the apoptosis. B, Molecular mechanism by which neurotrophins exert survival effect via promoting spontaneous activity. Activation of Trk receptor triggers Akt activation that is maintained by spontaneous activity-driven calcium influx through L-type channels. The sustained Akt, which requires signal from integrin β1 signaling, is necessary and sufficient for neurotrophin-dependent survival.
Since apoptosis was first reported in the neonatal hippocampus (Ferrer et al., 1994), this developmental death has been confirmed by other groups (White et al., 1998; Wakselman et al., 2008). Our results, obtained in vivo and in vitro, are consistent with these findings: by analyzing numbers of c-cas3+ neurons, we confirmed the extent of apoptosis reported previously. Further, consistent with previous results (Ferrer et al., 1994), we did not observe apoptotic neurons after P10 (data not shown). Together, these results establish that developmental death eliminates a large proportion of neurons from the hippocampus, indicating that this process plays an important role in controlling numbers of surviving neurons in this brain structure.
In contrast to the dramatic loss of neurons observed in the PNS, genetic deletion of neurotrophins and their receptors have little effect on neuron number in the CNS, including hippocampus (Jones et al., 1994; Minichiello and Klein, 1996; Alcántara et al., 1997; Rauskolb et al., 2010). These studies have led to the suggestion that neurotrophins may not be involved in the survival of CNS neurons. However, our results obtained in vivo and in vitro demonstrate that neurotrophins are required for the survival of hippocampal neurons and that neurotrophins support neuronal survival by promoting network activity. In pathological neuronal loss of CNS neurons, neurotrophins may play a major neuroprotective role (Nagahara et al., 2009; Nikolaev et al., 2009). Whether or not the survival mechanism presented here is conserved in neurodegenerative diseases remains to be elucidated.
Depolarizing GABA during development promotes neuronal differentiation through the action of BDNF (Marty et al., 1996). Transcription and release of BDNF is stimulated by GABA (Berninger et al., 1995; Kuczewski et al., 2008). These results are consistent with the commonly held model that neural activity causes neurotrophin release and this promotes survival. Our data show that neurotrophins cannot exert their effects on survival in the absence of neuroactivity, and thus assign a central role for activity in neurotrophin-mediated survival. We report two effects of BDNF on neuronal activity, an increase in the frequency of calcium oscillations (ENOs) and the recruitment of silent neurons to active networks. Precise mechanisms for these effects remain to be elucidated. One possibility is that BDNF alters excitability of the neurons. However, young hippocampal neurons do not show changes in either K+- or Na+-currents after BDNF treatment (Li et al., 1998). Another possibility is that BDNF alters synaptic properties (Li et al., 1998; Wardle and Poo, 2003). The neurotrophic effects of BDNF emphasize the importance of a rapid change in the activity state of hippocampal neurons at this stage of their maturation.
We show that the serine-threonine kinase Akt is necessary and sufficient for neuronal survival. Akt activation by receptor tyrosine kinases is generally rapid (Franke et al., 1995; Yoshizaki et al., 2007), but this is sufficient to induce survival responses (Androutsellis-Theotokis et al., 2006). Our data show that the kinetics of Akt signaling is longer-lasting in neurons than in other cell types, and the long-lasting Akt-activation is required for neuronal survival. We found that phosphorylation of Ser-473 Akt induced by calcium influx through L-type channels specifically required integrin β1. The mechanisms of Akt activation have been intensively studied in the context of cancer biology, where a kinase cascade activated by tyrosine kinase leads to phosphorylation of Akt at Thr-308. This event is required for subsequent phosphorylation of Akt at Ser-473 (Balendran et al., 1999; Toker and Newton, 2000). The mTOR complex 2 (mTORC2), a signaling complex related to the well defined, rapamycin-sensitive mTORC1, phosphorylates Akt at Ser-473 and plays a specific role in cell survival (Jacinto et al., 2006). Our data suggest that both phosphorylation events are required for neuronal survival and reveal a novel mechanism, assigning a central role to neuronal activity in activating this process.
Our study revealed a specific role for the integrin β1 subunit in the survival signaling of neonatal neurons in hippocampus. The ligand that activates integrin β1 to sustain Akt phosphorylation remains unknown. One candidate ligand is laminin, an extracellular-matrix protein that binds to and is expressed by neonatal neurons (Hynes, 2002; Sharif et al., 2004). Alternatively, the unknown ligand may be provided by non-neuronal cells. Contact with astrocytes, for example, facilitates synapse formation in young hippocampal neurons via signaling from integrin β1 (Hama et al., 2004). Further investigations of the spatial and temporal distribution of integrin β1 and its ligands may help to reveal mechanisms by which neurocircuitry is sculpted through neuronal death.
Our data show that it is important for a neuron to participate in an active network to survive the neonatal period, and that depolarizing GABA plays a key role in this mechanism. Depolarizing GABA is critical for the spontaneous network activity that is believed to establish the circuitry of the neonatal hippocampus (Ben-Ari et al., 1989). An appropriate balance between excitatory and inhibitory inputs is critical for normal adult brain function. Our results suggest that neurotrophin-mediated activity may ensure that neurons only survive when they are appropriately linked to a functional network with strong GABAergic input.
Footnotes
This research was supported by the Intramural Research Program of the National Institutes of Health, National Institute of Neurological Disorders and Stroke. We thank Drs. Antonella Antignani and Richard Youle for providing LFn-Bcl-XL and PA, Drs. Joby Joseph and Mark Stopfer for advice on statistics and electrophysiological recording, and Drs. Arek Szklarczyk, Steve W. Poser, and Peter H. Maughan for the culture preparation. We also thank Drs. Moses Chao, Michael Sendtner, and Mu-Ming Poo for helpful advice on the manuscript.
References
- Alcántara S, Frisén J, del Río JA, Soriano E, Barbacid M, Silos-Santiago I. TrkB signaling is required for postnatal survival of CNS neurons and protects hippocampal and motor neurons from axotomy-induced cell death. J Neurosci. 1997;17:3623–3633. doi: 10.1523/JNEUROSCI.17-10-03623.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Androutsellis-Theotokis A, Leker RR, Soldner F, Hoeppner DJ, Ravin R, Poser SW, Rueger MA, Bae SK, Kittappa R, McKay RD. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature. 2006;442:823–826. doi: 10.1038/nature04940. [DOI] [PubMed] [Google Scholar]
- Balendran A, Casamayor A, Deak M, Paterson A, Gaffney P, Currie R, Downes CP, Alessi DR. PDK1 acquires PDK2 activity in the presence of a synthetic peptide derived from the carboxyl terminus of PRK2. Curr Biol. 1999;9:393–404. doi: 10.1016/s0960-9822(99)80186-9. [DOI] [PubMed] [Google Scholar]
- Barde YA. Neurotrophins: a family of proteins supporting the survival of neurons. Prog Clin Biol Res. 1994;390:45–56. [PubMed] [Google Scholar]
- Ben-Ari Y, Cherubini E, Corradetti R, Gaiarsa JL. Giant synaptic potentials in immature rat CA3 hippocampal neurones. J Physiol. 1989;416:303–325. doi: 10.1113/jphysiol.1989.sp017762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Ari Y, Khazipov R, Leinekugel X, Caillard O, Gaiarsa JL. GABAA, NMDA and AMPA receptors: a developmentally regulated ‘ménage à trois’. Trends Neurosci. 1997;20:523–529. doi: 10.1016/s0166-2236(97)01147-8. [DOI] [PubMed] [Google Scholar]
- Berninger B, Marty S, Zafra F, da Penha Berzaghi M, Thoenen H, Lindholm D. GABAergic stimulation switches from enhancing to repressing BDNF expression in rat hippocampal neurons during maturation in vitro. Development. 1995;121:2327–2335. doi: 10.1242/dev.121.8.2327. [DOI] [PubMed] [Google Scholar]
- Bibel M, Barde YA. Neurotrophins: key regulators of cell fate and cell shape in the vertebrate nervous system. Genes Dev. 2000;14:2919–2937. doi: 10.1101/gad.841400. [DOI] [PubMed] [Google Scholar]
- Blondel O, Collin C, McCarran WJ, Zhu S, Zamostiano R, Gozes I, Brenneman DE, McKay RD. A glia-derived signal regulating neuronal differentiation. J Neurosci. 2000;20:8012–8020. doi: 10.1523/JNEUROSCI.20-21-08012.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavis P, Westbrook G. Integrins mediate functional pre- and postsynaptic maturation at a hippocampal synapse. Nature. 2001;411:317–321. doi: 10.1038/35077101. [DOI] [PubMed] [Google Scholar]
- Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain. 2006;129:1659–1673. doi: 10.1093/brain/awl082. [DOI] [PubMed] [Google Scholar]
- Cunningham MG, McKay RD. A hypothermic miniaturized stereotaxic instrument for surgery in newborn rats. J Neurosci Methods. 1993;47:105–114. doi: 10.1016/0165-0270(93)90026-n. [DOI] [PubMed] [Google Scholar]
- Elmariah SB, Oh EJ, Hughes EG, Balice-Gordon RJ. Astrocytes regulate inhibitory synapse formation via Trk-mediated modulation of postsynaptic GABAA receptors. J Neurosci. 2005;25:3638–3650. doi: 10.1523/JNEUROSCI.3980-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Tortosa A, Blanco R, Martín F, Serrano T, Planas A, Macaya A. Naturally occurring cell death in the developing cerebral cortex of the rat. Evidence of apoptosis-associated internucleosomal DNA fragmentation. Neurosci Lett. 1994;182:77–79. doi: 10.1016/0304-3940(94)90210-0. [DOI] [PubMed] [Google Scholar]
- Franke TF, Yang SI, Chan TO, Datta K, Kazlauskas A, Morrison DK, Kaplan DR, Tsichlis PN. The protein kinase encoded by the Akt proto-oncogene is a target of the PDGF-activated phosphatidylinositol 3-kinase. Cell. 1995;81:727–736. doi: 10.1016/0092-8674(95)90534-0. [DOI] [PubMed] [Google Scholar]
- Garaschuk O, Hanse E, Konnerth A. Developmental profile and synaptic origin of early network oscillations in the CA1 region of rat neonatal hippocampus. J Physiol. 1998;507:219–236. doi: 10.1111/j.1469-7793.1998.219bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gary DS, Milhavet O, Camandola S, Mattson MP. Essential role for integrin linked kinase in Akt-mediated integrin survival signaling in hippocampal neurons. J Neurochem. 2003;84:878–890. doi: 10.1046/j.1471-4159.2003.01579.x. [DOI] [PubMed] [Google Scholar]
- Gould E, Woolley CS, McEwen BS. Naturally occurring cell death in the developing dentate gyrus of the rat. J Comp Neurol. 1991;304:408–418. doi: 10.1002/cne.903040306. [DOI] [PubMed] [Google Scholar]
- Hama H, Hara C, Yamaguchi K, Miyawaki A. PKC signaling mediates global enhancement of excitatory synaptogenesis in neurons triggered by local contact with astrocytes. Neuron. 2004;41:405–415. doi: 10.1016/s0896-6273(04)00007-8. [DOI] [PubMed] [Google Scholar]
- Helfrich MH, Nesbitt SA, Dorey EL, Horton MA. Rat osteoclasts adhere to a wide range of RGD (Arg-Gly-Asp) peptide-containing proteins, including the bone sialoproteins and fibronectin, via a beta 3 integrin. J Bone Miner Res. 1992;7:335–343. doi: 10.1002/jbmr.5650070314. [DOI] [PubMed] [Google Scholar]
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell. 2002;110:673–687. doi: 10.1016/s0092-8674(02)00971-6. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Jones KR, Fariñas I, Backus C, Reichardt LF. Targeted disruption of the BDNF gene perturbs brain and sensory neuron development but not motor neuron development. Cell. 1994;76:989–999. doi: 10.1016/0092-8674(94)90377-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AD, Takeuchi F, Roth RA. Akt, a pleckstrin homology domain containing kinase, is activated primarily by phosphorylation. J Biol Chem. 1996;271:21920–21926. doi: 10.1074/jbc.271.36.21920. [DOI] [PubMed] [Google Scholar]
- Koopman G, Reutelingsperger CP, Kuijten GA, Keehnen RM, Pals ST, van Oers MH. Annexin V for flow cytometric detection of phosphatidylserine expression on B cells undergoing apoptosis. Blood. 1994;84:1415–1420. [PubMed] [Google Scholar]
- Kuczewski N, Porcher C, Ferrand N, Fiorentino H, Pellegrino C, Kolarow R, Lessmann V, Medina I, Gaiarsa JL. Backpropagating action potentials trigger dendritic release of BDNF during spontaneous network activity. J Neurosci. 2008;28:7013–7023. doi: 10.1523/JNEUROSCI.1673-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levi-Montalcini R. The nerve growth factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- Li YX, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. J Neurosci. 1998;18:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XH, Collier RJ, Youle RJ. Inhibition of axotomy-induced neuronal apoptosis by extracellular delivery of a Bcl-XL fusion protein. J Biol Chem. 2001;276:46326–46332. doi: 10.1074/jbc.M108930200. [DOI] [PubMed] [Google Scholar]
- Marty S, Berninger B, Carroll P, Thoenen H. GABAergic stimulation regulates the phenotype of hippocampal interneurons through the regulation of brain-derived neurotrophic factor. Neuron. 1996;16:565–570. doi: 10.1016/s0896-6273(00)80075-6. [DOI] [PubMed] [Google Scholar]
- Mendrick DL, Kelly DM. Temporal expression of VLA-2 and modulation of its ligand specificity by rat glomerular epithelial cells in vitro. Lab Invest. 1993;69:690–702. [PubMed] [Google Scholar]
- Minichiello L, Klein R. TrkB and TrkC neurotrophin receptors cooperate in promoting survival of hippocampal and cerebellar granule neurons. Genes Dev. 1996;10:2849–2858. doi: 10.1101/gad.10.22.2849. [DOI] [PubMed] [Google Scholar]
- Murase S, McKay RD. A specific survival response in dopamine neurons at most risk in Parkinson's disease. J Neurosci. 2006;26:9750–9760. doi: 10.1523/JNEUROSCI.2745-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagahara AH, Merrill DA, Coppola G, Tsukada S, Schroeder BE, Shaked GM, Wang L, Blesch A, Kim A, Conner JM, Rockenstein E, Chao MV, Koo EH, Geschwind D, Masliah E, Chiba AA, Tuszynski MH. Neuroprotective effects of brain-derived neurotrophic factor in rodent and primate models of Alzheimer's disease. Nat Med. 2009;15:331–337. doi: 10.1038/nm.1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagai M, Re DB, Nagata T, Chalazonitis A, Jessell TM, Wichterle H, Przedborski S. Astrocytes expressing ALS-linked mutated SOD1 release factors selectively toxic to motor neurons. Nat Neurosci. 2007;10:615–622. doi: 10.1038/nn1876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolaev A, McLaughlin T, O'Leary DD, Tessier-Lavigne M. APP binds DR6 to trigger axon pruning and neuron death via distinct caspases. Nature. 2009;457:981–989. doi: 10.1038/nature07767. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Pfaff M, McLane MA, Beviglia L, Niewiarowski S, Timpl R. Comparison of disintegrins with limited variation in the RGD loop in their binding to purified integrins alpha IIb beta 3, alpha V beta 3 and alpha 5 beta 1 and in cell adhesion inhibition. Cell Adhes Commun. 1994;2:491–501. doi: 10.3109/15419069409014213. [DOI] [PubMed] [Google Scholar]
- Rauskolb S, Zagrebelsky M, Dreznjak A, Deogracias R, Matsumoto T, Wiese S, Erne B, Sendtner M, Schaeren-Wiemers N, Korte M, Barde YA. Global deprivation of brain-derived neurotrophic factor in the CNS reveals an area-specific requirement for dendritic growth. J Neurosci. 2010;30:1739–1749. doi: 10.1523/JNEUROSCI.5100-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharif KA, Baker H, Gudas LJ. Differential regulation of laminin b1 transgene expression in the neonatal and adult mouse brain. Neuroscience. 2004;126:967–978. doi: 10.1016/j.neuroscience.2004.03.064. [DOI] [PubMed] [Google Scholar]
- Silos-Santiago I, Fagan AM, Garber M, Fritzsch B, Barbacid M. Severe sensory deficits but normal CNS development in newborn mice lacking TrkB and TrkC tyrosine protein kinase receptors. Eur J Neurosci. 1997;9:2045–2056. doi: 10.1111/j.1460-9568.1997.tb01372.x. [DOI] [PubMed] [Google Scholar]
- Stanco A, Szekeres C, Patel N, Rao S, Campbell K, Kreidberg JA, Polleux F, Anton ES. Netrin-1-alpha3beta1 integrin interactions regulate the migration of interneurons through the cortical marginal zone. Proc Natl Acad Sci U S A. 2009;106:7595–7600. doi: 10.1073/pnas.0811343106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staniszewska I, Sariyer IK, Lecht S, Brown MC, Walsh EM, Tuszynski GP, Safak M, Lazarovici P, Marcinkiewicz C. Integrin alpha9 beta1 is a receptor for nerve growth factor and other neurotrophins. J Cell Sci. 2008;121:504–513. doi: 10.1242/jcs.000232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens LE, Sutherland AE, Klimanskaya IV, Andrieux A, Meneses J, Pedersen RA, Damsky CH. Deletion of beta 1 integrins in mice results in inner cell mass failure and peri-implantation lethality. Genes Dev. 1995;9:1883–1895. doi: 10.1101/gad.9.15.1883. [DOI] [PubMed] [Google Scholar]
- Toker A, Newton AC. Cellular signaling: pivoting around PDK-1. Cell. 2000;103:185–188. doi: 10.1016/s0092-8674(00)00110-0. [DOI] [PubMed] [Google Scholar]
- Ullian EM, Christopherson KS, Barres BA. Role for glia in synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Wakselman S, Béchade C, Roumier A, Bernard D, Triller A, Bessis A. Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci. 2008;28:8138–8143. doi: 10.1523/JNEUROSCI.1006-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wardle RA, Poo MM. Brain-derived neurotrophic factor modulation of GABAergic synapses by postsynaptic regulation of chloride transport. J Neurosci. 2003;23:8722–8732. doi: 10.1523/JNEUROSCI.23-25-08722.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White FA, Keller-Peck CR, Knudson CM, Korsmeyer SJ, Snider WD. Widespread elimination of naturally occurring neuronal death in Bax-deficient mice. J Neurosci. 1998;18:1428–1439. doi: 10.1523/JNEUROSCI.18-04-01428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshizaki H, Mochizuki N, Gotoh Y, Matsuda M. Akt-PDK1 complex mediates epidermal growth factor-induced membrane protrusion through Ral activation. Mol Biol Cell. 2007;18:119–128. doi: 10.1091/mbc.E06-05-0467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifel LS, Kuruvilla R, Ginty DD. Functions and mechanisms of retrograde neurotrophin signalling. Nat Rev Neurosci. 2005;6:615–625. doi: 10.1038/nrn1727. [DOI] [PubMed] [Google Scholar]