

Abstract
Hypohidrotic ectodermal dysplasia (HED) is a type of genodermatosis characterized by the abnormal development of sweat glands, teeth, and hair. The most prevalent form of HED is X-linked hypohidrotic ectodermal dysplasia (XLHED), which is associated with mutations in the EDA gene. The aim of this case report was to describe a family with XLHED with emphasis on differences in orofacial features between members. Family members were systematically evaluated to characterize the pattern of inheritance and clinical features. Dental examination included evaluation of agenesis and abnormal teeth structure. The pedigree of the last seven generations of the family was constructed. Clinical examination and medical history revealed five males affected by HED and nine female as heterozygous carriers. The males exhibited the classic phenotype of XLHED, with dental abnormalities, hypohydrosis, and craniofacial dysmorphologies. The heterozygous carriers of the X-linked gene defect principally exhibited dental agenesis of the lateral maxillary incisors. Careful clinical examination, including dental evaluation, is an important way to detect heterozygous carriers of X-linked HED. Heterozygous parents of patients with HED may also show some features of the disorder. The identification of female carriers results in genetic counseling being offered to affected families, as well as providing adequate treatment as necessary and long-term follow-up of these patients.
Keywords: Hypohidrotic ectodermal dysplasia, Heterozygous carriers, Orofacial abnormalities, Dental anomaly
Introduction
Hypohidrotic ectodermal dysplasias (HED; OMIM 305100) form a large and complex group of congenital disorders, involving all the Mendelian modes of inheritance, and are characterized by the pathological development and morphogenesis of ectodermal structures [1–3]. The most prevalent form of HED is X-linked hypohidrotic ectodermal dysplasia (XLHED); however, some families show autosomal dominant or recessive inheritance forms of the disorder [4, 5]. The incidence of HED is estimated to be 1 per 100,000 births. It is characterized by absence or a diminished number of eccrine sweat glands, missing and malformed teeth, and thin and sparse hair. The severity of symptoms can vary between and within families [5, 6].
The oral phenotype includes multiple congenitally missing teeth, root and crown dysmorphism, mainly conical-shaped crowns, and reduced saliva flow [7, 8]. Microdontia is frequently observed in affected individuals [9]. Moderate to severe taurodontism is known to preferentially affect the second primary mandibular molars in some individuals with HED [10].
The disease is generally much more apparent in affected men than in carrier women. Female carriers who present with symptoms of XLHED may have defective dentition and stripes of hypotrichosis along the limbs and the back; most females only have mild or “partial” involvement and are generally not referred to a physician [11]. The gene responsible for XLHED is localized at Xq12-q13.1 that is associated with the EDA gene, which codes for a transmembrane protein that is expressed by keratinocytes, hair follicles, teeth, and sweat glands. This gene may play a key role in epithelial-mesenchymal signaling, leading to the clinical aspects of the disorder [5, 12–14].
The diagnosis of the majority of HED cases within the first year of life should be possible with the identification of recurrent episodes of unexplained hyperthermia, dry and scaly skin, delayed teething, ceruminal clots and recurrent airway infections, and other characteristics (thin, lightly pigmented scalp hair, and decreased sweating). With such key findings, clinicians can place a patient in a syndrome family [1–3, 15]. Skin biopsy may show absence or hypoplasia of sweat glands and a reduction of hair follicles and sebaceous glands. Radiographs generally showed hypodontia and dental abnormalities [16]. Specification of mutations can then be done by molecular analysis [11, 12, 17].
This study describes the clinical features of HED, with emphasis on its orofacial manifestations, by presenting a family affected by XLHED and briefly reviewing the literature on this genetic disorder.
Patients and Methods
The proband, a 5-year-old boy, was identified during routine clinical examination at the Center for Rehabilitation of Craniofacial Anomalies, Minas Gerais State, Brazil. The diagnosis was established after clinical examination, and we identify that the boy had hypohidrosis, heat intolerance, hypotrichosis, and missing teeth, and his scalp hair, eyelashes, and eyebrows were sparse and thin, characteristics classically ascribed to HED [1–3, 15]. After evaluation of this first case, we invited the family to the Center. The dermatological and craniofacial features of other family members were clinically evaluated in order to characterize the pattern of inheritance and clinical features. In this family, there was no history of either exposure to known teratogenic agents or maternal diseases. All pregnancies were described as normal, and no perinatal complications were mentioned.
Dental examination included the evaluation of missing teeth and any abnormal structures. The pedigree of the family was constructed using the last seven generations. Panoramic radiographs were also obtained. Informed consent was obtained from the legal guardians of the children and other participants, and the study was approved by the Institutional Ethics Committee.
Results
Analysis of the last seven generations revealed clinical features of HED. No patients had known consanguineous marriages. The family pedigree revealed an X-linked autosomal recessive pattern of transmission. Within the one family, five males were affected by HED and nine females were heterozygous carriers (Fig. 1). The clinical features of the male HED subjects and the female carriers are presented in Table 1.
Fig. 1.

Family pedigree. Family pedigree with HED. X-linked members revealed non-consanguineous marriages. Squares and circles indicate men and women, respectively. Blackened squares indicate the affected men, and circles shaded in black and white represent female carriers. An arrow indicates the proband (VII-4). An asterisk indicates the cases described
Table 1.
Clinical features of X-linked hypohidrotic ectodermal dysplasia
| Clinical findings | I-2 | II-2 | II-5 | VI-2 | VI-3 | VI-4 | VI-10 | VI-12 | VI-15 | VI-16 | VII-1 | VII-3 | VIII-4 | VIII-5 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Hypohidrosis | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Hypotrichosis | ||||||||||||||
| Mild | + | − | − | − | − | − | + | − | − | − | − | + | − | − |
| Severe | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Teeth | ||||||||||||||
| Presence one tooth | − | − | − | − | − | − | − | − | − | − | − | − | + | + |
| Conical teeth | + | + | + | + | + | + | + | + | + | + | + | + | − | − |
| Ag. lateral incisors | + | + | + | + | + | + | + | + | + | + | + | + | − | − |
| Ag. other teeth | + | + | + | − | − | − | − | + | − | + | − | − | − | − |
| Lower ears | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Heat intolerance | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Everted lips | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Scanty saliva | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Fragile, convex nails | − | + | + | − | − | − | + | + | − | − | − | + | + | + |
| Prominent forehead | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Dry, thin skin | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Saddle-shaped nose | − | + | + | − | − | − | − | + | − | − | − | − | + | + |
| Gender | F | M | M | F | F | F | F | M | F | F | F | F | M | M |
Ag Agenesis
Clinical examination of the males revealed the typical features of HED. The proband (family member VII-4) presented with hypohidrosis, hypotrichosis, and missing teeth. The scalp hair, eyelashes, and eyebrows were sparse and thin. The boy was also heat intolerant. He had a characteristic facial appearance, including a prominent forehead, saddle-back nose, protruding lips, and convex nails (Fig. 2). Clinical examination of the parents revealed agenesis of the lateral maxillary incisors and conical teeth in the mother (family member VI-15). No other features of DEH were observed in his parents. A second child was a 4-year-old male (family member VII-5), who was clinically similar to the first case (Fig. 3). The boy had a characteristic facial appearance, including a prominent forehead, saddle-back nose, and protruding lips. His mother had agenesis of the lateral maxillary incisors, mandibular incisors, and mandibular molar (family member VI-16). Panoramic radiograph of the proband showed the presence of a single tooth forming in the mandibular canine region (Fig. 4a). In the second boy, oral examination and panoramic radiography showed the presence of one tooth in the maxilla (Fig. 4b).
Fig. 2.

Proband case of X-linked hypohidrotic ectodermal dysplasia. The proband (family member VII-4). a Five-year-old male patient with the characteristic facial appearance, including a prominent forehead, saddle-shaped nose, prominent lips, and sparse, dry scalp hair, eyelashes, and eyebrows. b Apparent anodontia. c, d, eArrows indicates changes in extremities. c Hand with convex nails. d Foot with nail dysplasia. e Dehydrated foot
Fig. 3.

Second case of X-linked hypohidrotic ectodermal dysplasia. The second case (family member VII-5). a, b A 4-year-old boy, clinically similar to the proband case. Characteristic facial appearance, including a prominent forehead, saddle-back nose, and prominent lips. c Oral examination showed the presence of one tooth in the maxilla. d Hand with convex nails (arrow)
Fig. 4.
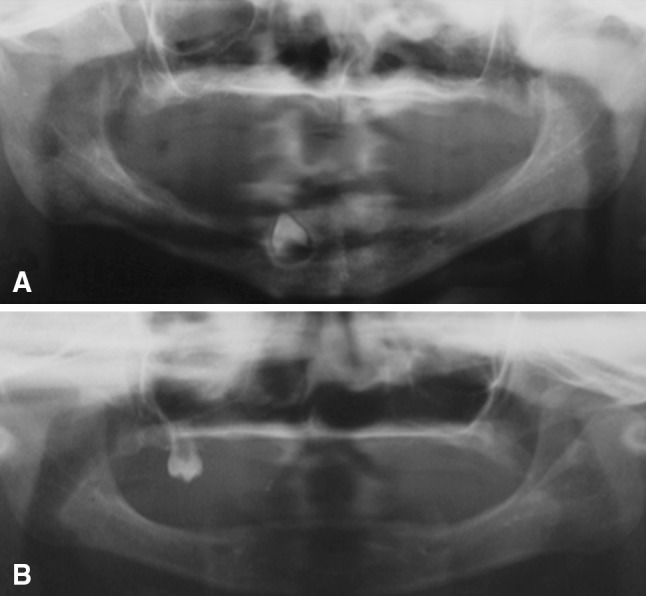
Panoramic radiographic of two cases of X-linked hypohidrotic ectodermal dysplasia. Panoramic radiograph of the proband (VII-4) (a) and of the second boy (VII-5) (b). The congenital absence of the majority of teeth is apparent. In the first case, only one tooth is developing in the mandibular canine region. In the second, only one molar is present in the maxilla
Clinical examination of female members showed principally the presence of hypodontia. Figure 5 shows a 5-year-old girl of this family (family member VII-3). Unlike her cousins, she had a normal face and hair. However, oral examination showed agenesis of the lateral maxillary incisors and conical teeth. Moreover, we noted areas of the body without hair, as well as convex nails. The same characteristics were noted in her mother (family member VI-10). Interestingly, evaluation of other women in this family also showed a mild HED phenotype (Table 1), especially with oral findings, as hypodontia.
Fig. 5.

Female carrier of X-linked hypohidrotic ectodermal dysplasia. Female carrier of XLHED (family member VII-3). a Five-year-old girl with the clinical features described for females in this family. She has a normal face and hair but agenesis of the lateral maxillary incisors and conical teeth. b Hand with convex nails (arrow)
We noted that females showed hypodontia, principally with agenesis of the lateral maxillary incisors, but that this was variable, with other teeth being absent in some cases. Normal levels of sweating were reported. The facial aspect was normal in all women evaluated. Based on the pedigree, we suggest that the HED was X-linked, manifesting in males but with a mild phenotype in females.
Initial dental treatment consisted of oral hygiene orientation for the patients and family. After this initial period, fissure sealants were applied and restoration conducted in the girl. For the other female members, restoration, periodontal, and endodontics treatment were considered. Surgery and prosthetic therapy were considered for the boys. The first patient is now rehabilitated with prosthesis for the maxillary and mandibular edentulous arches. The family also received genetic counseling.
Discussion
The clinical diagnosis of HED is easy, especially after the first year of life when medical history and clinical findings become highly characteristic [18]. After the initial diagnosis, the pattern of inheritance should be determined, since X-linked hemizygous males and autosomal recessive forms are not phenotypically distinguishable [19]. One of the main tasks in the case of a patient with HED is to find other possible carriers of the disorder in the patient’s family and to analyze the pattern of inheritance [18]. Clinical analyses of the families with XLHED are useful for checking carrier status and also provide information for genetic counseling and diagnosis of other affected members. Therefore, early diagnosis can maximize treatment by ensuring prompt referral to a specialist unit, thereby enabling long-term treatment planning.
In a female heterozygous for the XLHED gene, the presence of two different cell lines due to random inactivation (lyonization) of one of the two X chromosomes during embryogenesis results in much variability in the degree of clinical expression of the disorder. Thus, heterozygous females may show no clinical evidence of the disorder [20] or present only minor symptoms [21]. Dental abnormalities (hypodontia, microdontia, cone-shaped incisors), mild hypohidrosis, and mild hypotrichosis are the most commonly described signs in female carriers of XLHED [22]. It is widely believed that these linear characteristics of XLHED in female carriers of EDA mutations are the result of preferential inactivation of one X chromosome carrying the wild-type allele [23].
In the present study, clinical examination and a medical history were sufficient to identify five males affected by HED. Three mothers of males with HED and six additional females were heterozygous carriers of the X-linked gene defect. Teeth abnormalities were the most constant marker of the disorder in our nine heterozygous females with XLHED. Dental abnormalities can be considered a key feature since they are easily noted at a young age and should lead to diagnostic suspicion [18, 24]. Variability in dental phenotype has been described in heterozygous HED females, with moderate oligodontia, cone-shaped incisors, and delayed dental eruption [8, 18, 25].
Dental agenesis is found in heterozygous HED females, particularly involving the maxillary lateral incisors, mandibular incisors, and first molars [8, 18, 25]. Interestingly, the higher frequency of tooth agenesis observed in the most severe phenotypes is associated with homozygous patients [7, 17]. In this case report, we found two hemizygous male subjects each with only one tooth and heterozygous females with agenesis predominantly of the lateral maxillary incisors. These dental phenotypic manifestations thus play a crucial role in the detection and diagnosis of female carriers [26].
In our family, heterozygous subjects had either no typical facial features of HED or only mild facial signs of the disorder. Agenesis of the lateral maxillary incisors was present in all heterozygous female carriers. In two cases, we observed areas of sparse body hair and convex nails. Scalp hair is typically thin and light in heterozygous subjects [18]; however, the degree of hypotrichosis is highly variable [18, 27]. In this study, hypohidrosis was not apparent, except in male subjects, but hypohidrosis is only partial in heterozygous subjects and may not be prominent [18]. Nail dysplasia was observed in one male with HED and in two females, but not in other heterozygous carriers. Some studies have suggested that the variability in the phenotype of XLHED is associated with different mutations in the EDA gene [13, 14]. The genetic variability in this condition may lead to variability in its characteristics, including different dental phenotypes [26].
The treatment for a patient with ectodermal dysplasia varies and generally depends on age, dental agenesis, degree of malformation of teeth, the growth and development of the stomatognathic system of the patient, and the patient’s motivation [28]. Prosthodontic treatment for children with ectodermal dysplasia includes a removable partial denture or complete denture, over-denture, and implants [29]. In this case report, early prosthetic treatment was given to the two boys, as recommend for cooperative children. This early restoration of facial appearance is essential for normal psychological development [28].
References
- 1.Priolo M, Lagana C. Ectodermal dysplasias: a new clinical-genetic classification. J Med Genet. 2001;38:579–585. doi: 10.1136/jmg.38.9.579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lamartine J. Towards a new classification of ectodermal dysplasias. Clin Exp Dermatol. 2003;28:351–355. doi: 10.1046/j.1365-2230.2003.01319.x. [DOI] [PubMed] [Google Scholar]
- 3.Itin PH. Rationale and background as basis for a new classification of the ectodermal dysplasias. Am J Med Genet A. 2009;149A:1973–1976. doi: 10.1002/ajmg.a.32739. [DOI] [PubMed] [Google Scholar]
- 4.Lind KL, Stecksen-Blicks C, Lejon K, Schmitt-Egenolf M. EDAR mutation in autosomal dominant hypohidrotic ectodermal dysplasia in two Swedish families. BMC Med Genet. 2006;7:80. doi: 10.1186/1471-2350-7-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hout AH, Oudesluijs GG, Venema A, Verheij JB, Mol BG, Rump P, et al. Mutation screening of the ectodysplasin-A receptor gene EDAR in hypohidrotic ectodermal dysplasia. Eur J Hum Genet. 2008;16:673–679. doi: 10.1038/sj.ejhg.5202012. [DOI] [PubMed] [Google Scholar]
- 6.Reed WB, Lopez DA, Landing B. Clinical spectrum of anhidrotic ectoderma dysplasia. Arch Dermatol. 1970;102:134–143. doi: 10.1001/archderm.1970.04000080006002. [DOI] [PubMed] [Google Scholar]
- 7.Prager TM, Finke C, Miethke RR. Dental findings in patients with ectodermal dysplasia. J Orofac Orthop. 2006;67:347–355. doi: 10.1007/s00056-006-0619-4. [DOI] [PubMed] [Google Scholar]
- 8.Lexner MO, Bardow A, Hertz JM, Nielsen LA, Kreiborg S. Anomalies of tooth formation in hypohidrotic ectodermal dysplasia. Int J Paediatr Dent. 2007;17:10–18. doi: 10.1111/j.1365-263X.2006.00801.x. [DOI] [PubMed] [Google Scholar]
- 9.Ruhin B, Martinot V, Lafforgue P, Catteau B, Manouvrier-Hanu S, Ferri J. Pure ectodermal dysplasia: retrospective study of 16 cases and literature review. Cleft Palate Craniofac J. 2001;38:504–518. doi: 10.1597/1545-1569(2001)038<0504:PEDRSO>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 10.Glavina D, Majstorović M, Lulić-Dukić O, Jurić H. Hypohidrotic ectodermal dysplasia: dental features and carrier detection. Coll Antropol. 2001;25:303–310. [PubMed] [Google Scholar]
- 11.Mikkola ML. Molecular aspects of hypohidrotic ectodermal dysplasia. Am J Med Genet A. 2009;149A:2031–2036. doi: 10.1002/ajmg.a.32855. [DOI] [PubMed] [Google Scholar]
- 12.Cluzeau C, Hadj-Rabia S, Jambou M, Mansour S, Guigue P, Masmoudi S, et al. Only four Genes (EDA1, EDAR, EDARADD, and WNT10A) account for 90% of hypohidrotic/anhidrotic ectodermal dysplasia cases. Hum Mutat. 2011;32:70–72. doi: 10.1002/humu.21384. [DOI] [PubMed] [Google Scholar]
- 13.Zhang J, Han D, Song S, Wang Y, Zhao H, Pan S, et al. Correlation between the phenotypes and genotypes of X-linked hypohidrotic ectodermal dysplasia and non-syndromic hypodontia caused by ectodysplasin-A mutations. Eur J Med Genet. 2011;54:377–382. doi: 10.1016/j.ejmg.2011.03.005. [DOI] [PubMed] [Google Scholar]
- 14.Cañueto J, Zafra-Coboa MI, Ciria S, Unamunoa P, González-Sarmiento R. A novel EDA gene mutation in a Spanish family with X-linked hypohidrotic ectodermal dysplasia. Actas Dermosifiliogr. 2011 doi: 10.1016/j.ad.2011.04.004. [DOI] [PubMed] [Google Scholar]
- 15.Blüschke G, Nüsken KD, Schneider H. Prevalence and prevention of severe complications of hypohidrotic ectodermal dysplasia in infancy. Early Human Dev. 2010;86:397–399. doi: 10.1016/j.earlhumdev.2010.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Ryan FS, Mason C, Harper JI. Ectodermal dysplasia—an unusual dental presentation. J Clin Pediatr Dent. 2005;30:55–57. [PubMed] [Google Scholar]
- 17.Visinoni AF, Lisboa-Costa T, Pagnan NAB, Chautard-Freire-Maia EA. Ectodermal dysplasias: clinical and molecular review. Am J Med Genet Part A. 2009;149A:1980–2002. doi: 10.1002/ajmg.a.32864. [DOI] [PubMed] [Google Scholar]
- 18.Cambiaghi S, Restano L, Pakkonen K, Caputo R, Kere J. Clinical findings in mosaic carriers of hypohidrotic ectodermal dysplasia. Arch Dermatol. 2000;136:217–224. doi: 10.1001/archderm.136.2.217. [DOI] [PubMed] [Google Scholar]
- 19.Munoz F, Lestringant G, Sybert V, Frydman M, Alswaini A, Frossard PM, et al. Definitive evidence for an autosomal recessive form of hypohidrotic ectodermal dysplasia clinically indistinguishable from the more common X-linked disorder. Am J Hum Genet. 1997;61:94–100. doi: 10.1086/513905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Clarke A, Phillips DIM, Brown R, Harper PS. Clinical aspects of X-linked hypohidrotic ectodermal dysplasia. Arch Dis Child. 1987;62:989–996. doi: 10.1136/adc.62.10.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freire-Maia N, Pinheiro M. Carrier detection in Christ-Siemens-Touraine syndrome (X-linked hypohidrotic ectodermal dysplasia) Am J Hum Genet. 1982;34:672–674. [PMC free article] [PubMed] [Google Scholar]
- 22.Kerr CB, Wells RS, Cooper KE. Gene effect in carriers of anhidrotic ectodermal dysplasia. J Med Genet. 1966;3:169–176. doi: 10.1136/jmg.3.3.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gunadi, Miura K, Ohta M, Sugano A, Lee MJ, Sato Y, et al. Two novel mutations in the ED1 gene in Japanese families with X-linked hypohidrotic ectodermal dysplasia. Pediatr Res. 2009; 65:453–7. [DOI] [PubMed]
- 24.Nakata M, Koshiba H, Eto K, Nance WE. A genetic study of anodontia in X-linked hypohidrotic ectodermal dysplasia. Am J Hum Genet. 1980;32:908–919. [PMC free article] [PubMed] [Google Scholar]
- 25.Nishibu A, Hashiguchi T, Yotsumoto S, Takahashi M, Nakamura K, Kanzaki T, et al. A frameshift mutation of the ED1 gene in sibling cases with X-linked hypohidrotic ectodermal dysplasia. Dermatology. 2003;207:178–181. doi: 10.1159/000071790. [DOI] [PubMed] [Google Scholar]
- 26.Clauss F, Manière MC, Obry F, Waltmann E, Hadj-Rabia S, Bodemer C, et al. Dento-craniofacial phenotypes and underlying molecular mechanisms in hypohidrotic ectodermal dysplasia (HED): a review. J Dent Res. 2008;87:1089–1099. doi: 10.1177/154405910808701205. [DOI] [PubMed] [Google Scholar]
- 27.Sybert VP. Hypohidrotic ectodermal dysplasia: argument against an autosomal recessive form clinically indistinguishable from X-linked hypohidrotic ectodermal dysplasia (Christ-Siemens-Touraine syndrome) Pediatr Dermatol. 1989;6:76–81. doi: 10.1111/j.1525-1470.1989.tb01002.x. [DOI] [PubMed] [Google Scholar]
- 28.Bhargava A, Sharma A, Popli S, Bhargava R. Prosthodontic management of a child with ectodermal dysplasia: a case report. J Indian Prosthodont Soc. 2010;10:137–140. doi: 10.1007/s13191-010-0026-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tarjan I, Gabris K, Rozsa N. Early prosthetic treatment of patient with ectodermal dysplasia: a clinical report. J Prosthet Dent. 2005;93:419–424. doi: 10.1016/j.prosdent.2005.01.012. [DOI] [PubMed] [Google Scholar]