

Summary
Genetic screens for cell division cycle mutants in the filamentous fungus Aspergillus nidulans led to the discovery of never-in-mitosis A (NIMA), a serine/threonine kinase that is required for mitotic entry. Since that discovery, NIMA-related kinases, or NEKs, have been identified in most eukaryotes, including humans where eleven genetically distinct proteins named NEK1 to NEK11 are expressed. Although there is no evidence that human NEKs are essential for mitotic entry, it is clear that several NEK family members have important roles in cell cycle control. In particular, NEK2, NEK6, NEK7 and NEK9 contribute to the establishment of the microtubule-based mitotic spindle, whereas NEK1, NEK10 and NEK11 have been implicated in the DNA damage response. Roles for NEKs in other aspects of mitotic progression, such as chromatin condensation, nuclear envelope breakdown, spindle assembly checkpoint signalling and cytokinesis have also been proposed. Interestingly, NEK1 and NEK8 also function within cilia, the microtubule-based structures that are nucleated from basal bodies. This has led to the current hypothesis that NEKs have evolved to coordinate microtubule-dependent processes in both dividing and non-dividing cells. Here, we review the functions of the human NEKs, with particular emphasis on those family members that are involved in cell cycle control, and consider their potential as therapeutic targets in cancer.
Key words: NEK, NIMA, Mitosis, Centrosome, Microtubule, Mitotic spindle, DNA damage response, Cilia
Introduction
Maintaining genome stability is crucial to the health of an organism. The vast majority of human cancer cells are aneuploid, meaning they have too few or, more commonly, too many chromosomes. Furthermore, many cancers exhibit chromosome instability, whereby there is frequent loss or gain of chromosomes at each cell division. Together, these defects provide tumours with a rapid evolutionary potential that can lead to the acquisition of malignant properties and drive selection of drug resistance. Typically, aneuploidy and chromosome instability arise as a result of errors in the process of chromosome segregation that occurs on the microtubule-based spindle that assembles during mitosis. Not surprisingly, cells have developed complex, and often redundant, mechanisms that enable them to undergo mitosis without errors. Reversible phosphorylation is among the best understood of the mechanisms of mitotic control. Cell-cycle-dependent protein kinases, along with their counteracting phosphatases, regulate the phosphorylation status of many hundreds of substrate proteins that, in turn, dictate the events that orchestrate mitosis. To date, a relatively small number of protein kinase families that regulate mitosis have been identified. These include the cyclin-dependent kinases (CDKs), Aurora kinases and Polo-like kinases (PLKs). However, there is another, less-well-characterised protein kinase family, whose members have key roles in mitosis: the NIMA-related kinases or NEKs (Moniz et al., 2011; O'Connell et al., 2003; O'Regan et al., 2007; Quarmby and Mahjoub, 2005). Here, we review our current knowledge of how NEKs contribute to the process of cell division. Moreover, because targeting mitosis is a proven approach to cancer treatment, we also debate the potential use of NEKs as therapeutic targets in human cancer.
The NIMA-related kinase family
The founding member of the NEK family is the never-in-mitosis A (NIMA) protein of Aspergillus nidulans, which was identified by Ron Morris and colleagues in a genetic screen for cell division cycle mutants (Oakley and Morris, 1983). Loss-of-function mutations in nimA cause G2 arrest, whereas overexpression leads to cells attempting to enter mitosis prematurely (Osmani et al., 1991; Osmani et al., 1988). It has been subsequently discovered that degradation of NIMA is essential for mitotic exit, which puts it on a par with the Cdc2–cyclin-B complex as a master regulator of mitotic progression in Aspergillus (Pu and Osmani, 1995). Aspergillus express a single NIMA-related gene, as do the yeasts Saccharomyces cerevisiae (called kin3) and Schizosaccharomyces pombe (called fin1; note that this is unrelated to the budding yeast protein Fin1 that localises to the mitotic spindle). However, although these yeast NEKs contribute to various aspects of cell cycle progression, including chromatin condensation, spindle assembly and cytokinesis, they are not required for viability (De Souza et al., 2000; Grallert and Hagan, 2002; Grallert et al., 2004; Krien et al., 1998; Wu et al., 1998).
NEKs have now been identified in a wide range of organisms from protists, such as Chlamydomonas, Plasmodium and Tetrahymena, to multicellular eukaryotes, including Drosophila, Xenopus, mice and humans. Human cells express eleven genes that encode NEK1 to NEK11 (Fig. 1). The defining feature of this protein family is an N-terminal catalytic domain that contains all the motifs that are typical of a serine/threonine kinase and shares closer sequence similarity with Aspergillus NIMA than any other class of protein kinase. NEK10 breaks this rule by having a centrally located kinase domain, but with regards to its amino acid sequence it clearly belongs to the NEK family. Generally, the NEK kinase domains are only moderately conserved, with ∼40–50% identity on the amino acid level both to the kinase domain of NIMA and, on the whole, to each other. NEK6 and NEK7 are unusual in this respect, because their kinase domains share more than 85% sequence identity. All eleven human NEKs contain a His-Arg-Asp (HRD) motif within the catalytic domain, which is usually found in kinases that are positively regulated through phosphorylation (Johnson et al., 1996), and they all possess a serine or threonine residue within the activation loop, which is a probable site for an activating modification. In some NEKs, this residue is autophosphorylated, whereas in others it is targeted by an upstream kinase (Belham et al., 2003; Bertran et al., 2011; Rellos et al., 2007; Roig et al., 2002). In terms of a phosphorylation consensus sequence, early studies found that NIMA has a strong preference for phenylalanine at position −3 (i.e. FxxS/T, where ‘x’ is any amino acid) (Lu et al., 1994). More recent studies have indicated that human NEKs have a similar preference, with both NEK2 and NEK6 preferring a hydrophobic residue, ideally phenylalanine or leucine, at the −3 position (F/LxxS/T) (Alexander et al., 2011; Lizcano et al., 2002). However, these are not strict requirements, as phosphorylation sites that do not fall into this motif have been mapped on NEK substrates.
Fig. 1.

The human NIMA-related protein kinase (NEK) family. (A) A schematic view of the eleven human NEKs, highlighting their domain organisation. Shown are the kinase domains (purple), coiled-coils (green), degradation motifs (red), RCC1 (regulator of chromatin condensation 1) domains (light blue) and armadillo repeats (yellow). A summary of what is known about the activation, localisation and function of the kinases is included. aa, amino acids. (B) Crystal structure of human NEK7 (PDB code 2WQN). Tyr97, which points down into the active site, is coloured orange and ADP is coloured red. (C) Crystal structure of human NEK2 (PDB code 2W5A). Tyr70 in the ‘upward conformation’ is coloured orange and ADP is coloured red. (D) Magnified view of NEK2 bound to a potent and selective ‘hybrid’ inhibitor that induces an inactive conformation of the activation loop (PDB code 4A4X). Atoms in the inhibitor are coloured as follows: carbon, grey; nitrogen, blue; sulphur, yellow; oxygen, red; fluorine, cyan. The ATP-binding pocket of NEK2 has a bulky gatekeeper residue (Met86) and a phenylanine residue at the base (Phe148). This is a rare combination, which severely constrains the design of ATP-competitive inhibitors. However, it is present in several NEKs.
In contrast with the conserved catalytic domains, the C-terminal regions of the NEKs are highly divergent in length, sequence and domain organisation (Fig. 1). The one relatively common feature is an oligomerisation motif, usually a coiled-coil, which promotes autophosphorylation and activation. Autophosphorylation can occur within the activation loop of the kinase domain. However, autophosphorylation is also likely to occur elsewhere in the protein, as supported by the finding that both NEK8 and NEK9 autophosphorylate in the non-catalytic C-terminal region to regulate their localisation and/or activation (Bertran et al., 2011; Zalli et al., 2012). Several NEKs, including Aspergillus NIMA and vertebrate NEK2, have also been shown to contain destruction motifs within the non-catalytic regions that target the protein for degradation (Hames et al., 2001; Pu and Osmani, 1995). NEK2 contains both a KEN (Lys-Glu-Asn) box and a C-terminal methionine-arginine dipeptide (MR)-tail, which allow recognition by the anaphase-promoting complex/cyclosome (APC/C). Unusually, the MR-tail mediates a direct interaction with core subunits of the APC/C, thereby triggering NEK2 degradation in early mitosis in a manner that is independent of the spindle assembly checkpoint (SAC) (Hayes et al., 2006). NEK6 and NEK7 notably lack a C-terminal domain. They consist solely of a kinase domain with a short N-terminal extension that might be important for substrate recognition (Vaz Meirelles et al., 2010). Indeed, the non-catalytic regions of the other NEKs almost certainly contribute to substrate recognition, as well as regulation, and this is illustrated, for example, by the recognition of NEK6 and NEK7 by a sequence in the C-terminus of NEK9 (Roig et al., 2002).
Mitotic functions for human NEKs
Overexpression of NIMA not only causes Aspergillus cells to initiate mitotic events during any stage of the cell cycle, but also induces aspects of mitotic entry when introduced into fission yeast, Xenopus oocytes or human cells (Lu and Hunter, 1995; O'Connell et al., 1994). This observation provided the first clue that NEKs might have roles in cell cycle control in higher eukaryotes and effort is now being invested to explore these functions (Fig. 2). Furthermore, details about the pathways that control the activity of cell-cycle-regulated NEKs are emerging and are summarised in Fig. 3.
Fig. 2.
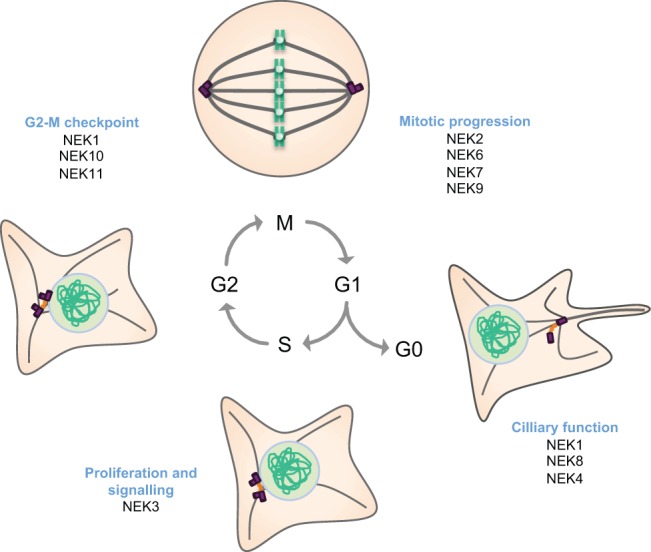
Roles for NEKs in cell cycle progression. Human NEKs contribute to different events during cell cycle progression and differentiation. Our current understanding is that during mitosis, NEK2, NEK6, NEK7 and NEK9 cooperate to ensure formation of a robust bipolar spindle that is capable of satisfying the SAC. They might also contribute to other aspects of mitotic progression, including chromatin condensation, nuclear envelope breakdown and cytokinesis. Beyond mitosis, NEK3 contributes to prolactin-dependent signalling and proliferation, whereas NEK1, NEK10 and NEK11 have all been implicated in DDR signalling, particularly at the G2-M transition. Finally, there is evidence that NEK1, NEK8 and possibly NEK4, have key roles in cilia in post-mitotic cells.
Fig. 3.

Pathways regulating NEKs in cell cycle control. (A) During interphase, NEK2 exists in a complex with protein phosphatase 1 (PP1) and mammalian STE20-like protein kinase 2 (MST2), and is maintained in an inactive state through dephosphorylation by PP1 (Helps et al., 2000; Mardin et al., 2010). Following the onset of mitosis, PLK1 phosphorylates MST2, thereby preventing PP1 from binding to the MST2–NEK2 complex (Mardin et al., 2011). This allows NEK2 to autophosphorylate and become active. NEK2 then phosphorylates the inter-centriolar linker proteins C-Nap1 and rootletin, causing their displacement from the centrosome. (B) NEK2 might also localise to the kinetochore of unaligned sister chromatids during mitosis and carry out a role in the SAC through phosphorylating HEC1 on Ser165 and interacting with MAD1 (Du et al., 2008; Lou et al., 2004; Wei et al., 2011). (C) At the onset of mitosis, CDK1 phosphorylates a number of sites in NEK9, which primes NEK9 for subsequent phosphorylation and activation by PLK1 (Bertran et al., 2011). Activated NEK9 can then phosphorylate NEK6 and NEK7, which subsequently phosphorylate components (Eg5, microtubules and the γ-TuRC) that are necessary for proper mitotic spindle formation (Sdelci et al., 2011). (D) In response to UV-induced DNA damage, NEK10 forms a trimeric complex with MEK1 and RAF1. This causes activation of MEK1, which then phosphorylates and activates ERK1/2. This leads to cell cycle arrest; however, the pathway by which this occurs has not yet been clarified. DNA breaks caused by IR activate the kinases ATM and ATR, which phosphorylate and activate CHK1. CHK1 phosphorylates NEK11 on Ser273, thereby activating it, and CDC25A on Ser76, thereby priming DSG motifs (e.g. Ser82) in CDC25A for phosphorylation by NEK11. This promotes binding of β-TrCP, which leads to the ubiquitylation and degradation of CDC25A, which, in turn, prevents CDK1–cyclin-B activation and mitotic entry. MTs, microtubules.
The role of NEKs in mitotic entry
No single human NEK is absolutely required for mitotic entry. Nonetheless, it has now been well established that NEK2, NEK6, NEK7 and NEK9 participate in the architectural changes that take place as cells move from interphase into mitosis. This includes functions in centrosome separation and mitotic spindle assembly and, potentially, chromatin condensation, nuclear pore complex (NPC) disassembly and nuclear envelope breakdown.
Of the human NEKs, NEK2 is the most closely related to Aspergillus NIMA and is the first NEK that has been studied in any depth. Like NIMA, NEK2 is cell cycle regulated, and its expression and activity peak in S and G2 phase. NEK2 is a core component of the human centrosome (Andersen et al., 2003; Fry et al., 1998b), where it regulates a key step in the centrosome cycle, namely centrosome disjunction (O'Regan et al., 2007). Importantly, many NEKs, from fungi to humans, are found at the respective microtubule-organising centres (MTOCs) (for examples, see De Souza et al., 2000; Krien et al., 2002; Mahjoub et al., 2002; Prigent et al., 2005; Wloga et al., 2006). This crucial piece of evidence has led to the hypothesis that NEKs exert their functions through regulation of centrosomes and the microtubule structures that they organise, and that this might not be restricted to dividing cells (Box 1).
Box 1. NEKs in ciliary function and ciliopathies.
Roles for NEKs in cilia were first revealed through studies in ciliated unicellular eukaryotes, such as Chlamydomonas and Tetrahymena (Bradley and Quarmby, 2005; Mahjoub et al., 2002; Wloga et al., 2006). Compared with non-ciliated fungi, the complement of genes encoding NEKs in these organisms has expanded considerably, and functional studies have suggested that these NEKs might be crucial in regulating ciliary length (Parker et al., 2007). In vertebrates, primary cilia have key roles in regulating signalling pathways that are initiated at the cell surface, and it is now understood that defective cilia underlie a wide range of inherited human genetic disorders, which are collectively referred to as ciliopathies (Bettencourt-Dias et al., 2011). A typical characteristic of these disorders is polycystic kidney disease (PKD), and studies on two mouse PKD models have led to the identification of mutations in the Nek1 and Nek8 genes (Liu et al., 2002; Upadhya et al., 2000; Vogler et al., 1999). Since then, mutations in NEK1 and NEK8 have also been identified in human ciliopathy patients; these NEKs are thought to have roles in the post-mitotic process of cilia assembly and/or function (Otto et al., 2008; Quarmby and Mahjoub, 2005; Thiel et al., 2011). Interestingly, NEK4 has also recently been implicated in cilium stability (Coene et al., 2011). Centrosomes and microtubules are intimately involved both in the assembly of the mitotic spindle and in ciliogenesis, which provides a plausible explanation for why NEKs have evolved to contribute to both processes. It has even been suggested that NEKs might somehow coordinate these processes to either induce cilia formation following cell cycle exit or to stimulate cilia resorption on cell cycle re-entry (Quarmby and Mahjoub, 2005; Spalluto et al., 2012). Moreover, besides defective ciliary signalling, aberrant cell cycle regulation or spindle orientation could directly contribute to PKD. Hence, some of the disease symptoms that result from the mutation or loss of NEK1 and NEK8 could result from the loss of a cell cycle function of these two kinases.
In human cells, centrosomes are composed of two microtubule-based barrels, called centrioles, to which proteins involved in microtubule nucleation are recruited. During interphase, the two centrioles are held in close proximity through a loose tether of filamentous proteins. This tether, or inter-centriolar linker, extends between the proximal ends of the mother and daughter centrioles and remains in place during S and G2 while the two centrioles duplicate. The role of NEK2 is to disassemble the tether at the onset of mitosis to facilitate centrosome separation and the establishment of the mitotic spindle. The organisation of the tether and how it is regulated by NEK2 have not yet been fully elucidated. However, two major components of the linker, which are related in sequence, are the large coiled-coil proteins, C-Nap1 (also known as CEP250) and rootletin (Bahe et al., 2005; Faragher and Fry, 2003; Fry et al., 1998a; Yang et al., 2006). These can be phosphorylated by NEK2, and the current hypothesis is that their phosphorylation triggers the dissolution of the tether. However, it is likely that there are other components of this linker structure that might also be regulated by NEK2, including both CEP68 and β-catenin (Bahmanyar et al., 2008; Graser et al., 2007).
Whereas disjunction of centrosomes by NEK2 facilitates spindle pole separation at the onset of mitosis, NEK6, NEK7 and NEK9 have other important roles in generating the mitotic spindle (O'Regan et al., 2007; Sdelci et al., 2011). These three proteins act in concert, whereby NEK9 is an upstream activator of NEK6 and NEK7 (Belham et al., 2003). NEK9 was originally identified through its physical association with NEK6. This interaction is more prominent during mitosis and requires NEK6 to be active (Belham et al., 2003; Rapley et al., 2008; Roig et al., 2002). Furthermore, NEK9 can phosphorylate NEK6 in vitro on a site in the activation loop that is important for NEK6 activity. Thus, on a first level, NEK9 acts as an upstream kinase for NEK6. On the basis of the high sequence similarity between NEK6 and NEK7, it has been assumed that NEK9 activates NEK7 in the same manner (Belham et al., 2003). However, on a second level, direct interaction with the non-catalytic domain of NEK9 appears to allosterically activate both NEK6 and NEK7 by disrupting an auto-inhibitory conformation of these kinases (Richards et al., 2009). The fact that NEK6 activity promotes interaction with NEK9 also raises the possibility of a feedback mechanism, whereby NEK6 (and NEK7) phosphorylates NEK9, although this remains to be investigated.
The kinase activity of all three of these NEKs is elevated in mitosis and early functional studies using overexpressed proteins and antibody microinjection provided evidence that these proteins had a role in spindle formation (Roig et al., 2002; Yissachar et al., 2006). Such a role has been confirmed through RNA interference (RNAi) depletion studies, which have revealed that the loss of NEK6, NEK7 or NEK9 leads to failure of centrosome separation in prophase and/or formation of weak mitotic spindles with reduced microtubule density and interpolar distances (Bertran et al., 2011; Kim et al., 2007; O'Regan and Fry, 2009). Importantly, these changes activate the SAC and thereby lead to mitotic arrest with cells frequently initiating apoptosis as a result. The simplest explanation for these spindle defects would be a reduction in microtubule nucleation from the centrosomes or spindle poles in mitosis. Indeed, NEK9 associates with multiple components of the γ-tubulin ring complex (γ-TuRC), which initiates microtubule nucleation, and phosphorylates the γ-TuRC adaptor protein, NEDD1/GCP-WD, whereas NEK7 might be required to recruit γ-tubulin to the poles (Kim et al., 2007; Roig et al., 2005; Sdelci et al., 2012). However, NEK6 does not obviously concentrate at spindle poles, but does weakly associate with the microtubules of the mitotic spindle itself, and both NEK6 and NEK7 co-sediment with microtubules (O'Regan and Fry, 2009). Moreover, depletion of NEK9 from Xenopus egg extracts prevents microtubule aster formation through either the centrosome- or the chromatin-mediated pathway, which suggests that NEK9 activity is not restricted to centrosomes (Roig et al., 2005). Thus, we speculate that these kinases might control microtubule nucleation not only at spindle poles but also within the spindle itself, possibly by regulating the augmin complexes that specifically recruit γ-TuRCs to spindle fibres (Goshima and Kimura, 2010).
Another route through which these kinases might control spindle formation is phosphorylation of microtubule-associated proteins, such as Eg5 (also known as kinesin-like protein KIF11), a member of the BimC kinesin family. Eg5 is a plus-end-directed motor that crosslinks and slides microtubules across one another in an anti-parallel manner, thereby driving spindle poles apart. Recruitment of Eg5 to spindle microtubules depends on its phosphorylation by CDK1 (Blangy et al., 1995; Sawin and Mitchison, 1995). However, Eg5 is also phosphorylated by NEK6, and this appears to be required for its function in centrosome separation. This could explain why depletion of either NEK6 or NEK9 can lead to monopolar spindle formation (Bertran et al., 2011; Rapley et al., 2008). It is also worth bearing in mind that NEK6 and NEK7 are capable of phosphorylating tubulin in vitro, which raises the prospect of direct regulation of microtubule dynamics through tubulin phosphorylation (O'Regan and Fry, 2009).
Beyond spindle formation, NEK2, NEK6, NEK7 and NEK9 might also have additional functions in mitotic progression. Data supporting a role for NEK2 in chromatin condensation have come from studies on mouse meiotic spermatocytes (Di Agostino et al., 2004). In these cells, chromatin condensation, as well as the activation of NEK2 and phosphorylation of the chromatin architecture protein HMGA2, is under the control of the mitogen-activated protein kinase (MAPK) pathway. NEK2 can interact with HMGA2 and phosphorylate it in vitro, which decreases the affinity of HMG2A for DNA. Thus, NEK2 might contribute to the release of HMGA2 from chromatin as cells progress from the pachytene stage of G2 into M phase. Interestingly, the NEK2C splice variant has a functional nuclear localisation signal that is absent from other splice variants, which supports the idea of a specific nuclear function for this isoform (Wu et al., 2007). However, the way in which NEK2 is activated by the MAPK pathway is not known. NEKs might also contribute to disassembly of nuclear pore complexes (NPCs) and nuclear envelope breakdown, as phosphorylation and release of the NPC component, Nup98, is driven by both CDK1 and the mitotic NEKs (Laurell et al., 2011). Moreover, NEK9 interacts with BICD2, a protein that associates with dynein and facilitates nuclear envelope breakdown at mitosis onset (Holland et al., 2002).
NEKs and cytokinesis
As mentioned above, Fin1 has a key role in regulating cytokinesis in fission yeast (Grallert et al., 2004). Meanwhile, in Drosophila, NEK2 localises to the midbody of late mitotic cells, and its overexpression results in the mislocalisation of actin and anillin at ectopic sites of cleavage furrow formation (Prigent et al., 2005). There is some evidence that vertebrate NEKs might also be involved in the final stages of cell division. First, NEK2B, a short NEK2 splice variant that lacks the APC/C-mediated degradation motifs, persists into late mitosis, and its depletion by RNAi results in delayed mitotic exit (Fletcher et al., 2005). Second, when NEK6- or NEK7-depleted cells succeed in progressing beyond metaphase, they frequently fail to complete abscission (Kim et al., 2007; O'Regan and Fry, 2009). Consistent with this, abscission defects are the most common phenotype in cells expressing hypomorphic mutants of NEK6 or NEK7 with reduced kinase activity. NEK6 and NEK7 concentrate at the midbody in late mitotic cells and the kinase activity of NEK6 is maximal during cytokinesis (Kim et al., 2007; O'Regan and Fry, 2009; Rapley et al., 2008). Furthermore, Nek7-knockout mice die in late embryogenesis or early post-natal stages, and fibroblasts derived from Nek7−/− embryos show defects that are indicative of cytokinesis failure (Salem et al., 2010). Mechanistically, it is possible that NEK6 and NEK7 regulate the localisation or activity of factors that are directly required for cytokinesis. Alternatively, these kinases could regulate the dynamics of central spindle microtubules in a similar manner to their role in the regulation of microtubules during spindle assembly (O'Regan and Fry, 2009).
The role of NEKs in cell cycle checkpoints
Cell cycle progression is monitored by a series of checkpoints that arrest the cell cycle in response to DNA damage. Whereas some NEKs, such as NEK2 and NEK6, are checkpoint targets that are inhibited by DNA damage (Fletcher et al., 2004; Lee et al., 2008), others have more integral roles in DNA-damage-response (DDR) signalling. NEK1 is involved in both the sensing and the repair of DNA strand breaks at the G1-S and G2-M transitions (Chen et al., 2011; Chen et al., 2008; Pelegrini et al., 2010; Polci et al., 2004). Depleting cells of NEK1 causes failure of checkpoint kinase 1 and 2 (CHK1 and CHK2; also known as CHEK1 and CHEK2) activation in response to ultraviolet (UV) light and ionising radiation (IR), which places NEK1 early in this pathway. However, NEK1 activation is not dependent on the ataxia telangiectasia mutated (ATM) or ataxia telangiectasia and Rad3-related protein (ATR) kinases, which suggests that NEK1 might act as an independent transducer of the damage signal.
NEK10 and NEK11 have both been implicated in the G2-M DDR checkpoint. In response to UV irradiation, but not to mitogenic growth factors, NEK10 forms a trimeric complex with MEK1 and RAF1, where RAF1 is required for the interaction between NEK10 and MEK1 (Moniz and Stambolic, 2011). Although NEK10 does not alter RAF1 kinase activity, it does promote MEK1 activation, which leads to phosphorylation of extracellular-signal-regulated kinase 1/2 (ERK1/2) and G2-M arrest. Indeed, NEK10 depletion impairs activation of MEK1 and/or ERK1/2 in response to UV treatment. NEK11 exhibits a cell-cycle-dependent expression pattern, and its expression remains high from S phase to the G2-M transition (Noguchi et al., 2002). However, NEK11 activity specifically increases in response to DNA replication inhibitors and genotoxic stresses, and this activation is lost following the inhibition of the ATM and ATR kinases (Melixetian et al., 2009; Noguchi et al., 2002). Following exposure to IR, ATM and ATR phosphorylate CHK1, which then activates NEK11 by phosphorylating it on Ser273, while also phosphorylating CDC25A on Ser76 (Melixetian et al., 2009). Phosphorylation of CDC25A on Ser76 primes it for further phosphorylation by active NEK11 within Asp-Ser-Gly (DSG) motifs of CDC25A, notably in the motif surrounding Ser82. This leads to binding of the SCF(β-TrCP) ubiquitin ligase, which targets CDC25A for proteasomal degradation and thereby causes G2-M arrest (Melixetian et al., 2009).
NEK2 might also have a role in the SAC, as it can interact with SAC components and phosphorylate the kinetochore complex protein HEC1 (also known as NDC80) (Du et al., 2008; Lou et al., 2004; Wei et al., 2011). Specifically, HEC1 is phosphorylated on Ser165 when present on the kinetochores of misaligned chromosomes and it has been suggested that this phosphorylation event is mediated by NEK2. Although not required for kinetochore localisation of Hec1, phosphorylation at this site might regulate kinetochore–microtubule binding (Du et al., 2008). However, to date, there is little evidence that manipulation of NEK2 expression or activity obviously interferes with SAC integrity.
Do ‘mitotic’ NEKs also function in interphase?
As alluded to above, some members of the NEK family have roles in non-dividing cells in regulating aspects of ciliary function. This is true both for unicellular organisms, such as Chlamydomonas and Tetrahymena, and for mammals, where mutations in NEK1 and NEK8 can lead to ciliopathy-related disorders. This aspect of NEK function is discussed in more detail in Box 1 and in the excellent review by Quarmby and Mahjoub (Quarmby and Mahjoub, 2005). However, the focus here is on those NEKs implicated in cell cycle control.
Recent data has suggested that NEK7 might have a role in regulating the centrosome cycle during interphase. NEK7 can be weakly detected at interphase centrosomes (Kim et al., 2007; Yissachar et al., 2006), although it was not detected in a proteomic analysis of centrosomes, and recombinant proteins do not obviously concentrate in this location unless they are fused to a centrosome-targeting motif (Kim et al., 2011). Nevertheless, it has been proposed that NEK7 contributes to centrosome duplication because its depletion leads to loss of centrosomes, whereas its overexpression induces formation of additional centrosomes in a kinase-dependent manner (Kim et al., 2011). NEK7 also promotes the recruitment of pericentriolar material (PCM) to centrosomes in G1 and S phase and this could explain its contribution to centrosome duplication, because the amount of PCM directly influences duplication efficiency (Loncarek et al., 2008). This result is intriguing, because it suggests that NEK7 has interphase functions that are dependent on its kinase activity, when the current view is that it is specifically activated in mitosis. Whether this is achieved through a basal level of activity that is present during interphase that is independent of NEK9 or whether there are alternative interphase-specific NEK7 activators that are perhaps localised to specific sites, remains to be seen. Interphase roles for NEK2 have also been suggested. First, in response to G1-S arrest, autophosphorylated NEK2 can phosphorylate NEK11, which results in it opening up and thus the activation of an otherwise inhibited conformation of NEK11 (Noguchi et al., 2004). Second, NEK2 phosphorylates centrobin and limits the recruitment of this daughter centriole-specific protein, which is essential for centrosome duplication, to the centrosome (Jeong et al., 2007). However, the mechanistic details for these putative roles remain unknown.
Allosteric regulatory mechanisms and chemical inhibitors
Crystal structures of the NEK2 and NEK7 catalytic domains have been determined (Rellos et al., 2007; Richards et al., 2009; Westwood et al., 2009). These structures have revealed inactive states of these proteins that have provided insights into their conformational flexibility and regulatory mechanisms (Fig. 1). Furthermore, they have enabled the generation of the first selective inhibitors that target NEK2 (Henise and Taunton, 2011; Innocenti et al., 2012; Whelligan et al., 2010). Crystal structures were particularly important in revealing the changes in NEK2 conformation that are induced by the inhibitors. In addition, they made it possible to design compounds that have improved potency against NEK2 and selectivity over other mitotic kinases. Two of the inhibitor scaffolds bind reversibly to the kinase, whereas a third one reacts to form a covalent linkage with Cys22 within the active site, thus irreversibly inhibiting catalytic activity. Consistent with RNAi studies, the use of the irreversible inhibitor indicates that NEK2 does not have an essential role in the mitotic progression of A549 cells. Further studies with the available inhibitors will enable the precise roles of NEK2 during mitosis in different cell types to be delineated, and the experience of designing NEK2 inhibitors will facilitate the generation of chemical inhibitors targeting other members of the NEK family.
The structure of NEK7, either bound to ADP or with a vacant active site, has revealed a new inhibited conformation for the kinase, in which the side-chain of Tyr97, which is located within the N-terminal lobe, points downwards into the active site (Fig. 1B) (Richards et al., 2009). This ‘Tyr-down’ conformation blocks a number of key interactions that are required for an active kinase. Indeed, mutation of this residue to alanine activates the kinase. Even the mutation of Tyr97 to a phenylalanine residue, which retains the aromatic ring but can no longer form a stabilising hydrogen bond with the backbone of Leu180, leads to some activation. Strikingly, the addition of a C-terminal fragment of NEK9 stimulates the kinase activity of wild-type NEK7, but not the already active NEK7 Y97A mutant. Similar results were obtained with NEK6, which has a tyrosine residue at the equivalent position (Tyr108). On the basis of these data, it has been proposed that NEK9 induces an allosteric activation of NEK7 (and NEK6) that is independent of its ability to activate these kinases through activation loop phosphorylation. Analysis of the kinase domain of NEK2 structure has revealed that it, too, has a tyrosine at this position (Fig. 1C, Tyr70). In the presence of ADP, the side-chain of this residue points upwards and out of the active site, that is, it is not inhibitory. Nonetheless, structures of NEK2 in the presence of ATP-competitive inhibitors have revealed a Tyr-down conformation, which indicates that this residue is flexible and can be switched up or down (Richards et al., 2009; Whelligan et al., 2010). Eight out of eleven NEKs, and ∼10% of human kinases in total, have a tyrosine residue at this position in their catalytic domains, thus it is possible that this mode of regulation applies to a number of these enzymes as well.
Structural studies of the non-catalytic domains of NEKs are yet to really begin but have the potential to reveal many more important modes of regulation. Biophysical and nuclear magnetic resonance (NMR)-based studies have been performed on the coiled-coil motif of NEK2, which lies just downstream of the catalytic domain (Croasdale et al., 2011). This motif promotes homodimerisation and activation of NEK2, probably by trans-autophosphorylation (Fry et al., 1999). According to sequence analysis, this motif adopts a rather unusual leucine zipper that could dimerise through one of two registers, and the NMR data strongly support a model whereby this motif interchanges between these two registers on a relatively slow timescale (17 s−1). On one hand, this argues that it will be challenging to obtain a crystal structure of the full-length protein; on the other hand, it opens up the possibility that interactions or post-translational modifications could regulate NEK2 by favouring adoption of one register over the other.
NEKs as anti-cancer targets
Microtubule poisons such as taxanes and vinca alkaloids are effective therapies in the treatment of many cancers, including breast, ovarian and lung tumours (Jordan and Wilson, 2004). However, their clinical use is limited by side effects, such as neuropathy, poor responses in a subset of patients and drug resistance. Microtubule poisons induce mitotic arrest by interfering with microtubule dynamics, which eventually leads to cell death. Patients would clearly benefit from compounds that trigger a similar cellular response but do not affect the functions of microtubules in nerve cells, and such a treatment could be used either to complement, or as an alternative to, microtubule poisons (Jackson et al., 2007). Members of the NEK family are attractive drug targets because they are involved in specific aspects of microtubule function and/or the DNA damage checkpoint, which is another precedented target pathway for cancer drug discovery (Box 2).
Box 2. Mitotic kinases as anti-cancer targets.
There is an ever-present need for new chemotherapeutics to provide effective therapies for cancer patients. As a consequence, there is currently considerable interest in mitotic kinases as potential cancer drug targets. Intensive medicinal chemistry research has resulted in the development of a number of selective inhibitors for CDK1, PLK1, Aurora A and Aurora B. Results from clinical trials on these compounds have shown modest anti-tumour activity. To allow further progress to be made in the development of anti-mitotic cancer drugs, it is crucial to explain why these compounds have not lived up to the promise from studies in cell and animal models. The fact that mitotic kinase inhibitors cause haematological toxicity shows that they are active against their targets in patient bone marrow cells, and, indeed, haematological malignancies have shown the most encouraging responses to mitotic kinase inhibitors. It has therefore been suggested that targeting mitosis alone might not form a sufficient basis for the effective therapy of solid tumours, which have a very low rate of cell duplication (Komlodi-Pasztor et al., 2011). Furthermore, mitotic kinases have varied and complex roles in cell cycle checkpoint and cell death pathways, and cells treated with mitotic kinase inhibitors have diverse fates (Gascoigne and Taylor, 2008). Because the functions of many NEKs are poorly characterised, it is not yet clear how we could define the desired selectivity profile of a successful and safe NEK inhibitor, although we might predict that it would be undesirable to inhibit NEKs that have been implicated in ciliary function. Nevertheless, the development of selective compounds for mechanistic studies is clearly a high priority if we are to understand how NEK inhibitors might be developed as effective clinical treatments.
To date, the potential of NEKs as anti-cancer targets is relatively unexplored and only limited target validation studies have been carried out on a few members of the family. NEK2 is a potential drug target in breast cancer (Hayward et al., 2004; Tsunoda et al., 2009; Wu et al., 2008), as well as cholangiocarcinoma and colorectal cancer (Kokuryo et al., 2007; Suzuki et al., 2010). The availability of NEK2-selective inhibitors should enable further target validation studies in these and other cancers. There is also evidence in support of NEK6 as a tumour-promoting protein and potential cancer drug target. NEK6 activity promotes anchorage-independent growth; depletion of NEK6 leads to death in cancer cell lines but is tolerated by normal fibroblasts (Nassirpour et al., 2010). Overexpression of NEK6 also inhibits p53-dependent cellular senescence (Jee et al., 2010). The molecular mechanisms that underlie these observations are unknown, and could reflect a non-mitotic function of NEK6. For example, NEK6 might affect the transcriptional programme of a cancer cell through phosphorylation of STAT3 on Ser727, an event that is required for maximal transcriptional activity (Jeon et al., 2010). Although kinases are generally regarded as druggable, developing NEK6-selective compounds will be a challenge, because the active site is identical to that of NEK7. Furthermore, these two kinases are particularly insensitive to ATP-competitive inhibitors, which suggests that an alternative approach to drug design might be required.
Conclusions and future perspectives
It has now been well established that some human NEKs have roles in mitotic progression, whereas others contribute to cilia function (Quarmby and Mahjoub, 2005). The common theme that underlies these two processes is the regulation of microtubule-dependent processes, MTOCs or microtubules themselves (Fig. 4). Phylogenetic analysis has indicated that the last common ancestor of eukaryotes, which was a ciliated cell, probably expressed as many as five NEKs, of which some regulated cell division and others regulated cilia (Parker et al., 2007). This analysis has also indicated that the NEKs can be divided into sub-families that might predict functional similarity. For example, NEK1, NEK3 and NEK5 fall into the same sub-family, whereas NEK2 is indeed probably the orthologue of NIMA and the yeast NEKs. The hypothesis is that those organisms without cilia, for example, Aspergillus, have lost the cilia-related NEKs, whereas organisms with complex cilia arrangements, for example, Tetrahymena, have expanded the repertoire of cilia-related NEKs. Human cells, of course, possess NEKs that are devoted to each process. However, it is interesting to consider that some NEKs might directly coordinate the two events, as seems to be the case for some of the Chlamydomonas NEKs (Bradley and Quarmby, 2005; Mahjoub et al., 2002). For example, NEK7 is intimately linked with mitosis, but the percentage of MEFs derived from Nek7-knockout mice that bear primary cilia is reduced compared with that in wild-type cells (Salem et al., 2010). NEK3 has a role in prolactin-mediated signalling, but also potentially regulates the acetylation status of microtubules in neurons (Chang et al., 2009; Miller et al., 2007). Furthermore, there is good evidence that NEK1 has roles in both the DDR and cilia function, and NEK2 has recently been proposed to promote cilium disassembly at the onset of mitosis (Spalluto et al., 2012). Another mitotic kinase, Aurora A, coordinates cilia resorption with mitotic entry, and this requires activation of Aurora A by the focal adhesion scaffolding protein NEDD9 (also known as HEF1 and Cas-L), which also happens be a regulator of NEK2 (Pugacheva and Golemis, 2005; Pugacheva et al., 2007).
Fig. 4.

Potential roles for NEKs in microtubule organisation. This diagram illustrates how NEKs might have evolved to contribute to different aspects of microtubule organisation in cells. (A) NEKs can regulate assembly of the microtubule-based mitotic spindle. NEK2 promotes loss of cohesion between the interphase centrosomes, whereas NEK6 and NEK7, under the control of NEK9, contribute to robust bipolar spindle assembly. (B) Some NEKs, such as NEK1 and NEK8, have roles in cilia, possibly through regulation of axonemal microtubules and/or basal bodies to control ciliary length. However, they might impact on ciliary function through alternative routes such as regulating the stability of proteins such as polycystin 2 (Yim et al., 2011). (C) We speculate that NEKs could have direct roles in regulating microtubules themselves. For example, NEK6 and NEK7 could phosphorylate α- or β-tubulin or microtubule-associated proteins, such as +TIP tracking proteins, to regulate microtubule dynamics, or they could modulate the activity of microtubule-based motor proteins, such as Eg5, or tubulin-modifying enzymes, such as those that polyglutamylate (indicated by ‘Glu’) the C-terminal tails of tubulin.
From a cancer biology perspective, overexpression of the mitotic NEKs in various tumour types suggests that they could act as important prognostic biomarkers. Furthermore, from what we currently know about their functions, inhibition of the mitotic or DDR NEKs has the potential to selectively interfere with the proliferation of cancer cells. However, greater structural detail of the NEKs is clearly required to understand their mechanisms of activation and to design specific inhibitors. Meanwhile, the development of knockout animals for the different NEKs will be an important step in defining their key physiological roles, as well as the potential side effects of NEK inhibitors. Finally, defining therapeutic windows, synthetic lethal interactions and potential combination treatments is a huge challenge that would clearly benefit from the development of cell-permeable inhibitors as tools. In summary, there is still a very long way to go in terms of understanding the basic biology of this kinase family. Only with the development of this understanding will the mechanisms through which NEKs contribute to human disease become clear and only then will we be in a position to fully exploit them as therapeutic targets.
Acknowledgments
We thank Sharon Yeoh for comments on the manuscript and sincerely apologise to authors whose work we have not had space to cite.
Footnotes
Funding
The work of A.M.F. is supported by the Wellcome Trust; Cancer Research UK; and the Association for International Cancer Research (AICR). R.B. acknowledges support from Cancer Research UK; and the Royal Society. The authors are members of the Leicester Cancer Research UK Experimental Cancer Medicine Centre.
References
- Alexander J., Lim D., Joughin B. A., Hegemann B., Hutchins J. R., Ehrenberger T., Ivins F., Sessa F., Hudecz O., Nigg E. A.et al. (2011). Spatial exclusivity combined with positive and negative selection of phosphorylation motifs is the basis for context-dependent mitotic signaling. Sci. Signal. 4, ra42 10.1126/scisignal.2001796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen J. S., Wilkinson C. J., Mayor T., Mortensen P., Nigg E. A., Mann M. (2003). Proteomic characterization of the human centrosome by protein correlation profiling. Nature 426, 570–574 10.1038/nature02166 [DOI] [PubMed] [Google Scholar]
- Bahe S., Stierhof Y. D., Wilkinson C. J., Leiss F., Nigg E. A. (2005). Rootletin forms centriole-associated filaments and functions in centrosome cohesion. J. Cell Biol. 171, 27–33 10.1083/jcb.200504107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahmanyar S., Kaplan D. D., Deluca J. G., Giddings T. H., Jr, O’Toole E. T., Winey M., Salmon E. D., Casey P. J., Nelson W. J., Barth A. I. (2008). beta-Catenin is a Nek2 substrate involved in centrosome separation. Genes Dev. 22, 91–105 10.1101/gad.1596308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belham C., Roig J., Caldwell J. A., Aoyama Y., Kemp B. E., Comb M., Avruch J. (2003). A mitotic cascade of NIMA family kinases. Nercc1/Nek9 activates the Nek6 and Nek7 kinases. J. Biol. Chem. 278, 34897–34909 10.1074/jbc.M303663200 [DOI] [PubMed] [Google Scholar]
- Bertran M. T., Sdelci S., Regué L., Avruch J., Caelles C., Roig J. (2011). Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. EMBO J. 30, 2634–2647 10.1038/emboj.2011.179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettencourt–Dias M., Hildebrandt F., Pellman D., Woods G., Godinho S. A. (2011). Centrosomes and cilia in human disease. Trends Genet. 27, 307–315 10.1016/j.tig.2011.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blangy A., Lane H. A., d’Hérin P., Harper M., Kress M., Nigg E. A. (1995). Phosphorylation by p34cdc2 regulates spindle association of human Eg5, a kinesin-related motor essential for bipolar spindle formation in vivo. Cell 83, 1159–1169 10.1016/0092-8674(95)90142-6 [DOI] [PubMed] [Google Scholar]
- Bradley B. A., Quarmby L. M. (2005). A NIMA-related kinase, Cnk2p, regulates both flagellar length and cell size in Chlamydomonas. J. Cell Sci. 118, 3317–3326 10.1242/jcs.02455 [DOI] [PubMed] [Google Scholar]
- Chang J., Baloh R. H., Milbrandt J. (2009). The NIMA-family kinase Nek3 regulates microtubule acetylation in neurons. J. Cell Sci. 122, 2274–2282 10.1242/jcs.048975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Chen P. L., Chen C. F., Jiang X., Riley D. J. (2008). Never-in-mitosis related kinase 1 functions in DNA damage response and checkpoint control. Cell Cycle 7, 3194–3201 10.4161/cc.7.20.6815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y., Chen C. F., Riley D. J., Chen P. L. (2011). Nek1 kinase functions in DNA damage response and checkpoint control through a pathway independent of ATM and ATR. Cell Cycle 10, 655–663 10.4161/cc.10.4.14814 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coene K. L., Mans D. A., Boldt K., Gloeckner C. J., van Reeuwijk J., Bolat E., Roosing S., Letteboer S. J., Peters T. A., Cremers F. P.et al. (2011). The ciliopathy-associated protein homologs RPGRIP1 and RPGRIP1L are linked to cilium integrity through interaction with Nek4 serine/threonine kinase. Hum. Mol. Genet. 20, 3592–3605 10.1093/hmg/ddr280 [DOI] [PubMed] [Google Scholar]
- Croasdale R., Ivins F. J., Muskett F., Daviter T., Scott D. J., Hardy T., Smerdon S. J., Fry A. M., Pfuhl M. (2011). An undecided coiled coil: the leucine zipper of Nek2 kinase exhibits atypical conformational exchange dynamics. J. Biol. Chem. 286, 27537–27547 10.1074/jbc.M110.196972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Souza C. P., Osmani A. H., Wu L. P., Spotts J. L., Osmani S. A. (2000). Mitotic histone H3 phosphorylation by the NIMA kinase in Aspergillus nidulans. Cell 102, 293–302 10.1016/S0092-8674(00)00035-0 [DOI] [PubMed] [Google Scholar]
- Di Agostino S., Fedele M., Chieffi P., Fusco A., Rossi P., Geremia R., Sette C. (2004). Phosphorylation of high-mobility group protein A2 by Nek2 kinase during the first meiotic division in mouse spermatocytes. Mol. Biol. Cell 15, 1224–1232 10.1091/mbc.E03-09-0638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du J., Cai X., Yao J., Ding X., Wu Q., Pei S., Jiang K., Zhang Y., Wang W., Shi Y.et al. (2008). The mitotic checkpoint kinase NEK2A regulates kinetochore microtubule attachment stability. Oncogene 27, 4107–4114 10.1038/onc.2008.34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faragher A. J., Fry A. M. (2003). Nek2A kinase stimulates centrosome disjunction and is required for formation of bipolar mitotic spindles. Mol. Biol. Cell 14, 2876–2889 10.1091/mbc.E03-02-0108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fletcher L., Cerniglia G. J., Nigg E. A., Yend T. J., Muschel R. J. (2004). Inhibition of centrosome separation after DNA damage: a role for Nek2. Radiat. Res. 162, 128–135 10.1667/RR3211 [DOI] [PubMed] [Google Scholar]
- Fletcher L., Cerniglia G. J., Yen T. J., Muschel R. J. (2005). Live cell imaging reveals distinct roles in cell cycle regulation for Nek2A and Nek2B. Biochim. Biophys. Acta 1744, 89–92 10.1016/j.bbamcr.2005.01.007 [DOI] [PubMed] [Google Scholar]
- Fry A. M., Mayor T., Meraldi P., Stierhof Y. D., Tanaka K., Nigg E. A. (1998a). C-Nap1, a novel centrosomal coiled-coil protein and candidate substrate of the cell cycle-regulated protein kinase Nek2. J. Cell Biol. 141, 1563–1574 10.1083/jcb.141.7.1563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A. M., Meraldi P., Nigg E. A. (1998b). A centrosomal function for the human Nek2 protein kinase, a member of the NIMA family of cell cycle regulators. EMBO J. 17, 470–481 10.1093/emboj/17.2.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fry A. M., Arnaud L., Nigg E. A. (1999). Activity of the human centrosomal kinase, Nek2, depends on an unusual leucine zipper dimerization motif. J. Biol. Chem. 274, 16304–16310 10.1074/jbc.274.23.16304 [DOI] [PubMed] [Google Scholar]
- Gascoigne K. E., Taylor S. S. (2008). Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell 14, 111–122 10.1016/j.ccr.2008.07.002 [DOI] [PubMed] [Google Scholar]
- Goshima G., Kimura A. (2010). New look inside the spindle: microtubule-dependent microtubule generation within the spindle. Curr. Opin. Cell Biol. 22, 44–49 10.1016/j.ceb.2009.11.012 [DOI] [PubMed] [Google Scholar]
- Grallert A., Hagan I. M. (2002). Schizosaccharomyces pombe NIMA-related kinase, Fin1, regulates spindle formation and an affinity of Polo for the SPB. EMBO J. 21, 3096–3107 10.1093/emboj/cdf294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grallert A., Krapp A., Bagley S., Simanis V., Hagan I. M. (2004). Recruitment of NIMA kinase shows that maturation of the S. pombe spindle-pole body occurs over consecutive cell cycles and reveals a role for NIMA in modulating SIN activity. Genes Dev. 18, 1007–1021 10.1101/gad.296204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graser S., Stierhof Y. D., Nigg E. A. (2007). Cep68 and Cep215 (Cdk5rap2) are required for centrosome cohesion. J. Cell Sci. 120, 4321–4331 10.1242/jcs.020248 [DOI] [PubMed] [Google Scholar]
- Hames R. S., Wattam S. L., Yamano H., Bacchieri R., Fry A. M. (2001). APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J. 20, 7117–7127 10.1093/emboj/20.24.7117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes M. J., Kimata Y., Wattam S. L., Lindon C., Mao G., Yamano H., Fry A. M. (2006). Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat. Cell Biol. 8, 607–614 10.1038/ncb1410 [DOI] [PubMed] [Google Scholar]
- Hayward D. G., Clarke R. B., Faragher A. J., Pillai M. R., Hagan I. M., Fry A. M. (2004). The centrosomal kinase Nek2 displays elevated levels of protein expression in human breast cancer. Cancer Res. 64, 7370–7376 10.1158/0008-5472.CAN-04-0960 [DOI] [PubMed] [Google Scholar]
- Helps N. R., Luo X., Barker H. M., Cohen P. T. (2000). NIMA-related kinase 2 (Nek2), a cell-cycle-regulated protein kinase localized to centrosomes, is complexed to protein phosphatase 1. Biochem. J. 349, 509–518 10.1042/0264-6021:3490509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henise J. C., Taunton J. (2011). Irreversible Nek2 kinase inhibitors with cellular activity. J. Med. Chem. 54, 4133–4146 10.1021/jm200222m [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland P. M., Milne A., Garka K., Johnson R. S., Willis C., Sims J. E., Rauch C. T., Bird T. A., Virca G. D. (2002). Purification, cloning, and characterization of Nek8, a novel NIMA-related kinase, and its candidate substrate Bicd2. J. Biol. Chem. 277, 16229–16240 10.1074/jbc.M108662200 [DOI] [PubMed] [Google Scholar]
- Innocenti P., Cheung K. M., Solanki S., Mas–Droux C., Rowan F., Yeoh S., Boxall K., Westlake M., Pickard L., Hardy T.et al. (2012). Design of potent and selective hybrid inhibitors of the mitotic kinase Nek2: structure-activity relationship, structural biology, and cellular activity. J. Med. Chem. 55, 3228–3241 10.1021/jm201683b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson J. R., Patrick D. R., Dar M. M., Huang P. S. (2007). Targeted anti-mitotic therapies: can we improve on tubulin agents? Nat. Rev. Cancer 7, 107–117 10.1038/nrc2049 [DOI] [PubMed] [Google Scholar]
- Jee H. J., Kim A. J., Song N., Kim H. J., Kim M., Koh H., Yun J. (2010). Nek6 overexpression antagonizes p53-induced senescence in human cancer cells. Cell Cycle 9, 4703–4710 10.4161/cc.9.23.14059 [DOI] [PubMed] [Google Scholar]
- Jeon Y. J., Lee K. Y., Cho Y. Y., Pugliese A., Kim H. G., Jeong C. H., Bode A. M., Dong Z. (2010). Role of NEK6 in tumor promoter-induced transformation in JB6 C141 mouse skin epidermal cells. J. Biol. Chem. 285, 28126–28133 10.1074/jbc.M110.137190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong Y., Lee J., Kim K., Yoo J. C., Rhee K. (2007). Characterization of NIP2/centrobin, a novel substrate of Nek2, and its potential role in microtubule stabilization. J. Cell Sci. 120, 2106–2116 10.1242/jcs.03458 [DOI] [PubMed] [Google Scholar]
- Johnson L. N., Noble M. E., Owen D. J. (1996). Active and inactive protein kinases: structural basis for regulation. Cell 85, 149–158 10.1016/S0092-8674(00)81092-2 [DOI] [PubMed] [Google Scholar]
- Jordan M. A., Wilson L. (2004). Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 4, 253–265 10.1038/nrc1317 [DOI] [PubMed] [Google Scholar]
- Kim S., Lee K., Rhee K. (2007). NEK7 is a centrosomal kinase critical for microtubule nucleation. Biochem. Biophys. Res. Commun. 360, 56–62 10.1016/j.bbrc.2007.05.206 [DOI] [PubMed] [Google Scholar]
- Kim S., Kim S., Rhee K.2011). NEK7 is essential for centriole duplication and centrosomal accumulation of pericentriolar material proteins in interphase cells. J. Cell Sci. 124, 3760–3770 10.1242/jcs.078089 [DOI] [PubMed] [Google Scholar]
- Kokuryo T., Senga T., Yokoyama Y., Nagino M., Nimura Y., Hamaguchi M. (2007). Nek2 as an effective target for inhibition of tumorigenic growth and peritoneal dissemination of cholangiocarcinoma. Cancer Res. 67, 9637–9642 10.1158/0008-5472.CAN-07-1489 [DOI] [PubMed] [Google Scholar]
- Komlodi–Pasztor E., Sackett D., Wilkerson J., Fojo T. (2011). Mitosis is not a key target of microtubule agents in patient tumors. Nat. Rev. Clin. Oncol. 8, 244–250 10.1038/nrclinonc.2010.228 [DOI] [PubMed] [Google Scholar]
- Krien M. J., Bugg S. J., Palatsides M., Asouline G., Morimyo M., O’Connell M. J. (1998). A NIMA homologue promotes chromatin condensation in fission yeast. J. Cell Sci. 111, 967–976 [DOI] [PubMed] [Google Scholar]
- Krien M. J., West R. R., John U. P., Koniaras K., McIntosh J. R., O’Connell M. J. (2002). The fission yeast NIMA kinase Fin1p is required for spindle function and nuclear envelope integrity. EMBO J. 21, 1713–1722 10.1093/emboj/21.7.1713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurell E., Beck K., Krupina K., Theerthagiri G., Bodenmiller B., Horvath P., Aebersold R., Antonin W., Kutay U. (2011). Phosphorylation of Nup98 by multiple kinases is crucial for NPC disassembly during mitotic entry. Cell 144, 539–550 10.1016/j.cell.2011.01.012 [DOI] [PubMed] [Google Scholar]
- Lee M. Y., Kim H. J., Kim M. A., Jee H. J., Kim A. J., Bae Y. S., Park J. I., Chung J. H., Yun J. (2008). Nek6 is involved in G2/M phase cell cycle arrest through DNA damage-induced phosphorylation. Cell Cycle 7, 2705–2709 10.4161/cc.7.17.6551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S., Lu W., Obara T., Kuida S., Lehoczky J., Dewar K., Drummond I. A., Beier D. R. (2002). A defect in a novel Nek-family kinase causes cystic kidney disease in the mouse and in zebrafish. Development 129, 5839–5846 10.1242/dev.00173 [DOI] [PubMed] [Google Scholar]
- Lizcano J. M., Deak M., Morrice N., Kieloch A., Hastie C. J., Dong L., Schutkowski M., Reimer U., Alessi D. R. (2002). Molecular basis for the substrate specificity of NIMA-related kinase-6 (NEK6). Evidence that NEK6 does not phosphorylate the hydrophobic motif of ribosomal S6 protein kinase and serum- and glucocorticoid-induced protein kinase in vivo. J. Biol. Chem. 277, 27839–27849 10.1074/jbc.M202042200 [DOI] [PubMed] [Google Scholar]
- Loncarek J., Hergert P., Magidson V., Khodjakov A. (2008). Control of daughter centriole formation by the pericentriolar material. Nat. Cell Biol. 10, 322–328 10.1038/ncb1694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Y., Yao J., Zereshki A., Dou Z., Ahmed K., Wang H., Hu J., Wang Y., Yao X. (2004). NEK2A interacts with MAD1 and possibly functions as a novel integrator of the spindle checkpoint signaling. J. Biol. Chem. 279, 20049–20057 10.1074/jbc.M314205200 [DOI] [PubMed] [Google Scholar]
- Lu K. P., Hunter T. (1995). Evidence for a NIMA-like mitotic pathway in vertebrate cells. Cell 81, 413–424 10.1016/0092-8674(95)90394-1 [DOI] [PubMed] [Google Scholar]
- Lu K. P., Kemp B. E., Means A. R. (1994). Identification of substrate specificity determinants for the cell cycle-regulated NIMA protein kinase. J. Biol. Chem. 269, 6603–6607 [PubMed] [Google Scholar]
- Mahjoub M. R., Montpetit B., Zhao L., Finst R. J., Goh B., Kim A. C., Quarmby L. M. (2002). The FA2 gene of Chlamydomonas encodes a NIMA family kinase with roles in cell cycle progression and microtubule severing during deflagellation. J. Cell Sci. 115, 1759–1768 [DOI] [PubMed] [Google Scholar]
- Mardin B. R., Lange C., Baxter J. E., Hardy T., Scholz S. R., Fry A. M., Schiebel E. (2010). Components of the Hippo pathway cooperate with Nek2 kinase to regulate centrosome disjunction. Nat. Cell Biol. 12, 1166–1176 10.1038/ncb2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mardin B. R., Agircan F. G., Lange C., Schiebel E. (2011). Plk1 controls the Nek2A-PP1γ antagonism in centrosome disjunction. Curr. Biol. 21, 1145–1151 10.1016/j.cub.2011.05.047 [DOI] [PubMed] [Google Scholar]
- Melixetian M., Klein D. K., Sørensen C. S., Helin K. (2009). NEK11 regulates CDC25A degradation and the IR-induced G2/M checkpoint. Nat. Cell Biol. 11, 1247–1253 10.1038/ncb1969 [DOI] [PubMed] [Google Scholar]
- Miller S. L., Antico G., Raghunath P. N., Tomaszewski J. E., Clevenger C. V. (2007). Nek3 kinase regulates prolactin-mediated cytoskeletal reorganization and motility of breast cancer cells. Oncogene 26, 4668–4678 10.1038/sj.onc.1210264 [DOI] [PubMed] [Google Scholar]
- Moniz L. S., Stambolic V. (2011). Nek10 mediates G2/M cell cycle arrest and MEK autoactivation in response to UV irradiation. Mol. Cell. Biol. 31, 30–42 10.1128/MCB.00648-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moniz L., Dutt P., Haider N., Stambolic V. (2011). Nek family of kinases in cell cycle, checkpoint control and cancer. Cell Div. 6, 18 10.1186/1747-1028-6-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassirpour R., Shao L., Flanagan P., Abrams T., Jallal B., Smeal T., Yin M. J. (2010). Nek6 mediates human cancer cell transformation and is a potential cancer therapeutic target. Mol. Cancer Res. 8, 717–728 10.1158/1541-7786.MCR-09-0291 [DOI] [PubMed] [Google Scholar]
- Noguchi K., Fukazawa H., Murakami Y., Uehara Y. (2002). Nek11, a new member of the NIMA family of kinases, involved in DNA replication and genotoxic stress responses. J. Biol. Chem. 277, 39655–39665 10.1074/jbc.M204599200 [DOI] [PubMed] [Google Scholar]
- Noguchi K., Fukazawa H., Murakami Y., Uehara Y. (2004). Nucleolar Nek11 is a novel target of Nek2A in G1/S-arrested cells. J. Biol. Chem. 279, 32716–32727 10.1074/jbc.M404104200 [DOI] [PubMed] [Google Scholar]
- O’Connell M. J., Norbury C., Nurse P. (1994). Premature chromatin condensation upon accumulation of NIMA. EMBO J. 13, 4926–4937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Connell M. J., Krien M. J., Hunter T. (2003). Never say never. The NIMA-related protein kinases in mitotic control. Trends Cell Biol. 13, 221–228 10.1016/S0962-8924(03)00056-4 [DOI] [PubMed] [Google Scholar]
- O’Regan L., Blot J., Fry A. M. (2007). Mitotic regulation by NIMA-related kinases. Cell Div. 2, 25 10.1186/1747-1028-2-25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Regan L., Fry A. M. (2009). The Nek6 and Nek7 protein kinases are required for robust mitotic spindle formation and cytokinesis. Mol. Cell. Biol. 29, 3975–3990 10.1128/MCB.01867-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakley B. R., Morris N. R. (1983). A mutation in Aspergillus nidulans that blocks the transition from interphase to prophase. J. Cell Biol. 96, 1155–1158 10.1083/jcb.96.4.1155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmani S. A., Pu R. T., Morris N. R. (1988). Mitotic induction and maintenance by overexpression of a G2-specific gene that encodes a potential protein kinase. Cell 53, 237–244 10.1016/0092-8674(88)90385-6 [DOI] [PubMed] [Google Scholar]
- Osmani A. H., McGuire S. L., Osmani S. A. (1991). Parallel activation of the NIMA and p34cdc2 cell cycle-regulated protein kinases is required to initiate mitosis in A. nidulans. Cell 67, 283–291 10.1016/0092-8674(91)90180-7 [DOI] [PubMed] [Google Scholar]
- Otto E. A., Trapp M. L., Schultheiss U. T., Helou J., Quarmby L. M., Hildebrandt F. (2008). NEK8 mutations affect ciliary and centrosomal localization and may cause nephronophthisis. J. Am. Soc. Nephrol. 19, 587–592 10.1681/ASN.2007040490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J. D., Bradley B. A., Mooers A. O., Quarmby L. M. (2007). Phylogenetic analysis of the Neks reveals early diversification of ciliary-cell cycle kinases. PLoS ONE 2, e1076 10.1371/journal.pone.0001076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelegrini A. L., Moura D. J., Brenner B. L., Ledur P. F., Maques G. P., Henriques J. A., Saffi J., Lenz G. (2010). Nek1 silencing slows down DNA repair and blocks DNA damage-induced cell cycle arrest. Mutagenesis 25, 447–454 10.1093/mutage/geq026 [DOI] [PubMed] [Google Scholar]
- Polci R., Peng A., Chen P. L., Riley D. J., Chen Y. (2004). NIMA-related protein kinase 1 is involved early in the ionizing radiation-induced DNA damage response. Cancer Res. 64, 8800–8803 10.1158/0008-5472.CAN-04-2243 [DOI] [PubMed] [Google Scholar]
- Prigent C., Glover D. M., Giet R. (2005). Drosophila Nek2 protein kinase knockdown leads to centrosome maturation defects while overexpression causes centrosome fragmentation and cytokinesis failure. Exp. Cell Res. 303, 1–13 [DOI] [PubMed] [Google Scholar]
- Pu R. T., Osmani S. A. (1995). Mitotic destruction of the cell cycle regulated NIMA protein kinase of Aspergillus nidulans is required for mitotic exit. EMBO J. 14, 995–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva E. N., Golemis E. A. (2005). The focal adhesion scaffolding protein HEF1 regulates activation of the Aurora-A and Nek2 kinases at the centrosome. Nat. Cell Biol. 7, 937–946 10.1038/ncb1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugacheva E. N., Jablonski S. A., Hartman T. R., Henske E. P., Golemis E. A. (2007). HEF1-dependent Aurora A activation induces disassembly of the primary cilium. Cell 129, 1351–1363 10.1016/j.cell.2007.04.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quarmby L. M., Mahjoub M. R. (2005). Caught Nek-ing: cilia and centrioles. J. Cell Sci. 118, 5161–5169 10.1242/jcs.02681 [DOI] [PubMed] [Google Scholar]
- Rapley J., Nicolàs M., Groen A., Regué L., Bertran M. T., Caelles C., Avruch J., Roig J. (2008). The NIMA-family kinase Nek6 phosphorylates the kinesin Eg5 at a novel site necessary for mitotic spindle formation. J. Cell Sci. 121, 3912–3921 10.1242/jcs.035360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rellos P., Ivins F. J., Baxter J. E., Pike A., Nott T. J., Parkinson D. M., Das S., Howell S., Fedorov O., Shen Q. Y.et al. (2007). Structure and regulation of the human Nek2 centrosomal kinase. J. Biol. Chem. 282, 6833–6842 10.1074/jbc.M609721200 [DOI] [PubMed] [Google Scholar]
- Richards M. W., O’Regan L., Mas–Droux C., Blot J. M., Cheung J., Hoelder S., Fry A. M., Bayliss R. (2009). An autoinhibitory tyrosine motif in the cell-cycle-regulated Nek7 kinase is released through binding of Nek9. Mol. Cell 36, 560–570 10.1016/j.molcel.2009.09.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig J., Mikhailov A., Belham C., Avruch J. (2002). Nercc1, a mammalian NIMA-family kinase, binds the Ran GTPase and regulates mitotic progression. Genes Dev. 16, 1640–1658 10.1101/gad.972202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roig J., Groen A., Caldwell J., Avruch J. (2005). Active Nercc1 protein kinase concentrates at centrosomes early in mitosis and is necessary for proper spindle assembly. Mol. Biol. Cell 16, 4827–4840 10.1091/mbc.E05-04-0315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salem H., Rachmin I., Yissachar N., Cohen S., Amiel A., Haffner R., Lavi L., Motro B. (2010). Nek7 kinase targeting leads to early mortality, cytokinesis disturbance and polyploidy. Oncogene 29, 4046–4057 10.1038/onc.2010.162 [DOI] [PubMed] [Google Scholar]
- Sawin K. E., Mitchison T. J. (1995). Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc. Natl. Acad. Sci. USA 92, 4289–4293 10.1073/pnas.92.10.4289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sdelci S., Bertran M. T., Roig J. (2011). Nek9, Nek6, Nek7 and the separation of centrosomes. Cell Cycle 10, 3816–3817 10.4161/cc.10.22.18226 [DOI] [PubMed] [Google Scholar]
- Sdelci S., Schütz M., Pinyol R., Bertran M. T., Regué L., Caelles C., Vernos I., Roig J. (2012). Nek9 phosphorylation of NEDD1/GCP-WD contributes to Plk1 control of γ-tubulin recruitment to the mitotic centrosome. Curr. Biol. 22, 1516–1523 10.1016/j.cub.2012.06.027 [DOI] [PubMed] [Google Scholar]
- Spalluto C., Wilson D. I., Hearn T. (2012). Nek2 localises to the distal portion of the mother centriole/basal body and is required for timely cilium disassembly at the G2/M transition. Eur. J. Cell Biol. 91, 675–686 10.1016/j.ejcb.2012.03.009 [DOI] [PubMed] [Google Scholar]
- Suzuki K., Kokuryo T., Senga T., Yokoyama Y., Nagino M., Hamaguchi M. (2010). Novel combination treatment for colorectal cancer using Nek2 siRNA and cisplatin. Cancer Sci. 101, 1163–1169 10.1111/j.1349-7006.2010.01504.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel C., Kessler K., Giessl A., Dimmler A., Shalev S. A., von der Haar S., Zenker M., Zahnleiter D., Stöss H., Beinder E.et al. (2011). NEK1 mutations cause short-rib polydactyly syndrome type majewski. Am. J. Hum. Genet. 88, 106–114 10.1016/j.ajhg.2010.12.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsunoda N., Kokuryo T., Oda K., Senga T., Yokoyama Y., Nagino M., Nimura Y., Hamaguchi M. (2009). Nek2 as a novel molecular target for the treatment of breast carcinoma. Cancer Sci. 100, 111–116 10.1111/j.1349-7006.2008.01007.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Upadhya P., Birkenmeier E. H., Birkenmeier C. S., Barker J. E. (2000). Mutations in a NIMA-related kinase gene, Nek1, cause pleiotropic effects including a progressive polycystic kidney disease in mice. Proc. Natl. Acad. Sci. USA 97, 217–221 10.1073/pnas.97.1.217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz Meirelles G., Ferreira Lanza D. C., da Silva J. C., Santana Bernachi J., Paes Leme A. F., Kobarg J. (2010). Characterization of hNek6 interactome reveals an important role for its short N-terminal domain and colocalization with proteins at the centrosome. J. Proteome Res. 9, 6298–6316 10.1021/pr100562w [DOI] [PubMed] [Google Scholar]
- Vogler C., Homan S., Pung A., Thorpe C., Barker J., Birkenmeier E. H., Upadhya P. (1999). Clinical and pathologic findings in two new allelic murine models of polycystic kidney disease. J. Am. Soc. Nephrol. 10, 2534–2539 [DOI] [PubMed] [Google Scholar]
- Wei R., Ngo B., Wu G., Lee W. H. (2011). Phosphorylation of the Ndc80 complex protein, HEC1, by Nek2 kinase modulates chromosome alignment and signaling of the spindle assembly checkpoint. Mol. Biol. Cell 22, 3584–3594 10.1091/mbc.E11-01-0012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwood I., Cheary D. M., Baxter J. E., Richards M. W., van Montfort R. L., Fry A. M., Bayliss R. (2009). Insights into the conformational variability and regulation of human Nek2 kinase. J. Mol. Biol. 386, 476–485 10.1016/j.jmb.2008.12.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whelligan D. K., Solanki S., Taylor D., Thomson D. W., Cheung K-M., Boxall K., Mas–Droux C., Barillari C., Burns S., Grummitt C. G.et al. (2010). Aminopyrazine inhibitors binding to an unusual inactive conformation of the mitotic kinase Nek2: SAR and structural characterization. J. Med. Chem. 53, 7682–7698 10.1021/jm1008727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wloga D., Camba A., Rogowski K., Manning G., Jerka–Dziadosz M., Gaertig J. (2006). Members of the NIMA-related kinase family promote disassembly of cilia by multiple mechanisms. Mol. Biol. Cell 17, 2799–2810 10.1091/mbc.E05-05-0450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Qiu X. L., Zhou L., Zhu J., Chamberlin R., Lau J., Chen P. L., Lee W. H. (2008). Small molecule targeting the Hec1/Nek2 mitotic pathway suppresses tumor cell growth in culture and in animal. Cancer Res. 68, 8393–8399 10.1158/0008-5472.CAN-08-1915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu L., Osmani S. A., Mirabito P. M. (1998). A role for NIMA in the nuclear localization of cyclin B in Aspergillus nidulans. J. Cell Biol. 141, 1575–1587 10.1083/jcb.141.7.1575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu W., Baxter J. E., Wattam S. L., Hayward D. G., Fardilha M., Knebel A., Ford E. M., da Cruz e Silva E. F., Fry A. M. (2007). Alternative splicing controls nuclear translocation of the cell cycle-regulated Nek2 kinase. J. Biol. Chem. 282, 26431–26440 10.1074/jbc.M704969200 [DOI] [PubMed] [Google Scholar]
- Yang J., Adamian M., Li T. (2006). Rootletin interacts with C-Nap1 and may function as a physical linker between the pair of centrioles/basal bodies in cells. Mol. Biol. Cell 17, 1033–1040 10.1091/mbc.E05-10-0943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yim H., Sung C. K., You J., Tian Y., Benjamin T. (2011). Nek1 and TAZ interact to maintain normal levels of polycystin 2. J. Am. Soc. Nephrol. 22, 832–837 10.1681/ASN.2010090992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yissachar N., Salem H., Tennenbaum T., Motro B. (2006). Nek7 kinase is enriched at the centrosome, and is required for proper spindle assembly and mitotic progression. FEBS Lett. 580, 6489–6495 10.1016/j.febslet.2006.10.069 [DOI] [PubMed] [Google Scholar]
- Zalli D., Bayliss R., Fry A. M. (2012). The Nek8 protein kinase, mutated in the human cystic kidney disease nephronophthisis, is both activated and degraded during ciliogenesis. Hum. Mol. Genet. 21, 1155–1171 10.1093/hmg/ddr544 [DOI] [PMC free article] [PubMed] [Google Scholar]