

Summary
Here, we describe a phosphorylation-based reverse myristoyl switch for mammalian ZNRF2, and show that this E3 ubiquitin ligase and its sister protein ZNRF1 regulate the Na+/K+ pump (Na+/K+ATPase). N-myristoylation localizes ZNRF1 and ZNRF2 to intracellular membranes and enhances their activity. However, when ZNRF2 is phosphorylated in response to agonists including insulin and growth factors, it binds to 14-3-3 and is released into the cytosol. On membranes, ZNRF1 and ZNRF2 interact with the Na+/K+ATPase α1 subunit via their UBZ domains, while their RING domains interact with E2 proteins, predominantly Ubc13 that, together with Uev1a, mediates formation of Lys63-ubiquitin linkages. ZNRF1 and ZNRF2 can ubiquitylate the cytoplasmic loop encompassing the nucleotide-binding and phosphorylation regions of the Na+/K+ATPase α1 subunit. Ouabain, a Na+/K+ATPase inhibitor and therapeutic cardiac glycoside, decreases ZNRF1 protein levels, whereas knockdown of ZNRF2 inhibits the ouabain-induced decrease of cell surface and total Na+/K+ATPase α1 levels. Thus, ZNRF1 and ZNRF2 are new players in regulation of the ubiquitous Na+/K+ATPase that is tuned to changing demands in many physiological contexts.
Key words: Myristoyl switch, Protein trafficking, Ubiquitylation, Na+/K+ATPase, Na+ pump
Introduction
ZNRF1 and ZNRF2 are closely related E3 ubiquitin ligases of the RING superfamily (Araki and Milbrandt, 2003). Generally, RING E3 domains interact with ubiquitin-charged E2 proteins and transfer the ubiquitin to a lysine residue in the substrate protein, which docks elsewhere on the E3. In addition to C-terminal RING domains, ZNRF1 and ZNRF2 both contain a UBZ domain, which is a Cys2His2 zinc-finger structure that in other proteins binds to mono- and multi-ubiquitylated targets (Bienko et al., 2005; Bish and Myers, 2007; MacKay et al., 2010). The N-termini of both proteins are predicted to be N-myristoylated (Araki and Milbrandt, 2003). The two proteins differ in their N-terminal halves where ZNRF2 is rich in glycine and alanine. While ZNRF2 was reported to contain a MAGE domain in this region (Plans et al., 2006), the sequence similarity with several MAGE-containing proteins is not within the MAGE domain itself.
ZNRF1 was first identified as an mRNA induced in Schwann cells after peripheral nerve injury (Araki et al., 2001). ZNRF1 and ZNRF2 proteins are associated with presynaptic membranes, and the effects of overexpressing ligase-dead mutants in PC12 cells suggested a possible role in Ca2+-dependent exocytosis (Araki and Milbrandt, 2003). ZNRF1 has been reported to interact with tubulin, and target glutamine synthetase and protein kinase B (PKB/Akt) for ubiquitylation and proteasomal degradation (Saitoh and Araki, 2010; Wakatsuki et al., 2011; Yoshida et al., 2009). However, other than interacting with various E2 proteins in yeast-two hybrid experiments, the function of ZNRF2 is unknown (Markson et al., 2009; Plans et al., 2006; van Wijk et al., 2009).
Recently, ZNRF2 was found to be phosphorylated by a kinase downstream of phosphoinositide 3-kinase (PI3K), and then captured by the 14-3-3 family of phosphoprotein-binding proteins in HEK293 cells (Dubois et al., 2009). Phosphorylated Ser19, within a sequence [RTRAY(pS19)GS] that conforms to the consensus for basophilic protein kinases, including Akt/PKB, was identified as a 14-3-3 binding site. However, 14-3-3s are dimers that often dock onto two phosphorylated sites on their targets to influence their conformation and interactions. The biochemical data indicated that a second unknown 14-3-3-binding site exists on ZNRF2 (Dubois et al., 2009).
Signalling via PI3K-PKB regulates intracellular membrane homeostasis and trafficking events in diverse physiological contexts. Examples include insulin-stimulated transport of GLUT4 glucose transporter-carrying vesicles to the plasma membrane to allow glucose to enter muscles and adipose tissues (Rowland et al., 2011; Taniguchi et al., 2006), increased cell surface expression of the Na+/K+ATPase to clear postprandial blood K+ (Al-Khalili et al., 2003; Comellas et al., 2010) and membrane dynamics associated with inhibiting apoptosis and autophagy (Eisenberg-Lerner et al., 2009). However, too few downstream components have been defined to fully explain how the PI3K–PKB pathway regulates such membrane events, and ZNRF1 and ZNRF2 seemed attractive candidate mediators.
Here, we discover that as well as being a target of PI3K-PKB, ZNRF2 is phosphorylated in response to multiple stimuli, and these phosphorylations control the interactions of ZNRF2 with 14-3-3 proteins, and release the enzyme from intracellular membranes by triggering a ‘reverse myristoyl switch’. We also find that ZNRF1 and ZNRF2 dock onto and ubiquitylate the α1 subunit of Na+/K+ATPase, and ZNRF2 regulates the amount of this subunit at the plasma membrane.
Results
Multiple stimuli regulate the phosphorylation and 14-3-3 binding of endogenous ZNRF2
We found that ZNRF2 is a convergence point for multiple stimuli. The binding to 14-3-3 of endogenous ZNRF2, as well as ZNRF2-GFP from stably-transfected cells, was stimulated by IGF1, EGF, phorbol ester (PMA), the adenylate cyclase activator forskolin and AMPK activator A769662, though not the Ca2+ ionophore A23187, as determined by co-immunoprecipitation of ZNRF2 with endogenous 14-3-3s and direct binding of exogenous 14-3-3 to the immunoprecipitated ZNRF2 in far-western assays (Fig. 1A,B). The binding to 14-3-3 was abolished by in vitro dephosphorylation of ZNRF2 (Fig. 1C).
Fig. 1.

Phosphorylation-dependent binding of 14-3-3 to ZNRF2 in response to agonists. ZNRF2-GFP (A) and endogenous ZNRF2 (B), isolated from HEK293 cells that had been exposed to various stimuli and inhibitor combinations, were tested for binding to 14-3-3s in a far-western overlay and for co-immunoprecipitation with endogenous 14-3-3s. ZNRF2 expression and phosphorylation (pSer19, pSer82 and pSer145) was analyzed by western blotting of cell lysates (30 µg), as was the phosphorylation status of AMPK, PKB, ERK1/2, ACC and VASP. SS, serum-starved conditions. (C) ZNRF2-GFP, transiently expressed in HEK293 cells grown in the presence of 10% FBS, was immunoprecipitated and dephosphorylated with lambda-phosphatase (with or without 50 mM EDTA to inhibit dephosphorylation). Binding of 14-3-3 to ZNRF2 was analyzed by 14-3-3 far-western overlay. (D) ZNRF2-GFP isolated from transfected HEK293 cells stimulated with IGF1 in the presence or absence of the PI3K inhibitors (PI-103, Wortmannin, LY294002), PKB inhibitor (AKTi1/2), mTORC1 (rapamycin), p90RSK inhibitor (BI-D1870), PKC inhibitor (Gö6983), an ERK1/2 pathway inhibitor (PD184352) and an mTOR kinase inhibitor (Ku-0063794) was tested for binding to 14-3-3 in a far-western assay, and for co-immunoprecipitation with endogenous 14-3-3s (K19 antibody). Western blotting was performed as in B. (E) GFP-transfected cells were stimulated with PMA in the presence or absence of the PKC inhibitors (Gö6983 and Gö6976), p90RSK inhibitor (BI-D1870), ERK1/2 pathway inhibitors (PD184352 and U0126) and p38 MAPK inhibitors (SB203580 and Birb0796).
To identify relevant in vivo phosphorylations quantitatively, we used differential SILAC labelling of HEK293 cells and mass spectrometric analyses (MS) of immunoprecipitated endogenous ZNRF2. Phosphorylations of Ser19, Ser80 or 82, Ser116, Ser135 and Ser145 were identified. Ser19 was the major IGF1-stimulated site (4-fold increase over basal), whereas both Ser19 (3.71-fold) and Ser82/80 (15.77/8.73-fold) responded to PMA (Table 1; supplementary material Fig. S1; Fig. S2C,D).
Table 1. SILAC-labelled samples from supplementary material Fig. S2A were analyzed by Orbitrap MS.
| Fold ratio | ||||||
| Residue | IGF1/SS | PI-103/SS | IGF1/PI-103 | PMA/SS | Gö6983/SS | PMA/Gö6983 |
| Ser19 | 4.19 | 0.82 | 5.12 | 3.71 | 0.31 | 12.10 |
| Ser80 | 1.80 | 1.06 | 1.69 | 15.77 | 0.10 | 165.78 |
| Ser82 | 1.22 | 1.4 | 0.88 | 8.73 | 0.07 | 125.11 |
| Ser113 | 1.08 | 0.82 | 1.32 | - | - | - |
| Ser116 | - | - | - | 0.86 | 0.77 | 1.11 |
| Ser135 | 1.15 | 1.16 | 0.99 | 1.26 | 0.83 | 1.52 |
| Ser145 | 1.63 | 2.21 | 0.74 | 1.24 | 0.57 | 2.17 |
Data were quantified using MaxQuant (version 13.13.10) and are presented as a fold ratio of indicated treatments. SS, serum-starved conditions.
The MS analysis could not differentiate between phosphorylation of Ser80 or 82 and Ser19 or 21, even when overexpressed ZNRF2-GFP was analysed (supplementary material Fig. S2C,D). Therefore, bacterially-expressed GST-ZNRF2 was also phosphorylated in vitro by candidate kinases PKBα, SGK1, p90RSK and PKA using [32P-γ]ATP/Mg2+, followed by HPLC and solid-phase Edman sequencing (supplementary material Fig. S2A, performed as in Campbell and Morrice, 2002). These experiments confirmed that PKBα and SGK1 phosphorylated Ser19 only, whereas p90RSK and PKA phosphorylated Ser19 and Ser82. PKA phosphorylation of Ser145 was also detected by MS precursor ion scanning of these samples. We therefore raised antibodies to recognise Ser19, Ser82 and Ser145 only when these residues are phosphorylated. Using these antibodies, we confirmed the in vitro phosphorylations by AGC kinases (supplementary material Fig. S1B). AMPKα did not phosphorylate ZNRF2 in vitro (supplementary material Fig. S1B) even though AMPK activators enhanced 14-3-3 binding to ZNRF2 in cells (Fig. 1A).
Consistent with the MS analyses, these antibodies were used to show that Ser145 displays a high basal phosphorylation in unstimulated (serum-deprived) cells (Fig. 1A). IGF1 stimulated phosphorylation of Ser19, Ser145 and to a lesser extent Ser82, whereas PMA and forskolin increased the phosphorylation of Ser19, Ser82 and Ser145 (Fig. 1A).
The IGF1-stimulated phosphorylation of Ser19 and Ser145 was inhibited slightly by the mTOR inhibitor (Ku-0063794) and completely by the PI3K inhibitor PI-103. LY294002, a selective PI3K inhibitor, decreased 14-3-3 binding to ZNRF2, abolished the phosphorylation of Ser145 and decreased the phosphorylation of Ser19 (Fig. 1A,D). PMA-stimulated phosphorylations of Ser19, Ser82 and Ser145 were inhibited by non-specific PKC inhibitor Gö6983 and PKCα/β inhibitor Gö6976 (Fig. 1E). In contrast, forskolin stimulation of these phosphorylations was blocked by H-89, which inhibits kinases including PKA (Fig. 1A). The PKB inhibitor AKTi-1/2 decreased the phosphorylation of Ser19 (Fig. 1D). Inhibition of mTORC1 with rapamycin did not affect any IGF1-stimulated phosphorylations or 14-3-3 binding. MEK1/2 (PD184352) and p90RSK (BI-D1870) inhibitors did not affect any IGF1- or PMA-stimulated phosphorylations or 14-3-3 binding (Fig. 1D,E). Inhibiting mTOR with Ku-0063794 slightly decreased phosphorylation of Ser19 and Ser145, and binding to 14-3-3 (Fig. 1D).
These data suggest that Akt/PKB phosphorylates Ser19, PKC(s) phosphorylate Ser19 and Ser82, and PKA phosphorylates Ser19, Ser82 and Ser145.
Phosphorylated Ser19 and Ser82 of ZNRF2 bind to 14-3-3, while Ser145 also influences this interaction
The IGF1-stimulated binding to 14-3-3 was abolished by Ser19Ala mutation of ZNRF2-GFP and decreased by Ser82Ala mutation (Fig. 2A). Consistent with the major IGF1-induced 14-3-3-binding site being phosphorylated Ser19 (Fig. 1, Fig. 2A), PKBα and SGK1, which phosphorylate only this site in vitro (Fig. 2B), did not induce 14-3-3 binding to Ser19Ala-ZNRF2 (Fig. 2B).
Fig. 2.

Effect of mutations in ZNRF2 on its binding to 14-3-3. (A) Wild-type ZNRF2-GFP or single/double serine to alanine mutants were isolated from transfected HEK293 cells that had been stimulated with IGF1, PMA or forskolin as indicated. The ZNRF2-GFP proteins were tested for their phosphorylation and 14-3-3 binding, as for Fig. 1. (B) Similar to A, but including mutated GST-ZNRF2 phosphorylated with PKBα, SGK1, p90RSK, PKA, PKCα and PKCζ, as indicated. WT, wild type.
In contrast, binding to 14-3-3 in response to PMA and forskolin was partially decreased by mutation of either Ser19 or Ser82 (Fig. 2A). Consistent with these data, the candidate mediators of these signals, namely p90RSK, PKCα, PKCζ and PKA phosphorylated Ser19 and Ser82, and PKA also phosphorylated Ser145, and the resulting 14-3-3 binding was partially decreased by mutating either Ser19 or Ser82 (Fig. 2B). The double mutation of Ser19 and Ser82, or Ser19 and Ser145 abolished the 14-3-3 interaction with every stimulus tested. Although Ser145Ala mutation did not diminish 14-3-3 binding in a far-western assay (Fig. 2A,B), the mutation of this residue decreased co-immunoprecipitation of the endogenous 14-3-3s (Fig. 2A).
Ser19, Ser82 and Ser145 are conserved in ZNRF2 across vertebrates (supplementary material Fig. S2B). Of these phosphorylated sites, Ser19 and Ser82 conform to mode I 14-3-3-binding sites (http://scansite.mit.edu). Together, these data indicate that a 14-3-3 dimer docks onto phosphorylated Ser19 and Ser82, and that phosphorylation of Ser145 also affects this interaction, perhaps by inducing a conformational change in ZNRF2. All seven 14-3-3 isoforms were able to interact with ZNRF2, when expressed as GST-fusions in HEK293 cells stably expressing full-length ZNRF2-GFP (unpublished data).
ZNRF1 can also bind to 14-3-3, possibly via PMA- and forskolin-stimulated phosphorylation of Ser50 (supplementary material Fig. S2B, Fig. S3A,B), though a second phosphosite on ZNRF1 for binding to a 14-3-3 dimer has not yet been identified. While ZNRF1 has a serine residue that aligns with Ser19 of ZNRF2, it is not in the context of a 14-3-3-binding motif (supplementary material Fig. S2B).
ZNRF2 is targeted to intracellular membranes by N-myristoylation and released into the cytoplasm by phosphorylation of Ser19 and Ser145
The predicted N-myristoylation of ZNRF2 was confirmed by mass spectrometric analysis of ZNRF2-GFP from stably-transfected cells (Fig. 3A,B). Consistent with the N-myristoyl group acting as a membrane anchor, ZNRF2-GFP was located at internal membranes and plasma membrane (Fig. 3C), overlapping with the distribution of the Golgi marker TGN46 (supplementary material Fig. S4A). In contrast, ZNRF2 with the N-terminal glycine mutated to alanine [designated Gly2Ala (G2A)], or N-terminal GFP tag, displayed a diffuse intracellular distribution (Fig. 3C). Also, ZNRF2-GFP was partitioned between membrane and soluble fractions prepared by ultracentrifugation of detergent-free lysates of cells cultured in medium containing serum, whereas the non-myristoylated Gly2Ala mutant was exclusively in the soluble fraction (Fig. 3D).
Fig. 3.

N-myristoylation of ZNRF2 and its effect on phosphorylation, activity, subcellular localization and protein-protein interactions. (A) Alignment of the N-terminus of ZNRF2 homologues showing the myristoylation consensus motif. (B) Detection of myristic acid in ZNRF2 by mass spectrometric analysis (Orbitrap). MS data from a tryptic digest of ZNRF2-GFP isolated from HEK293 cells was searched using Mascot allowing for the myristoyl N-terminal glycine modification, and the scores for the peptide shown, found in two samples, were 43 and 66 with the significance cutoff (P<0.05) being 19. The difference between the expected mass of unmodified GAKQSGPAAANGR (1183.6058 Da) and observed mass (m/z = 697.9107:2+ = 1393.8068 Da) is 210.2010 Da, which corresponds to the mass of the myristoyl modification (210.1984 Da). The y-ions generated by MS2 fragmentation indicate the sequence from the C-terminal end of the peptide, whereas the b-ions are consistent with an N-myristoyl group. (C) Subcellular localization of ZNRF2 in HeLa and HEK293 cells transfected with GFP-tagged wild-type or Gly2Ala-ZNRF2 (G2A). Cells were stained with DAPI (to label nuclei). At least five sets of cells were analyzed and representative images are shown. Scale bar: 10 µm. (D) HEK293-Flp-In-Trex cells stably expressing GFP-tagged wild-type or Gly2Ala-ZNRF2 were fractionated as described in the Materials and Methods, and cell lysates were subjected to western blotting with the indicated antibodies.
The proportion of ZNRF2-GFP in the membrane fraction increased when cells were serum deprived; or when cells grown in serum were treated with PI-103, Gö6983 and H-89 before stimulation with IGF1, PMA and forskolin, respectively (Fig. 4A). Conversely, the amount of ZNRF2-GFP in the membrane fractions decreased when cells were stimulated with IGF1, PMA, forskolin and serum (Fig. 4A). Consistent with these fractionation data, live cell imaging showed that ZNRF2-GFP moved from intracellular membranes to the cytosol upon IGF1 stimulation (Fig. 5A), and returned to the membranes when cells were treated with PI3K inhibitors GDC-0941 (Fig. 4C) and PI-103 (supplementary material Fig. S4B), and PKB inhibitors MK-2206 (Fig. 4D) and AKTi-1/2 (supplementary material Fig. S4B), which decreased ZNRF2 phosphorylation at Ser19 and Ser145 (Fig. 4B). These membranal structures associated with dephosphorylated ZNRF2 co-localized with a Golgi tracker stain (Fig. 4C,D). Similarly, the association of ZNRF2 with membranes was lost when cells were stimulated with PMA or forskolin, and regained when cells were given the kinase inhibitors Gö6983 and H-89, respectively (Fig. 4A).
Fig. 4.

Reversible membrane-to-cytosol translocation of ZNRF2. (A) HEK293 cells exposed to various stimuli and inhibitor combinations were fractionated into cytosol (CE) and membrane (ME) extracts by ultracentrifugation and the extracts were analyzed by western blotting with the indicated antibodies. Ubiquitylation activity of endogenous ZNRF2 from cytosol and membrane fractions was analyzed in vitro using Ubc13-Uev1a as E2. (B) HEK293-Flp-In-TRex cells stably expressing ZNRF2-GFP were exposed to various PI3K and PKB inhibitors and phosphorylation ZNRF2 phosphorylation was analyzed by western blotting. (C,D) HEK293-Flp-In-TRex cells stably expressing ZNRF2-GFP were used for live cell imaging. Cells were treated with 1 µM GDC-0941 (C) or 10 µM MK-2206 for 60 min (D). Images were taken before and after the treatments. Scale bars: 10 µm.
Fig. 5.
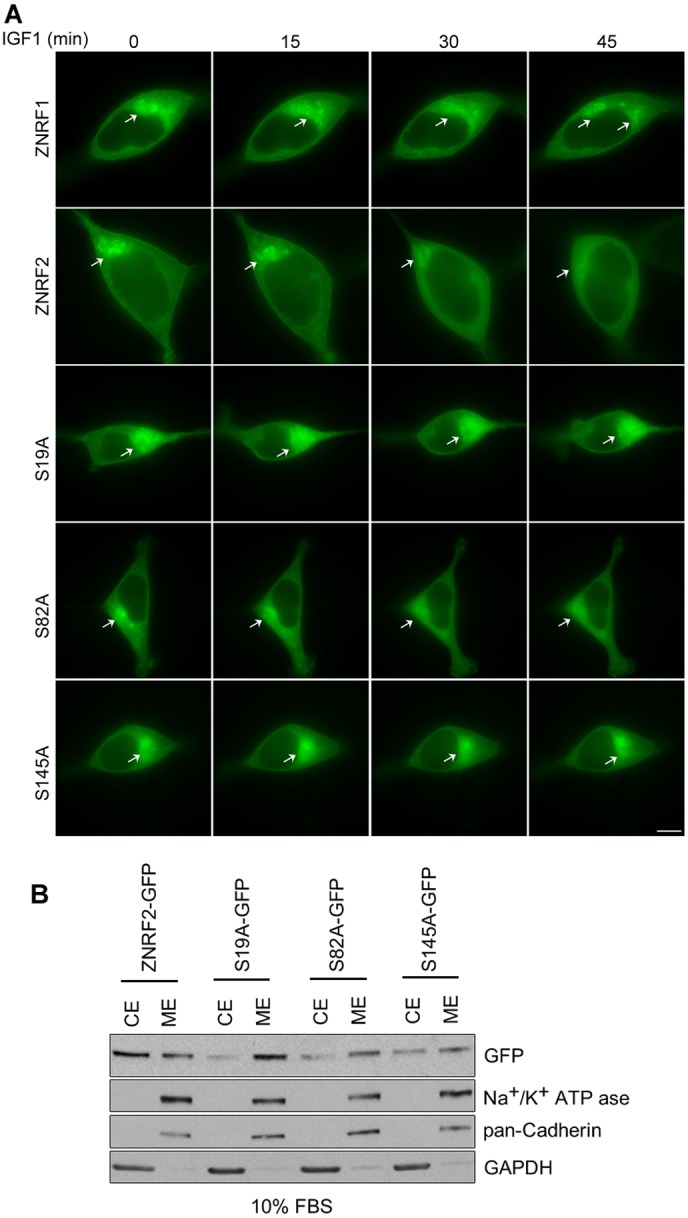
Effect of Ser19Ala mutation on the reversible membrane-to-cytosol translocation of ZNRF2. (A) As in Fig. 4C, except that before imaging the cells were serum starved for 12 h and then stimulated with IGF1. Images were taken before and after the stimulation. Scale bar: 10 µm. (B) HEK293-Flp-In-TRex cells stably expressing wild-type or the indicated mutants of ZNRF2-GFP were fractionated into cytosol (CE) and membrane (ME) extracts by ultracentrifugation, and the extracts were analyzed by western blotting with the indicated antibodies.
Interestingly, in cells growing under 10% FBS conditions, ZNRF2-Ser19Ala (and to a lesser extent ZNRF2-Ser82Ala and ZNRF2-Ser145Ala) showed greater membrane localization than the wild-type control (Fig. 5B; supplementary material Fig. S4E). Strikingly, ZNRF2-Ser19Ala stayed on membranes even when cells were stimulated with IGF1 (Fig. 5A). Together these findings show that as well as promoting binding to 14-3-3, the phosphorylation of Ser19 counteracts the N-myristoyl-mediated targeting of ZNRF2 to membranes.
Similar to ZNRF2, ZNRF1-GFP was in both membrane and soluble fractions of transfected cell lysates, while GFP-ZNRF1 lacking the N-myristoyl group was entirely soluble (supplementary material Fig. S3F). However, in contrast to ZNRF2, we observed no reversible membrane-to-cytosol shuttling upon IGF1 stimulation of either ZNRF1-GFP (Fig. 5A) or endogenous ZNRF1 (supplementary material Fig. S3C). ZNRF1-GFP showed a more membranal localization in cells growing under 10% FBS conditions, compared to ZNRF2, whose localization was more diffuse (supplementary material Fig. S4E).
ZNRF1 and ZNRF2 are co-purified with Ubc13 and Na+/K+ATPase
Towards understanding the cellular functions of ZNRF2 and ZNRF1, we aimed to identify their interacting proteins. N-myristoylated ZNRF2-GFP and ZNRF1-GFP, non-myristoylated GFP-ZNRF2 and GFP-ZNRF1, and GFP alone, were isolated from lysates of transiently-transfected HEK293 cells using GFP-Trap beads. After SDS-PAGE, strong bands at the molecular weights expected for the GFP-tagged proteins and GFP were observed in the respective lanes (Fig. 6A). Using mass spectrometry, the Na+/K+ATPase α1 catalytic subunit (ATP1A1) was found to co-purify with ZNRF2-GFP and ZNRF1-GFP, but not the N-terminally tagged proteins, indicating that N-myristoylation is important for these interactions. In contrast, Ubc13 (UBE2N) and Uev1a (MMS2) co-purified with ZNRF2 and ZNRF1, regardless of whether the GFP tag was on the N- or C-terminus (Fig. 6A). Ubc13 was previously identified as a ZNRF2 interactor in a yeast 2-hybrid screen (Plans et al., 2006), and forms a complex with Uev1a that directs the formation of Lys63-polyubiquitin chains. Other E2s, namely UBE2D1, UBE2D2 and UBE2D3 (also known as UbcH5A, B and C) were also present in the immunoprecipitates in lower amounts.
Fig. 6.

Interactions of ZNRF1 and ZNRF2 with Na+/K+ATPase subunit α1, Ubc13 and 14-3-3. (A) HEK293 cells were transiently transfected with either N- or C-terminally tagged ZNRF1 or ZNRF2. Proteins were immunoprecipitated from 50 mg of lysates, resolved on a 4-12% gradient NuPAGE gel and stained with colloidal Coomassie Brilliant Blue. The excised gel pieces were digested with trypsin and analyzed by mass spectrometry. Identified proteins are shown on the right-hand side of the gel. (B) Extracts of cells stably expressing GFP or C-terminally GFP-tagged ZNRF1 or ZNRF2 were subjected to immunoprecipitation and precipitates were analyzed by western blotting with the indicated antibodies. (C) Full-length ZNRF2 or ZNRF2 deletion fragments (the amino acid numbers of the fragment are indicated) with a C-terminal GFP tag were tested for the binding to endogenous Na+/K+ATPase α1, Ubc13 and 14-3-3s. (D) As in C, except that ZNRF1 plasmids were used. (E) Deletion fragments of Na+/K+ATPase subunit α with a C-terminal FLAG tag were tested for the binding to ZNRF1-GFP or ZNRF2-GFP. (F) Extracts of HEK293 cells were analyzed by size exclusion chromatography on a HiLoad 26/60 Superdex 200 column in buffer containing 0.2 M NaCl, and every third fraction was denatured and analyzed by western blotting with the indicated antibodies. The elution positions of Dextran blue (2000 kDa, in the void volume of this column), thyroglobulin (670 kDa), bovine gamma-globulin (158 kDa) and chicken ovalbumin (44 kDa) are shown.
These interactions were confirmed by western blotting, which also showed that more Na+/K+ATPase α1 subunit associated with ZNRF1-GFP than with ZNRF2-GFP, whereas the opposite was true for Ubc13 (Fig. 6B). We also noticed that while no Na+/K+ATPase α1 subunit binds to non-myristoylated ZNRF2 (Gly2Ala) (Fig. 6C), the lack of N-myristoylation lessened, but did not abolish the binding of ZNRF1 to Na+/K+ATPase α1 (Fig. 6D). The binding of ZNRF2 to 14-3-3 was also inhibited by lack of N-myristoylation, whether due to an N-terminal tag as before (supplementary material Fig. S4C,D), or Gly2Ala mutation (Fig. 6C).
Endogenous Na+/K+ATPase α1 subunit was co-immunoprecipitated with endogenous ZNRF1 and ZNRF2, though more lysate was needed to detect Na+/K+ATPase α1 subunit in immunoprecipitates of ZNRF2 compared with ZNRF1 (unpublished observations). In size-exclusion chromatography of HEK293 cell extracts, ZNRF2 migrated in three positions running with 17 to 44, 158 and 670 kDa markers, of which the latter two forms overlapped with peaks of Na+/K+ATPase α1 subunit (Fig. 6F). We also found that ZNRF1 and ZNRF2 bind to a FLAG-Thr339-Leu772 fragment that encompasses the cytoplasmic nucleotide-binding/phosphorylation (N/P) catalytic domains of the Na+/K+ATPase α1 subunit, but not to FLAG-Arg385-Arg596 fragment corresponding to the nucleotide-binding domain alone (Fig. 6E).
N-myristoylation and UBZ domains of ZNRF1 and ZNRF2 mediate interactions with the Na+/K+ATPase α1 subunit and the RING domains bind to Ubc13
We next identified the regions in ZNRF2 and ZNRF1 responsible for interacting with Na+/K+ATPase α1 subunit and Ubc13. Again, disrupting myristoylation, whether by Gly2Ala mutation, N-terminal deletion or an N-terminal tag, markedly decreased (ZNRF1) or abolished (ZNRF2) the interactions with Na+/K+ATPase α1 subunit (Fig. 6C,D). The interactions with Na+/K+ATPase α1 subunit also required the UBZ domains of ZNRF1 and ZNRF2, and a UBZ domain attached to even a minimal N-myristoylated region (residues 1 to 11) could co-purify some Na+/K+ATPase α1 subunit from transfected cell lysates (Fig. 6C,D). Although the UBZ domain is a Cys2His2 zinc finger (MacKay et al., 2010; Woodruff et al., 2010), replacing the zinc-coordinating cysteines with alanines did not prevent binding of either ZNRF2 (C160A/C163A) or ZNRF1 (C145A/C148A) to the Na+/K+ATPase α1 subunit. In contrast, the binding of Ubc13 required intact RING domains of ZNRF1 or ZNRF2, since point mutations that rendered the RING domains catalytically inactive (for example, Fig. 6D, Cys184Ala-ZNRF1) and/or lack of the RING domain (Fig. 6C, 1-180) abolished the binding to Ubc13.
ZNRF1 and ZNRF2 ubiquitylate the Na+/K+ATPase α1 subunit with certain specificity requirements
Endogenous and C-terminally-tagged forms of both ZNRF1 and ZNRF2 from HEK293 cells could form Lys63-linked polyubiquitin chains in vitro with Ubc13-Uev1a as E2 (Fig. 7A). However, both E3s also formed other types of ubiquitin chains with a variety of E2s in in vitro assays (supplementary material Fig. S5A,B). Therefore, while the Ubc13-Uev1a was selectively co-purified with both ZNRF1 and ZNRF2 (Fig. 6), we cannot rule out the possibility that these E3 proteins also operate with other E2s. Interestingly, the non-myristoylated forms of ZNRF1 and ZNRF2 (Gly2Ala mutant or with N-terminal GFP tag) isolated from HEK293 cells showed less E3 ubiquitin ligase activity in vitro than the proteins with C-terminal GFP tag when Ubc13-Uev1a complex was used as E2 (Fig. 7B). In agreement with these findings, endogenous ZNRF2 isolated from membrane fractions showed higher ubiquitylating activity than from cytosolic fractions, when Ubc13-Uev1a complex was used as E2 (Fig. 4A).
Fig. 7.

ZNRF1 can ubiquitylate Na+/K+ATPase α1 in vitro. (A) In vitro ubiquitylation reactions of ZNRF2 and ZNRF1 with (WT) ubiquitin, K48R ubiquitin or K63R ubiquitin were analyzed by western blotting (left) or Coomassie Blue staining (CBB, right). (B) The indicated proteins were immunoprecipitated from HEK293 cells and used for in vitro ubiquitylation reactions. (C) In vitro ubiquitylation reaction of FLAG-Na+/K+ATPase α1 purified from HEK293-Flp-In-TRex cells using GST-ZNRF1, GST-ZNRF2 or GST-ZNRF2-C199A. (D) As in C, except that the ZNRF2-GFP or GFP control was purified from HEK293-Flp-In-TRex cells treated with DMSO or PI-103 (1 µM, 30 min). UbcH5a was used as the E2. Reactions were analyzed by western blotting using anti-ubiquitin antibody (E) As in D, except Ubc13-Uev1a was used as the E2. The arrows indicate ubiquitylated FLAG-Na+/K+ATPase α1. Reactions were analyzed as in D. (F) The indicated fragments of Na+/K+ATPase α1 were purified either from HEK293 cells or E. coli and used for in vitro ubiquitylation reactions using either UbcH5a or Ubc13-Uev1a as the E2 and GFP-ZNRF2 as the E3. Reactions were analyzed by western blotting using K48- or K63-specific polyubiquitin chain antibodies.
In vitro, using UbcH5a as E2, bacterially-expressed GST-ZNRF1 could ubiquitylate purified FLAG-tagged full-length Na+/K+ATPase α1 subunit isolated from stably-transfected HEK293 cells (Fig. 7C). In contrast, bacterially-expressed GST-ZNRF2 did not ubiquitylate the FLAG-tagged forms of Na+/K+ATPase α1 (Fig. 7C). This finding raised the question whether ZNRF2 needs to be modified in mammalian cells before it can ubiquitylate Na+/K+ATPase α1. Indeed, when ZNRF2 was purified from HEK293 cells, whether grown in 10% FBS or treated with PI-103, it was able to ubiquitylate FLAG-tagged Na+/K+ATPase α1 isolated from stably-transfected HEK293 cells (Fig. 7D). Similarly, when Ubc13-Uev1a was used as E2, bacterially-expressed ZNRF1, and ZNRF2 isolated from mammalian cells, but not ligase-dead enzymes, could ubiquitylate FLAG-tagged full-length Na+/K+ATPase α1 isolated from mammalian cells (Fig. 7E).
Using UbcH5a as E2, bacterially-expressed GST-ZNRF1 (unpublished observations) and ZNRF2-GFP purified from HEK293 cells (Fig. 7E) could also ubiquitylate a FLAG-Thr339-Leu772 fragment that encompasses the cytoplasmic nucleotide-binding/phosphorylation (N/P) catalytic domains of the Na+/K+ATPase α1, whether purified from mammalian or bacterial cells. However, they could not ubiquitylate a FLAG-Arg385-Arg596 Na+/K+ATPase α1 fragment corresponding to the nucleotide-binding (N) domain alone (Fig. 7F). In contrast, the FLAG-Thr339-Leu772 (N/P) could not act as a substrate of ZNRF1 (unpublished observations) or ZNRF2-GFP (Fig. 7E) when Ubc13-Uev1a was used as the E2 (Fig. 7F).
ZNRF2 knockdown inhibits the ouabain-induced decrease in Na+/K+ATPase α1 plasma membrane and total levels, and ouabain also causes cellular depletion of ZNRF1 protein
Next, we aimed to investigate how ZNRF1 and ZNRF2 siRNA knockdown affected the Na+/K+ATPase. Consistent with previous reports (for example, Liu et al., 2004; Yoshimura et al., 2008), ouabain used at 50 nM (well below the IC50 for acute inhibition of the Na+/K+ATPase) caused a decrease in total Na+/K+ATPase α1 protein in HeLa cells (Fig. 8A,B) and also decreased the amount of Na+/K+ATPase α1 expressed at the cell surface, as detected in a cell-surface biotinylation assay (Fig. 8B,C). In contrast, in cells depleted for ZNRF2 via siRNA, the total and cell surface amounts of Na+/K+ATPase α1 were unchanged in response to ouabain treatment (Fig. 8A,B). It proved impossible to knockdown ZNRF1 without killing the cells. Interestingly however, 50 nM ouabain treatment caused a marked loss of cellular ZNRF1 protein (Fig. 8B; supplementary material Fig. S3G).
Fig. 8.

ZNRF2 knockdown rescues ouabain-induced decrease of total and plasma membrane levels of Na+/K+ATPase α1. (A) HeLa cells were transfected with control (scrambled) or ZNRF2 siRNA and 56 h post-transfection, cells were treated with 50 nM ouabain for 16 h. Na+/K+ATPase α1 levels and ZNRF2 knockdown levels were analyzed by western blotting of cell lysates. GAPDH was used as an internal control. (B) As in A, except membrane surface proteins were biotinylated, purified using streptavidin-agarose and analyzed by western blotting for Na+/K+ATPase α1 levels. Total lysate fractions were analyzed for ZNRF2 and ZNRF1 levels as in A. (C) Signals from three experiments similar to B, with ouabain concentrations from 50 to 70 nM for 12 to 15 h, were quantified using the Image J program. **P<0.005 (as shown by a Student's t-test). (D) Working model for the regulation of ZNRF2 and its potential role in the regulation of Na+/K+ATPase α1.
ZNRF1 and ZNRF2 proteins are differentially expressed in mouse tissues
Western blotting showed that ZNRF1 is most highly expressed in thymus and lung, and is also present in brain, liver, spleen and pancreas. In contrast, ZNRF2 is expressed most highly in lung, and is also present in brain, heart, lung, liver, pancreas, and other tissues (supplementary material Fig. S5C). ZNRF2 and ZNRF1 are differentially expressed in 22 cell lines, with the two E3s displaying parallel variation in expression across most of the cell lines tested (supplementary material Fig. S5D).
Discussion
Here we define the E3 ubiquitin ligase ZNRF2 as a convergence point for multiple regulatory inputs, and show that ZNRF1 and ZNRF2 ubiquitylate Na+/K+ATPase α1 and control its cell surface expression (Fig. 8D). Wherever possible, experiments have been performed with the endogenous proteins.
The Na+/K+ATPase pumps two Na+ out and three K+ into animal cells for every ATP hydrolyzed, generating an electrochemical gradient that supports membrane electrical excitability, solute transport and osmotic balance. This pump is the heaviest user of energy in the body, and its regulation in response to changing demands is critical to health. Developmental regulation occurs by tissue-specific expression of isoforms of the catalytic (α) subunit, and regulatory (β) and FXYD subunits (Crambert and Geering, 2003; Morth et al., 2011; Morth et al., 2007; Ogawa et al., 2009; Shinoda et al., 2009). The pump is also regulated acutely; for example the skeletal muscle Na+/K+ATPase activity is enhanced by insulin to clear blood K+ after a meal, and by adrenaline and contraction to prevent fatigue (Ewart and Klip, 1995; Hatou et al., 2010). Demands for the Na+/K+ATPase also change markedly during nerve excitation, renal blood filtration and breathing, and Na+/K+ATPases are highly regulated in context-dependent ways by adrenaline, dopamine, thyroid hormone, aldosterone, catecholamines and insulin (reviewed by Comellas et al., 2010; Ewart and Klip, 1995). Mechanistic details of these regulations are sketchy, though protein kinases including PKA, PKC and SGK have been found to phosphorylate the α and FXYD subunits, triggering trafficking of the Na+/K+ATPase between intercellular membranes and cell surface, as well as changing intrinsic activity of the pump (Fuller et al., 2009; Han et al., 2010; Lang et al., 2010; Poulsen et al., 2010).
It is therefore intriguing that among the multiple protein kinases that we find to converge on ZNRF2 are enzymes that are already implicated in regulating the Na+/K+ATPase. Specifically, cell-based experiments using different stimuli/inhibitor combinations, as well as in vitro phosphorylation by purified protein kinases, define ZNRF2 as a novel point of convergence of the PI3K/PKB, PKC and PKA pathways. We note that ZNRF1 has been reported to target PKB for ubiquitylation and proteasomal degradation (Wakatsuki et al., 2011). Conversely however, we found that ZNRF1 (probably Ser53) is a substrate of PKB in vitro and in cells in response to IGF1 (supplementary material Fig. S2E, Fig. S3). Though this PKB-mediated phosphorylation does not create a 14-3-3 binding site, ZNRF1 does bind to 14-3-3 via phosphoSer50 and further work is needed to characterise the ZNRF1–14-3-3 interaction more fully.
Functionally, the phosphorylations of ZNRF2 influence its interactions with both 14-3-3 proteins and intracellular membranes. Our data indicate that N-myristoylation is crucial for anchoring ZNRF2 on intracellular membranes, but cannot maintain a stable interaction with membranes when the Ser19 is phosphorylated and binds to 14-3-3. This regulation is reminiscent of ‘myristoyl-switching’ in which a myristoyl moiety is tucked into a hydrophobic pocket of a protein until a signal, such as Ca2+ binding, triggers a conformational change, exposing the myritoyl group for association with membranes or other proteins (Lim et al., 2011; Oakhill et al., 2010; Zozulya and Stryer, 1992). For ZNRF2, the term ‘reverse myristoyl switch’ is perhaps more apt, because the phospho-Ser19 trigger stops the membrane binding mediated by the N-myristoylation.
Presumably, when ZNRF2 is released from membranes, its myristoyl moiety would need a new hydrophobic environment. We hypothesize therefore that phosphorylation and/or 14-3-3 binding of Ser19 induces a structural change in ZNRF2, creating a hydrophobic pocket that encloses the N-myristoyl group intramolecularly. Consistent with this possibility, GNF-2, a small molecule binder of the N-myristoyl pocket of c-Abl (Choi et al., 2009; Hantschel et al., 2003) enhanced ZNRF2 targeting to membranes (supplementary material Fig. S6A). Further work is needed to determine whether GNF-2 exerts this action by binding directly to ZNRF2, though consistent with this notion, GNF-2 did not alter ZNRF2 phosphorylation (supplementary material Fig. S6B). Non-myristoylated forms of ZNRF1 and ZNRF2 have a lower in vitro E3 ubiquitin ligase activity than their myristoylated forms, suggesting that myristoylation may also enhance their catalytic activity via an intramolecular influence on conformation.
It is challenging to determine the specificities of E3 ubiquitin ligases with respect to their target substrates and their partnerships with the E2s that specify the ubiquitin chain types generated in vivo. In a few well-defined cases, polyubiquitylation of substrates by E3s requires two different E2 enzymes: one to mono-ubiquitylate the substrate and the second to elongate into a polyubiquitin chain using a defined lysine residue of ubiquitin (Windheim et al., 2008; Ye and Rape, 2009).
Here, we found that ZNRF1 and ZNRF2 can form free ubiquitin chains in vitro, with linkages that are specified by the different E2s. However, both ZNRF1 and ZNRF2 were co-immunoprecipitated from cell lysates with UBE2N and UBE2V1/Uev1b (human form of yeast Ubc13 and MMS2), which work together as an E2 that specifies the assembly of polyubiquitin chains linked through lysine 63.
The Na+/K+ATPase α1 subunit was also co-purified with endogenous ZNRF1 and ZNRF2 from HEK293 cells, requiring both membrane targeting and the UBZ domain of the E3 proteins. UBZ domains are Cys2His2 zinc fingers, which in other proteins bind to mono- or multi-ubiquitylated proteins, mainly in the context of DNA damage response processes (Ye and Rape, 2009). Curiously however, mutation of the zinc-coordinating cysteine residues did not affect the interaction of the UBZ of ZNRF1 with the Na+/K+ATPase α1, and we note that similarly the zinc was not required for the ubiquitin-binding activity of the Saccharomyces cerevisiae polymerase η UBZ domain (Woodruff et al., 2010).
In vitro, ZNRF1 and ZNRF2 were able to ubiquitylate full-length Na+/K+ATPase α1 using either Ubc13-Uev1a or UbcH5a as the E2 enzyme, but could only ubiquitylate a fragment encompassing the cytoplasmic nucleotide-binding/phosphorylation (N/P) region of the Na+/K+ATPase α1, when UbcH5a, and not Ubc13-Uev1a, was used as the E2. Based on these findings, our working hypothesis is that prior ubiquitylation ‘primes’ Na+/K+ATPase α1 for recognition by the UBZ domains of ZNRF1 and ZNRF2. The E3 proteins then work with an E2 to add polyubiquitin chains to the ‘primed’ Na+/K+ATPase α1.
Another key consideration is that while UbcH5a has no specificity for any particular lysine residue of ubiquitin, the E2 that co-purified with ZNRF1 and ZNRF2 was Ubc13-Uev1a, which specifically links via Lys63 of ubiquitin and would not be expected to attach the first ubiquitin to a substrate such as Na+/K+ATPase α1 (Eddins et al., 2006; Windheim et al., 2008). In addition to the putative ubiquitylation of Na+/K+ATPase α1 for docking of the UBZ domain of the E3s, it is therefore possible that a further ‘priming ubiquitin’ must be present on the Na+/K+ATPase α1 to act as substrate for the Ubc13-Uev1a and ZNRF1/2 partnership. Consistent with this theory, UbcH5a, but not Ubc13-Uev1a, supported ubiquitylation of the cytoplasmic N/P fragment of the Na+/K+ATPase α1 (Fig. 7), perhaps because this fragment lacked the priming ubiquitin required by Ubc13-Uev1a.
Ouabain, and related glycosides that inhibit the Na+/K+ATPase, are therapeutically valuable for increasing heart muscle contractile force, though these compounds also have molecular and cellular side effects. Our findings provide new insight into cellular actions of ouabain via ZNRF1 and ZNRF2. Knockdown of ZNRF2 in cells prevented the decrease in surface plasma membrane and total Na+/K+ATPase α1 protein levels that was induced by ouabain. Another intriguing observation was that low concentrations of the Na+/K+ATPase inhibitor caused a marked reduction in cellular ZNRF1 levels, even though we were unable to deplete ZNRF1 protein experimentally using siRNA without killing the cells. These findings implicate ZNRF1 and ZNRF2 in mediating the regulation of Na+/K+ATPase in cells, and indicate that there may be an intricate regulatory interplay between these proteins. It is therefore possible that the reason why ZNRF2 affected Ca2+-dependent exocytosis in PC12 cells (Araki and Milbrandt, 2003) is that the Na+/K+ATPase supports this process (reviewed by Lingrel, 2010). In contrast, we observed no obvious effect of ZNRF2 on the half-life of Na+/K+ATPase α1, nor on Na+/K+ATPase-dependent Rb+ uptake (supplementary material Fig. S6D,E,F). Of course, enzymatic activity would be affected as a secondary effect of changes in Na+/K+ATPase location, but given that the experiments in Fig. 8B require use of ouabain in vivo, we could not also test enzyme activity in these experiments.
Overall, our results define regulatory and functional roles for motifs and domains from the N- to the C-terminus of ZNRF1 and ZNRF2. N-myristoylation targets these proteins to membranes. The adjacent alanine- and glycine-rich region of ZNRF2 is phosphorylated and binds 14-3-3s in response to multiple extracellular stimuli, and these regulations oppose the effect of N-myristoylation by dissociating the ZNRF2 from membranes into the cytoplasm. On the membranes, both ZNRF1 and ZNRF2 interact via their UBZ domains with the cytoplasmic region of Na+/K+ATPase α1, which we hypothesise involves targeting to a pre-ubiquitylated or otherwise ‘primed’ form of Na+/K+ATPase α1. The RING finger then catalyses further ubiquitylation of the Na+/K+ATPase α1, possibly by polyubiquitin chain elongation involving Lys63 linkages specified by the Ubc13-Uev1a complex, which is co-purified in stoichiometric amounts when the E3s are immunoprecipitated from cell lysates. Our data further suggest that ZNRF1 and ZNRF2 influence the ouabain-induced endocytosis and/or degradation of the Na+/K+ATPase α1, and there is also a feedback in this system because ouabain causes cellular depletion of ZNRF1.
While our findings raise many new mechanistic questions, overall they identify ZNRF1 and ZNRF2 as new players in the regulation of the Na+/K+ATPase, the ubiquitous energy-demanding pump that has special roles in processes such as cellular nutrient uptake, renal Na+ reabsorption, neuronal excitability and muscle contraction. Genetic experiments in mice models should define the relative importance of these mechanisms in health and disease.
Materials and Methods
Materials
Digoxygenin-O-methylcarbonyl-ε-aminocaproic-acid-N-hydroxysuccinimide (DIG) ester and protease inhibitor cocktail tablets (no. 1697498 and no. 05892791001) were from Roche; polyethylenimine (PEI) was from Polysciences; Protein G-Sepharose, glutathione-Sepharose and enhanced chemiluminescence western blotting kit were from Amersham Bioscience; [32P-γ]ATP was from Perkin Elmer; dimethyl pimelimidate (DMP) and GNF-2 were from Sigma-Aldrich; BODIPY TR ceramide (D-7540), Precast NuPAGE polyacrylamide Bis-Tris gels, Colloidal Coomassie, LDS sample buffer, hygromycin, tetracycline, blasticidin and zeocin were from Invitrogen; sequencing-grade trypsin was from Promega; and microcystin-LR was kindly provided by Dr. Linda Lawton (Robert Gordon University, Aberdeen, UK).
Generation of stably-transfected HEK293 Flp-In T-REx and U2OS Flp-In stable lines
To ensure low-level uniform expression of recombinant proteins, we followed manufacturer's instructions (Invitrogen) to generate stable cell lines that express GFP-tagged forms of ZNRF1 and ZNRF2, and FLAG-tagged Na+/K+ATPase α1 subunit, from the pcDNA5-FRT-TO-GFP plasmid in a tetracycline-inducible manner. Protein expression was induced with 1 µg/ml of tetracycline. U2OS Flp-In cells, were transfected with pcDNA5-FRT-TO-GFP vectors containing ZNRF1 or ZNRF2 together with the plasmid pOG44 (Invitrogen) which encodes Flp recombinase.
Antibodies, cell lysis and immunoprecipitation
Anti-ZNRF2 was raised against bacterially-expressed GST-ZNRF2 (human) in sheep S617C at the PTU/BSC, Scottish National Blood Transfusion Service, Penicuik. After anti-GST antibodies were depleted from the 3rd bleed using immobilized GST, antibodies were affinity purified with immobilized immunogen. The antibodies that recognize phosphorylated sites on ZNRF2 were raised in sheep against the following synthetic phosphopeptides: CGRTRAYpSGSDLPS (residues 13 to 25 plus Cys for coupling, where pS represents pSer19; sheep S611C, 2nd bleed); CAAPRSRpSLGGAVG (residues 76 to 88 plus Cys, pSer82; S612C, 2nd bleed) and CPRLVIGpSLPAHLSP (residues 139 to 152, plus Cys, pSer145; S612C, 3rd bleed). Ubc13 antibody was raised against His-Ubc13 protein (S342B, 1st bleed). Monoclonal Na+/K+ATPase α1 (C464.6) antibody, anti-14-3-3 (K19) and GFP antibodies were from Santa Cruz. Anti-HA, anti-FLAG M2 and anti-FLAG agarose (A2220) were from Sigma. Antibodies that recognize pThr172 on AMPK, pSer473 on PKB, pSer80 on human ACC, pThr202/Tyr204 on ERK1/2, pThr389 on p70 S6 kinase and pSer157 on VASP were from Cell Signaling Technology. ZNRF1 antibodies were raised against bacterially-expressed GST-ZNRF1 (human) in sheep S219D or from Sigma (WH0084937M1, used for immunoprecipitation).
Digoxigenin (DIG)-14-3-3 probes were prepared as described previously (Moorhead et al., 1999), used in far-western assays (where DIG-14-3-3 takes the place of primary antibodies in western blots) and detected with a HRP-conjugated anti-DIG secondary (Roche). GFP-Trap®-agarose was from Chromotek.
Human HEK293 cells cultured on 10-cm-diameter dishes in DMEM (Dulbecco's modified Eagle's medium) containing 10% (v/v) FBS (foetal bovine serum) were used untransfected, stably-transfected, or 36 to 48 h after transient transfection (PEI method) with the plasmids indicated. Cells were serum-starved for 12 h (unstimulated), then stimulated with IGF1 (50 ng/ml for 20 min), PMA (100 ng/ml for 30 min), EGF (50 ng/ml for 15 min), forskolin (10 µM for 30 min), A769662 (50 µM for 60 min), A23187 (10 µM for 1 h), and protein phosphatase inhibitor calyculin A (50 ng/ml for 15 min). Where indicated, prior to stimulations, cells were incubated with PI-103 (1 µM for 30 min), LY294002 (100 µM for 1 h), Ku-0063794 (1 µM for 30 min), wortmannin (100 nM for 1 h), AKTi1/2 (10 µM for 1 h), rapamycin (50 nM for 30 min), Gö6983 (1 µM for 30 min), Gö6976 (1 µM for 30 min), PD184352 (2 µM for 1 h), U0126 (10 µM for 1 h), BI-D1870 (10 µM for 30 min), SB203580 (5 µM for 1 h), Birb0796 (100 nM for 1 h) and H-89 (30 µM for 30 min). After stimulations, the medium was aspirated and cells lysed in 0.35 ml of ice-cold Triton X-100 Lysis Buffer (TX-LB) comprised 25 mM Tris-HCl (pH 7.5), 1 mM EDTA, 1 mM EGTA, 1% Triton X-100, 50 mM NaF, 5 mM sodium pyrophosphate, 1 mM sodium orthovanadate, 1 mM benzamidine, 0.2 mM PMSF, 0.1% 2-mercaptoethanol, 1 µM microcystin-LR, 0.27 M sucrose and one mini CompleteTM proteinase inhibitor cocktail tablet (NO. 1697498, Roche) per 10 ml of lysis buffer. Lysates were clarified by centrifugation (16,000 g for 15 min at 4°C), snap frozen and stored at −80°C. Protein concentrations were determined with Coomassie Protein Assay Reagent (Thermo Scientific).
GFP-tagged proteins were isolated from 2-4 mg lysates using 10 µl of GFP–Trap®-agarose, incubated for 2 h at 4°C, and washed thrice with lysis buffer. Anti-FLAG immunoprecipitations used 10 µl anti-FLAG agarose for 2 h at 4°C. For immunoprecipitations of endogenous ZNRF1 and ZNRF2 and HA-proteins, and controls with pre-immune IgG, the antibodies were covalently coupled to Protein G-Sepharose (1 mg of antibody to 1 ml of resin) by cross-linking with dimethyl pimelimidate (Harlow and Lane, 1988). Immunoprecipitations of endogenous proteins used 2 µg of antibody/mg cell lysate for 4 to 16 h at 4°C.
Plasmids
Recombinant DNA procedures, restriction digests, ligations and PCR, were performed using standard protocols. All PCR reactions were carried out using KOD Hot Start DNA polymerase (Novagen). DNA sequencing was performed by The Sequencing Service, College of Life Sciences, University of Dundee (www.dnaseq.co.uk).
Vectors for expression of proteins in mammalian cells were pEGFPN1 or pcDNA5 FRT/TO (for stable integration of the plasmid and expression induced by tetracycline) modified to express GFP-tagged forms of the human ZNRF2 protein, generally with a C-terminal GFP tag unless stated. In-house numbers for the reference clones are as follows: ZNRF2-GFP (DU16338), ZNRF2 Ser19Ala-GFP (DU16339), ZNRF2 Ser82Ala-GFP (DU16471), ZNRF2 Ser145Ala-GFP (DU16841), ZNRF2 Ser19/Ser82Ala-GFP (DU16536), ZNRF2 Ser19/Ser145Ala-GFP (DU16532), ZNRF2 Gly2Ala-GFP (DU31836), ZNRF2 C199A-GFP (DU16691), ZNRF2-(1-156)-GFP (DU31834), ZNRF2-(1-180)-GFP (DU31835), ZNRF2-(156-242)-GFP (DU31853), ZNRF2(1-11)+(156-242)-GFP (DU31969), and GFP-ZNRF2 with N-terminal GFP tag (DU16513).
In addition, plasmids for expressing the following human proteins were prepared from the pcDNA5 FRT/TO vector: ZNRF2-GFP (DU31340), ZNRF2 Ser19Ala-GFP (DU36204), ZNRF2 Ser82Ala-GFP (DU36205), ZNRF2 Ser145Ala-GFP (DU36201), ZNRF2 Gly2Ala-GFP (DU36030), ZNRF2 C199A-GFP (DU31341), ZNRF2 C160A/C163A-GFP (DU31966), N-terminal GFP tag ZNRF2 (DU16513), ZNRF1-GFP (DU36220), GFP-ZNRF1 (DU33620), ZNRF1 C184A-GFP (DU36515), ZNRF1-C145A/C148A-GFP (DU36546), ZNRF1-(1-116)-(FRT/TO)-GFP (DU36528), ZNRF1-(1-140)-(FRT/TO)-GFP (DU36529), ZNRF1-(1-11)+(140-END)-(FRT/TO)-GFP (DU36547), ZNRF1-(140-end)-(FRT/TO)-GFP (DU36530), ATP1A1-FLAG (DU31843), ATP1A1 (R385-R596)-FLAG (DU31857) and ATP1A1 (T339-L772)-FLAG (DU31897).
Human 14-3-3 isoforms with N-terminal GST tags were expressed in mammalian cells from the following plasmids that were generated using the pEBG6P-1 vector: 14-3-3beta (DU31357), 14-3-3epsilon (DU2660), 14-3-3eta (DU32512), 14-3-3gamma (DU32507), 14-3-3sigma (DU31358), 14-3-3theta (DU32508) and 14-3-3zeta (DU3173).
The pGEX6P vector was used for the expressing the following human proteins with N-terminal GST tags in E. coli: GST-ZNRF2, GST-ZNRF2 Ser19Ala, GST-ZNRF2 Ser82Ala, GST-ZNRF2 Ser145Ala, GST-ZNRF2 Ser19/Ser82Ala, GST-ZNRF2 Ser19/Ser145Ala, and GST-ZNRF1. The pGEX6P vector was also used to express mouse ZNRF2 with an N-terminal GST tag in E. coli (DU36367), which was used for raising an antibody against mouse ZNRF2.
Mass spectrometry to identify proteins and phosphorylated residues
Protein identification was by in-gel digestion of Coomassie colloidal blue-stained protein gel bands for 16 h with 5 µg/ml trypsin, and analysis of the tryptic peptides by LC-MS on a Thermo LTQ-Orbitrap system. The MS data was analyzed through the Mascot search engine (www.matrixscience.com) against the human International Protein Index database. Tryptic phosphopeptides were identified by LC-MS on an ABI 4000 Q-TRAP system using precursor ion scanning in negative mode to search for release of the (PO3)− ion (−79 Da) allowing for +/−1 Da (Williamson et al., 2006), followed by MS2 analysis in positive mode. The resulting data files were searched against the appropriate sequence, using Mascot run on an in-house server, with a peptide mass tolerance of 1.2 Da, a fragment mass tolerance of 0.8 Da, and with variable modifications allowing for phosphorylation of serine/threonine or tyrosine and for methionine oxidation or dioxidation. MassFingerPrinting results from Mascot were viewed using a software package from ProteinGURU (http://www.proteinguru.com).
SILAC (stable isotope labelling by amino acids in cell culture) for HEK293 cells
SILAC DMEM (high glucose without NaHCO3, L-glutamine, arginine, lysine and methionine; Biosera no. A0347) was supplemented with methionine, glutamine, NaHCO3, 10% dialysed FBS (Hyclone) and the following combinations of unlabelled and isotopically-labelled arginine (84 µg/ml) and lysine (146 µg/ml): L-arginine and L-lysine (Sigma–Aldrich) for R0K0 (light); L-arginine-HCl (U-13C6) and L-lysine-2HCl (4,4,5,5,D4) for R6K4 (medium); and L-arginine-HCl (U-13C6, 15N4) and 13C-L-lysine-2HCl (U-13C6, 15N2) for R10K8 (heavy) (Cambridge Isotope Laboratory). The SILAC medium was filtered through a 0.22-µm filter (Millipore) and cells cultured for five passages in these media.
Tryptic peptides of proteins from SILAC-labelled cells were analyzed by Orbitrap mass spectrometry and raw mass spectrometric data files for each experiment were collated into a single quantitated dataset using the MaxQuant (version 1.0.13.13; http://www.maxquant.org/) and the Mascot search engine (Matrix Science, version 2.2.2) softwares. Enzyme specificity was set to that of trypsin, allowing for cleavage N-terminal to proline residues and between aspartic acid and proline residues. Other parameters were: (i) Variable modifications: methionine oxidation and protein N-acetylation; (ii) Fixed modifications: cysteine carbamidomethylation; (iii) Database: target-decoy human MaxQuant (ipi.HUMAN.v3.52.decoy) (containing 148,380 database entries); (iv) Labels: R6K4 or R10K8; (v) MS/MS tolerance: 0.5 Da; (vi) Minimum peptide length: 6; (vii) Top MS/MS peaks per 100 Da: 5; (viii) Maximum missed cleavages: 2; (ix) Maximum of labelled amino-acids: 3; (x) False Discovery Rate (FDR): 1%; (xi) Posterior Error Probability: 1; (xii) Minimum ratio count: 2. The protein ratios were calculated using all peptides. As well as taking the FDR into account, proteins were considered identified if they had at least one unique peptide and considered quantified if they had at least one quantified SILAC pair. Data quality was also assessed manually, and only high confidence results reported.
Immunofluorescence and microscopy of fixed cells
Cells grown on coverslips were fixed with 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, rinsed with PBS, stained with DAPI (4′,6-diamidino-2-phenylindole) and mounted. The slides were viewed under a Zeiss LSM700 microscope using an alphaPlan-Apochromat ×100 NA (numerical aperture) 1.46 objective.
Live cell imaging
Cells were grown on glass bottom dishes (WillCo). Prior to live cell imaging, culture medium was changed to Leibovitz's L-15 medium (Invitrogen). Images were collected using the DeltaVision microscope system (Applied Precision) with an Olympus IX70 microscope, using a Plan Apo 60X 1.4 numerical aperture optical lens (Olympus) and a cooled CCD camera (Coolsnap HQ, Photometrics) via SoftWoRx software (Applied Precision). Time-lapse images were collected every 3 min for 2 h, maintaining the temperature at 37°C in an environmental chamber (Solent Scientific, Segensworth, UK).
Subcellular fractionation by ultracentrifugation
Cells were lysed in detergent-free 50 mM Tris pH 7.5, 250 mM sucrose, 4 mM EDTA pH 7.0, 1 mM EGTA pH 7.0, ‘Complete’ protease inhibitor (Roche), 50 mM NaF, 10 mM glycerophosphate, 5 mM pyrophosphate, 1 mM orthovanadate, 1 µM microcystin, 1 mM benzamidine and 0.1 mM PMSF. The lysate was passed through a 21G needle ten times and clarified by centrifugation at 1000 g at 4°C for 5 min. The supernatant was spun at 150,000 g in a Beckman TLX ultracentrifuge, rotor TLS55 at 4°C for 30 min. The 150,000-g-pellet was washed twice with 1 ml detergent-free lysis buffer prior to resuspending in lysis buffer containing 0.1 M NaCl and 1% (v/v) Triton X-100. NaCl and Triton X-100 were added to the 150,000-g-supernatant to the same final concentrations as the membrane fractions. Both fractions were sonicated on ice for 1 min at 50% power using Vibracell sonicator (Sonics & Materials Inc., CN, USA).
Ubiquitylation assays
A mixture of 0.1 µM E1 ubiquitin ligase, 1 µM E2 (UbcH5a or Ubc13/UEV1a), 0.1 mM ubiquitin (Boston Biochem), 5 mM MgCl2, 2 mM ATP in 50 mM Tris-HCl (pH 7.5) and ZNRF2-GFP immunoprecipitated from 2 mg of cell lysate (15 µl GFP-Trap beads), was incubated in a total volume of 50 µl for 10–60 min at 30°C. Reactions were stopped with LDS sample dye (Invitrogen), products resolved by NuPAGE Bis-Tris 4–12% gradient gels (Invitrogen), transferred to nitrocellulose membranes and immunoblotted with anti-ubiquitin (DAKO). For ZNRF2 immunoprecipitated from HEK293 cell lysates, the non-specific protein kinase inhibitor staurosporine (75 µM) was also added to prevent phosphorylation of ZNRF2 during the assay by any protein kinases that might be present as trace contaminants.
In vitro phosphorylation of GST-ZNRF2
Bacterially-expressed GST-ZNRF2 (2 µg) was incubated with 10 mM MgCl2, and 0.1 mM [γ-32P]ATP (∼230 cpm/pmol) for the indicated times at 30°C. Kinases were used at 1 U/ml, where one unit of protein kinase activity is the amount of enzyme that catalyses the incorporation of 1 nmol phosphate into standard substrate in 1 min. Samples were subjected to SDS-PAGE followed by colloidal Coomassie staining and autoradiography. The ZNRF2 was excised, and stoichiometry of incorporation of 32P-radioactivity into protein quantified as described (Hastie et al., 2006).
Labelling of cell surface proteins by biotin
HeLa cells cultured in 100 mm dishes were washed three times with ice-cold PBS and incubated with 0.5% sulfo-NHS-SS-biotin (Pierce) in PBS containing 0.9 mM CaCl2 and 0.5 mM MgCl2 at 4°C for 60 min with gentle agitation. The biotinylation was terminated by the addition of PBS containing 50 mM glycine and 5 mg/ml bovine serum albumin. After repeated washes with PBS, the cells were harvested in modified RIPA buffer (50 mM Tris-HCl, pH 8.0, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate and 0.1% SDS and 0.5% Tween-20, protease inhibitor cocktail (no. 05892791001, Roche). Cell lysate (0.5–1 mg) was incubated with 25 µl streptavidin-agarose (Pierce) for 12 h, beads were washed 4 times in TX-LB and proteins were eluted in 50 mM DTT in LDS-PAGE sample buffer (total 50 µl) at 65°C while shaking at 1400 rpm for 15 min.
siRNA knockdowns
ZNRF2 and ZNRF1 siRNAs were synthesized by Eurofins MWG Operon. siRNA sequences used were: CAAUAUGCCUUGAAGAAUU and CAUAGACUCGUGGUUUGAA for ZNRF2; and GUGAGUGUGUGAUCUGCCU and CAUAGACUCGUGGUUUGAA for ZNRF1. HeLa cells were transfected at 60–70% confluency with 100 nM siRNA using Interferin (Polyplus).
Reproducibility
Except for mass spectrometry data, results shown are representative of at least three similar experiments.
Supplementary Material
Acknowledgments
We are grateful to the teams managed by James Hastie and Hilary McLauchlan of the DSTT, for protein production and antibody purification; to Bob Gourlay for help with mass spectrometry, the DNA Sequencing Service at the College of Life Sciences, University of Dundee; Kirsten McLeod and Janis Stark for tissue culture support; Ivan M. Muñoz for gel filtration samples; Axel Knebel for providing supplementary material Fig. S5A,B; and William Fuller, University of Dundee for helpful discussion.
Footnotes
Funding
This work was supported by the UK Medical Research Council [grant number MC_U127084354]; and pharmaceutical companies that support the Division of Signal Transduction Therapy (DSTT) at the University of Dundee (AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Merck-Serono and Pfizer). Deposited in PMC for release after 6 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.110296/-/DC1
References
- Al–Khalili L., Yu M., Chibalin A. V. (2003). Na+,K+-ATPase trafficking in skeletal muscle: insulin stimulates translocation of both alpha 1- and alpha 2-subunit isoforms. FEBS Lett. 536, 198–202 10.1016/S0014-5793(03)00047-4 [DOI] [PubMed] [Google Scholar]
- Araki T., Milbrandt J. (2003). ZNRF proteins constitute a family of presynaptic E3 ubiquitin ligases. J. Neurosci. 23, 9385–9394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araki T., Nagarajan R., Milbrandt J. (2001). Identification of genes induced in peripheral nerve after injury. Expression profiling and novel gene discovery. J. Biol. Chem. 276, 34131–34141 10.1074/jbc.M104271200 [DOI] [PubMed] [Google Scholar]
- Bienko M., Green C. M., Crosetto N., Rudolf F., Zapart G., Coull B., Kannouche P., Wider G., Peter M., Lehmann A. R.et al. (2005). Ubiquitin-binding domains in Y-family polymerases regulate translesion synthesis. Science 310, 1821–1824 10.1126/science.1120615 [DOI] [PubMed] [Google Scholar]
- Bish R. A., Myers M. P. (2007). Werner helicase-interacting protein 1 binds polyubiquitin via its zinc finger domain. J. Biol. Chem. 282, 23184–23193 10.1074/jbc.M701042200 [DOI] [PubMed] [Google Scholar]
- Campbell D. G., Morrice N. A. (2002). Identification of protein phosphorylation sites by a combination of mass spectrometry and solid phase Edman sequencing. J. Biomol. Tech. 13, 119–130 [PMC free article] [PubMed] [Google Scholar]
- Choi Y., Seeliger M. A., Panjarian S. B., Kim H., Deng X., Sim T., Couch B., Koleske A. J., Smithgall T. E., Gray N. S. (2009). N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J. Biol. Chem. 284, 29005–29014 10.1074/jbc.M109.026633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comellas A. P., Kelly A. M., Trejo H. E., Briva A., Lee J., Sznajder J. I., Dada L. A. (2010). Insulin regulates alveolar epithelial function by inducing Na+/K+-ATPase translocation to the plasma membrane in a process mediated by the action of Akt. J. Cell Sci. 123, 1343–1351 10.1242/jcs.066464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crambert G., Geering K. (2003). FXYD proteins: new tissue-specific regulators of the ubiquitous Na,K-ATPase. Sci. STKE 2003, RE1 10.1126/stke.2003.166.re1 [DOI] [PubMed] [Google Scholar]
- Dubois F., Vandermoere F., Gernez A., Murphy J., Toth R., Chen S., Geraghty K. M., Morrice N. A., MacKintosh C. (2009). Differential 14-3-3 affinity capture reveals new downstream targets of phosphatidylinositol 3-kinase signaling. Mol. Cell. Proteomics 8, 2487–2499 10.1074/mcp.M800544-MCP200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddins M. J., Carlile C. M., Gomez K. M., Pickart C. M., Wolberger C. (2006). Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat. Struct. Mol. Biol. 13, 915–920 10.1038/nsmb1148 [DOI] [PubMed] [Google Scholar]
- Eisenberg–Lerner A., Bialik S., Simon H. U., Kimchi A. (2009). Life and death partners: apoptosis, autophagy and the cross-talk between them. Cell Death Differ. 16, 966–975 10.1038/cdd.2009.33 [DOI] [PubMed] [Google Scholar]
- Ewart H. S., Klip A. (1995). Hormonal regulation of the Na(+)-K(+)-ATPase: mechanisms underlying rapid and sustained changes in pump activity. Am. J. Physiol. Cell Physiol. 269, C295–C311 [DOI] [PubMed] [Google Scholar]
- Fuller W., Howie J., McLatchie L. M., Weber R. J., Hastie C. J., Burness K., Pavlovic D., Shattock M. J. (2009). FXYD1 phosphorylation in vitro and in adult rat cardiac myocytes: threonine 69 is a novel substrate for protein kinase C. Am. J. Physiol. Cell Physiol. 296, C1346–C1355 10.1152/ajpcell.00523.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F., Bossuyt J., Martin J. L., Despa S., Bers D. M. (2010). Role of phospholemman phosphorylation sites in mediating kinase-dependent regulation of the Na+-K+-ATPase. Am. J. Physiol. Cell Physiol. 299, C1363–C1369 10.1152/ajpcell.00027.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantschel O., Nagar B., Guettler S., Kretzschmar J., Dorey K., Kuriyan J., Superti–Furga G. (2003). A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 112, 845–857 10.1016/S0092-8674(03)00191-0 [DOI] [PubMed] [Google Scholar]
- Harlow E., Lane D. (1988). Antibodies: a Laboratory Manual. Cold Spring Harbor, N.Y. New York: Cold Spring Harbor Laboratory Press [Google Scholar]
- Hastie C. J., McLauchlan H. J., Cohen P. (2006). Assay of protein kinases using radiolabeled ATP: a protocol. Nat. Protoc. 1, 968–971 10.1038/nprot.2006.149 [DOI] [PubMed] [Google Scholar]
- Hatou S., Yamada M., Akune Y., Mochizuki H., Shiraishi A., Joko T., Nishida T., Tsubota K. (2010). Role of insulin in regulation of Na+-/K+-dependent ATPase activity and pump function in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 51, 3935–3942 10.1167/iovs.09-4027 [DOI] [PubMed] [Google Scholar]
- Lang F., Strutz–Seebohm N., Seebohm G., Lang U. E. (2010). Significance of SGK1 in the regulation of neuronal function. J. Physiol. 588, 3349–3354 10.1113/jphysiol.2010.190926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim S., Strahl T., Thorner J., Ames J. B. (2011). Structure of a Ca2+-myristoyl switch protein that controls activation of a phosphatidylinositol 4-kinase in fission yeast. J. Biol. Chem. 286, 12565–12577 10.1074/jbc.M110.208868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingrel J. B. (2010). The physiological significance of the cardiotonic steroid/ouabain-binding site of the Na,K-ATPase. Annu. Rev. Physiol. 72, 395–412 10.1146/annurev-physiol-021909-135725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Kesiry R., Periyasamy S. M., Malhotra D., Xie Z., Shapiro J. I. (2004). Ouabain induces endocytosis of plasmalemmal Na/K-ATPase in LLC-PK1 cells by a clathrin-dependent mechanism. Kidney Int. 66, 227–241 10.1111/j.1523-1755.2004.00723.x [DOI] [PubMed] [Google Scholar]
- MacKay C., Déclais A. C., Lundin C., Agostinho A., Deans A. J., MacArtney T. J., Hofmann K., Gartner A., West S. C., Helleday T.et al. (2010). Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142, 65–76 10.1016/j.cell.2010.06.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markson G., Kiel C., Hyde R., Brown S., Charalabous P., Bremm A., Semple J., Woodsmith J., Duley S., Salehi–Ashtiani K.et al. (2009). Analysis of the human E2 ubiquitin conjugating enzyme protein interaction network. Genome Res. 19, 1905–1911 10.1101/gr.093963.109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorhead G., Douglas P., Cotelle V., Harthill J., Morrice N., Meek S., Deiting U., Stitt M., Scarabel M., Aitken A.et al. (1999). Phosphorylation-dependent interactions between enzymes of plant metabolism and 14-3-3 proteins. Plant J. 18, 1–12 10.1046/j.1365-313X.1999.00417.x [DOI] [PubMed] [Google Scholar]
- Morth J. P., Pedersen B. P., Toustrup–Jensen M. S., Sørensen T. L., Petersen J., Andersen J. P., Vilsen B., Nissen P. (2007). Crystal structure of the sodium-potassium pump. Nature 450, 1043–1049 10.1038/nature06419 [DOI] [PubMed] [Google Scholar]
- Morth J. P., Pedersen B. P., Buch–Pedersen M. J., Andersen J. P., Vilsen B., Palmgren M. G., Nissen P. (2011). A structural overview of the plasma membrane Na+,K+-ATPase and H+-ATPase ion pumps. Nat. Rev. Mol. Cell Biol. 12, 60–70 10.1038/nrm3031 [DOI] [PubMed] [Google Scholar]
- Oakhill J. S., Chen Z. P., Scott J. W., Steel R., Castelli L. A., Ling N., Macaulay S. L., Kemp B. E. (2010). β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc. Natl. Acad. Sci. USA 107, 19237–19241 10.1073/pnas.1009705107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa H., Shinoda T., Cornelius F., Toyoshima C. (2009). Crystal structure of the sodium-potassium pump (Na+,K+-ATPase) with bound potassium and ouabain. Proc. Natl. Acad. Sci. USA 106, 13742–13747 10.1073/pnas.0907054106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plans V., Scheper J., Soler M., Loukili N., Okano Y., Thomson T. M. (2006). The RING finger protein RNF8 recruits UBC13 for lysine 63-based self polyubiquitylation. J. Cell. Biochem. 97, 572–582 10.1002/jcb.20587 [DOI] [PubMed] [Google Scholar]
- Poulsen H., Morth P., Egebjerg J., Nissen P. (2010). Phosphorylation of the Na+,K+-ATPase and the H+,K+-ATPase. FEBS Lett. 584, 2589–2595 10.1016/j.febslet.2010.04.035 [DOI] [PubMed] [Google Scholar]
- Rowland A. F., Fazakerley D. J., James D. E. (2011). Mapping insulin/GLUT4 circuitry. Traffic 12, 672–681 10.1111/j.1600-0854.2011.01178.x [DOI] [PubMed] [Google Scholar]
- Saitoh F., Araki T. (2010). Proteasomal degradation of glutamine synthetase regulates schwann cell differentiation. J. Neurosci. 30, 1204–1212 10.1523/JNEUROSCI.3591-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinoda T., Ogawa H., Cornelius F., Toyoshima C. (2009). Crystal structure of the sodium-potassium pump at 2.4 A resolution. Nature 459, 446–450 10.1038/nature07939 [DOI] [PubMed] [Google Scholar]
- Taniguchi C. M., Emanuelli B., Kahn C. R. (2006). Critical nodes in signalling pathways: insights into insulin action. Nat. Rev. Mol. Cell Biol. 7, 85–96 10.1038/nrm1837 [DOI] [PubMed] [Google Scholar]
- van Wijk S. J., de Vries S. J., Kemmeren P., Huang A., Boelens R., Bonvin A. M., Timmers H. T. (2009). A comprehensive framework of E2-RING E3 interactions of the human ubiquitin-proteasome system. Mol. Syst. Biol. 5, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakatsuki S., Saitoh F., Araki T. (2011). ZNRF1 promotes Wallerian degeneration by degrading AKT to induce GSK3B-dependent CRMP2 phosphorylation. Nat. Cell Biol. 13, 1415–1423 10.1038/ncb2373 [DOI] [PubMed] [Google Scholar]
- Williamson B. L., Marchese J., Morrice N. A. (2006). Automated identification and quantification of protein phosphorylation sites by LC/MS on a hybrid triple quadrupole linear ion trap mass spectrometer. Mol. Cell. Proteomics 5, 337–346 10.1074/mcp.M500210-MCP200 [DOI] [PubMed] [Google Scholar]
- Windheim M., Peggie M., Cohen P. (2008). Two different classes of E2 ubiquitin-conjugating enzymes are required for the mono-ubiquitination of proteins and elongation by polyubiquitin chains with a specific topology. Biochem. J. 409, 723–729 10.1042/BJ20071338 [DOI] [PubMed] [Google Scholar]
- Woodruff R. V., Bomar M. G., D'Souza S., Zhou P., Walker G. C. (2010). The unusual UBZ domain of Saccharomyces cerevisiae polymerase η. DNA Repair (Amst.) 9, 1130–1141 10.1016/j.dnarep.2010.08.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Y., Rape M. (2009). Building ubiquitin chains: E2 enzymes at work. Nat. Rev. Mol. Cell Biol. 10, 755–764 10.1038/nrm2780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida K., Watanabe M., Hatakeyama S. (2009). ZNRF1 interacts with tubulin and regulates cell morphogenesis. Biochem. Biophys. Res. Commun. 389, 506–511 10.1016/j.bbrc.2009.09.011 [DOI] [PubMed] [Google Scholar]
- Yoshimura S. H., Iwasaka S., Schwarz W., Takeyasu K. (2008). Fast degradation of the auxiliary subunit of Na+/K+-ATPase in the plasma membrane of HeLa cells. J. Cell Sci. 121, 2159–2168 10.1242/jcs.022905 [DOI] [PubMed] [Google Scholar]
- Zozulya S., Stryer L. (1992). Calcium-myristoyl protein switch. Proc. Natl. Acad. Sci. USA 89, 11569–11573 10.1073/pnas.89.23.11569 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.