

Abstract
The cysteine endoproteases (EP)-A and EP-B were purified from green barley (Hordeum vulgare L.) malt, and their identity was confirmed by N-terminal amino acid sequencing. EP-B cleavage sites in recombinant type-C hordein were determined by N-terminal amino acid sequencing of the cleavage products, and were used to design internally quenched, fluorogenic peptide substrates. Tetrapeptide substrates of the general formula 2-aminobenzoyl-P2-P1-P1′-P2′-tyrosine(NO2)-aspartic acid, in which cleavage occurs between P1 and P1′, showed that the cysteine EPs preferred phenylalanine, leucine, or valine at P2. Arginine was preferred to glutamine at P1, whereas proline at P2, P1, or P1′ greatly reduced substrate kinetic specificity. Enzyme cleavage of C hordein was mainly determined by the primary sequence at the cleavage site, because elongation of substrates, based on the C hordein sequence, did not make them more suitable substrates. Site-directed mutagenesis of C hordein, in which serine or proline replaced leucine, destroyed primary cleavage sites. EP-A and EP-B were both more active than papain, mostly because of their much lower Km values.
Two EPs known to play a central role in the breakdown of barley (Hordeum vulgare L.) endosperm storage proteins (hordeins) are Cys EPs designated EP-A (Koehler and Ho, 1990a) and EP-B (Koehler and Ho, 1988). They are secreted by the scutellum and aleurone layer into the starchy endosperm during germination in response to GA3 (Koehler and Ho, 1990b; Marttila et al., 1995). EP-B has an apparent molecular mass of 30 kD, is identical to MEP-1 (Phillips and Wallace, 1989), and has a possible homolog with an apparent molecular mass of 31 kD (Zhang and Jones, 1996). The substrate specificity of this EP-B has been determined from the cleavage patterns of small proteins such as hordothionin, levitide, and cholecystokinin (Poulle and Jones, 1988; Zhang and Jones, 1996). There is little information about the substrate specificity of EP-A, although both EP-A and EP-B have been shown to digest hordein (Phillips and Wallace, 1989; Koehler and Ho 1990a, 1990b). Characterization of the substrate specificity of barley Cys EPs provides an essential basis for an understanding of the activation of β-amylase by MEP-1 (= EP-B) (Guerin et al., 1992) and limit dextrinase (Sissons, 1996) during germination, in which these enzymes are released (and activated) from bound and latent forms by proteolytic cleavage.
Hordeins are unusual proteins, the amino acid composition and water insolubility of which present special problems as protease substrates. Pro and Gln comprise 40% of the total amino acids, and Pro causes particular problems for EPs, especially when it is at the protease scissile bond P1-P1′. (The substrate positions are denoted Pi, … , P2, P1, P1′, P2′, … , Pj, in correspondence with the binding subsites Si, etc., according to Berger and Schechter 1970). Hordeins, which are stored as compact protein bodies within the vacuoles of endosperm cells, comprise four major classes: B, C, D, and γ. Purification of the hordein polypeptides reveals that they are complexed in larger aggregates by intermolecular disulfide bonds between Cys residues present in B, D, and γ hordein. Circular dichroism spectroscopy and small-angle x-ray-scattering studies of purified hordein polypeptides and synthetic oligopeptides indicate that C and D hordeins are rod-shaped molecules in an extended β-turn helix structure (Halford et al., 1992; I'Anson et al., 1992). The ability to express a recombinant C hordein and to refold it to its native conformation (Tamas et al., 1994) provides the first opportunity, to our knowledge, to study the degradation of a native substrate by a barley Cys EP. We have used this system to determine the Cys EP cleavage sites in C hordein. Furthermore, we have designed internally quenched, fluorogenic peptide substrates (Meldal and Breddam, 1991) based on the C hordein cleavage sequences to determine substrate specificity and the effect of increasing substrate length.
MATERIALS AND METHODS
Air-dried (45°C) green malt was obtained from Carlsberg A/S, Copenhagen, Denmark.
Chemicals
E-64, β-mercaptoethanol, papain (2× recrystallized), N-CBZ-Phe-Arg-AMC, Cys, and buffers were purchased from Sigma.
Synthesis of Internally Quenched Fluorogenic Substrates
Fluorogenic substrates were of the general structure Abz-(Xaa)n-Tyr(NO2)-Asp-OH, where the fluorescent group is Abz, the quencher is Tyr(NO2), Xaa is any of the genetically encoded amino acids, and n = 4 to 11 (Meldal and Breddam, 1991). Peptides were synthesized on solid support (Pega1900 resin) using an Applied Biosystems 432A peptide synthesizer. Normal amino acids were introduced as the Fmoc-Xaa-OH or as the N-Fmoc-protected acid under (1-H-benzotriazol-1-yl)-1,2,3,3-tetramethyluronium hexafluorophosphate activation. The fluorogenic amino acids were incorporated using Fmoc-Tyr(NO2) and t-butyloxycarbonyl-Abz-O-3,4-dihydro-4-oxo-1,2,3-benzotriazo-3-yl prepared as previously described (Meldal and Breddam, 1991). During each cycle the Fmoc group was removed by piperidine prior to the addition of each new amino acid. The final product was cleaved from the resin by treatment with 95% trifluoroacetic acid, washed with 95% acetic acid, concentrated in a rotary evaporator, washed with diethyl ether, and freeze dried. Purity and identity were confirmed by HPLC, amino acid analysis, and MALDI-TOF MS.
Purification of EP-A and EP-B
EP-A and EP-B were isolated from green malt by the modification of published methods (Phillips and Wallace, 1989; Koehler and Ho, 1988, 1990a). The 35 to 75% (NH4)2SO4 fraction was applied to an S-200HR column. The active fractions were pooled and dialyzed overnight against 20 mm NaC2H3O2, pH 3.5, before loading onto a SP-Sepharose column, and eluted with a 0 to 1 m NaCl gradient. Active fractions were pooled, adjusted to pH 6.0 using Mes buffer, and diluted before loading onto a Q-Sepharose column. After elution with a 0 to 500 mm NaCl gradient, active fractions were pooled and stored until required at −20°C, after the addition of glycerol to a final concentration of 40%.
Isolation of Recombinant C Hordein and Degradation by EP-B
C hordein, encoded by the λ-hor-17 clone (Entwistle, 1988) was isolated from Escherichia coli, as described by Tamas et al. (1994). Purified C hordein was dissolved in 0.1 m acetic acid, aliquoted, freeze dried, and kept at −20°C until required. C hordein was dissolved in assay buffer to a concentration of 10 μg/μL, and 27 μL of C hordein was incubated with 8 μL of assay buffer and 10 μL of 0.096 μm solution of EP-B at 40°C. A time course of C hordein degradation was determined by removing 5-μL aliquots at seven different time points and adding the aliquots to 1.5 μL of sample buffer containing SDS at 80°C. These samples were run on a 16% acrylamide gel and stained with Coomassie blue.
SDS-PAGE and Blotting
SDS-PAGE was performed under reducing conditions in a mini-gel apparatus (Protean II, Bio-Rad) using 16% acrylamide, high-Tris gels according to the method of Fling and Gregerson (1986). Protein-containing bands were visualized by staining with 0.03% Coomassie brilliant blue R250 dissolved in 10% TCA and 40% methanol, or by silver staining. For N-terminal amino acid sequencing, proteins were blotted onto a PVDF membrane (Immobilon-P, Millipore) using a semidry electroblotter (Aricos, Ølstykke, Denmark) and the Tris/6-aminohexanoic acid buffer system recommended by the manufacturer. Bands were visualized by staining for a few seconds in Coomassie brilliant blue (see above), and were then cut out and subjected to N-terminal amino acid sequencing with a model 470A sequenator coupled to a model 120A phenylthiohydantoin analyzer (both Applied Biosystems). Total proteolysis of C hordein was carried out at 40°C for 60 min, and the products were separated by HPLC before analysis by N-terminal amino acid sequencing or MALDI-TOF MS.
Determination of Enzyme Activity and Kinetic Constants
Enzymatic hydrolysis of the peptide substrates was followed by observation of the change in substrate fluorescence upon addition of enzyme with a luminescence spectrofluorimeter (emission at 420 nm, 10-nm slit; excitation at 320 nm, 10-nm slit; model LS50, Perkin Elmer) at 25°C. The substrates were dissolved in dimethylformamide at concentrations of 50 to 200 μm, and 10 μL of the substrate was added to a cuvette containing 2.4 mL of assay buffer (50 mm NaC2H3O2, pH 4.5, 2 mm Cys, and 2 mm β-mercaptoethanol). The initial fluorescence (Io) was measured and the enzyme solution (final concentration, Eo) was added. The concentration of active enzyme was determined by titration with E-64 according to the method of Barrett and Kirschke (1981). At each of the three substrate concentrations (so), the initial velocity for substrate cleavage (vo) was determined from the initial slope of the curve (emission versus time). The hydrolysis was allowed to proceed overnight and final fluorescence for total cleavage (I∞) was measured. Values were confirmed by measuring I∞ 24 h after the addition of subtilisin and/or trypsin to the same reaction mixture. The kcat/Km values were determined using the relation:
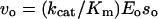 |
which is valid at low substrate concentrations (so ≪ Km) for systems that obey Michaelis-Menten kinetics.
The cleavage site for each substrate was determined by amino acid sequencing of the hydrolysis products obtained after the incubation of 1 μL of a concentrated solution of substrate with 1 μL of diluted enzyme in 45 μL of assay buffer for 3 h at 25°C.
Papain was activated in 100 mm sodium phosphate, pH 7.0, containing 10 mm Cys, 1 mm EDTA, and 0.01% Brij 35 for 5 min at 4°C. Assays were carried out in 50 mm sodium phosphate, pH 6.5, 2 mm Cys, 1 mm EDTA, and 0.08% Brij 35, as described by Ménard et al. (1990). N-CBZ-Phe-Arg-AMC was dissolved in dimethylformamide to make a stock at 7.7 mm, and assays were run at 25°C in a final volume of 2 mL, using an excitation at 360 nm and an emission at 460 nm. kcat/Km values were calculated as above using 10-nm slits and substrate concentrations of about 200 nm, whereas Km values were calculated from Hanes plots of s versus s/v (where s = substrate concentration and v = initial rate) using 5-nm slits and substrate concentrations between 1 and 150 μm. The spectrofluorimeter was calibrated with known concentrations of AMC.
Nucleotide Sequencing and Site-Directed Mutagenesis
The nucleotide sequences of the C hordein coding sequence obtained from Tamas et al. (1994) and the EP-A cDNA clone were determined by dideoxy chain termination using the TaQ Dye Deoxy Terminator Cycle Sequencing kit and a sequencer (model 373A, Applied Biosystems). Site-directed mutagenesis of the primary cleavage site near the N terminus was carried out by splice-overlap PCR using the appropriate forward primer.
RESULTS
Purification of EP-A and EP-B
A protein with an apparent molecular mass of 30 kD was isolated in pure form from green barley malt after extraction in 50 mm NaC2H3O2 buffer containing 2 mm Cys and 2 mm β-mercaptoethanol, and subsequent column chromatography. Its proteolytic activity during purification was followed by fluorometric assay with the internally quenched peptide substrate Abz-AFRFAA-Tyr(NO2)PPD. This activity was inhibited by the Cys endopeptidase inhibitors E-64 and leupeptin. Its identity as EP-B was established on the basis of its N-terminal amino acid sequence, which was identical to that reported by Koehler and Ho (1990a). A second peak of protease activity, which eluted after EP-B on SP Sepharose, was purified by elution from Q Sepharose. It had an apparent molecular mass of 37 kD, and was identified as EP-A from the agreement of its N-terminal amino acid sequence with the published sequence (Koehler and Ho, 1988).
Determination of Primary Cleavage Sites
Pure C hordein, expressed in E. coli and folded in vitro to its native conformation (Tamas et al., 1994), was used as a substrate to determine EP-B cleavage sites. Recombinant C hordein was incubated for different times with purified EP-B. Several discrete bands could be seen within the first 0.5 min of incubation, and the C hordein was fully degraded within 40 min (Fig. 1). The experiment was repeated on a larger scale, and the separated cleavage products were blotted onto a PVDF membrane for N-terminal amino acid sequencing to determine the sites within the C hordein sequence where EP-B cleaved most rapidly. The five major polypeptides had one of two N-terminal sequences: QPYPQNPYLPQKPFPVQ or QPFHTPQQYFPYLP. Comparison of these sequences with that of C hordein (Fig. 2) revealed that initial EP-B cleavage sites occurred after Q-20 and Q-37. The size distribution of the cleavage products predicts at least two further primary cleavage sites closer to the C terminus, to account for the smaller polypeptides with N termini identical to the larger ones.
Figure 1.

Coomassie blue-stained gel of the time course of recombinant C hordein digestion by purified EP-B. Numbers above each lane are minutes of incubation at 40°C. A similar gel was used to electroblot proteolytic bands onto PVDF for N-terminal amino acid sequencing to determine the original cleavage site in C hordein. C, Control (no EP-B); M, molecular mass markers.
Figure 2.

Amino acid sequence of the recombinant C hordein deduced from the nucleotide sequence, with the major cleavage sites shown in bold and the scissile bond indicated by an asterisk. The sequence differs from that published by Entwistle (1988), which contains a tandem repeat of the sequence PQQASPLQPQ.
Sequencing of peptides after exhaustive C hordein cleavage by EP-B followed by HPLC revealed three relatively abundant peptides with the same C-terminal sequence: … QPLPQPQQPFR-199, indicating cleavage after R-199. Most of the initial C hordein cleavage products can be explained as being the result of attack by EP-B at one or more of these specific cleavage sites (Fig. 2). In view of the nonrandom proteolytic degradation of C hordein by EP-B, the initial cleavage sites may be defined as primary cleavage sites.
Determination of Secondary Cleavage Sites
After 40 min, EP-B cleaved C hordein into fragments too small to be resolved by SDS-PAGE, and these products were separated by HPLC for subsequent identification by MS and/or amino acid sequencing. This allowed us to identify 44 fragments that ranged from 2 to 26 residues, and, thus, the position of a further 35 cleavage sites in the C hordein polypeptide (Fig. 3). The C hordein sequence has been shown to emphasize the octameric repeat PQQPXaaPQQ, in which Xaa is usually a hydrophobic residue such as Phe, Val, Leu, or Tyr. These secondary cleavage sites are listed in Table II and the most common one, PQ↓QP, is a reflection of the high Pro and Gln content of C hordein.
Figure 3.

C hordein sequence organized to show its intrinsic octameric repeats (PQQPXaaPQQ), where Xaa = F, L, Y, I, S, etc., together with primary (⇓) and secondary (↓) cleavage sites deduced from partial and total digestion with EP-B.
Table II.
Analysis of EP-B cleavage sites in C hordein
| Primary Cleavage Sites | Secondary Cleavage Sitesa | P2 | P1 | P1′ | P2′ b | Not Cleaved |
|---|---|---|---|---|---|---|
| ↓ | ↓ | |||||
| FR QQ | PQ QP (14) | P | Q | Q | P | LQ PQ (3) |
| FQ QP | PQ QS (2) | Q | R | S | S | LQ PH |
| VQ QP | PQ KP | F | L | F | L | LN PS |
| LQ QP | PQ NP | L | E | I | A | LF PQ |
| LQ SP | PQ QA | E | G | A | E | FL PQ |
| LI IP | PQ GS | T | H | E | F | VQ PQ |
| FH TP | V | I | G | I | LP QK | |
| QY FP | W | S | K | Q | FP VQ | |
| YQ IP | Y | Y | M | T | FP WQ | |
| PL QP | N | V | FP QP | |||
| PH QP | T | Y | FP QQ | |||
| EL FP | Y | |||||
| QQ YF | ||||||
| QQ AS | ||||||
| QQ SY | ||||||
| QQ II | ||||||
| MR QL | ||||||
| QG SE | ||||||
| PE EL | ||||||
| TQ QT | ||||||
| WS MV |
Numbers in parentheses indicates frequency of occurrence.
Listed in order of decreasing frequency.
Cleavage of Mutated C Hordeins by EP-B
To investigate whether the major cleavage sites were determined by primary sequence or secondary structure, site-directed mutagenesis was used to change the sequences around two C hordein cleavage sites (L-11 → S and L-19 → P), together with the introduction of a novel putative cleavage site by the substitution of Q-15 → L (Fig. 4). The expressed mutated C hordein DNA was purified and incubated with EP-B, and the primary cleavage products were separated by SDS-PAGE and sequenced. Cleavage occurred between Q-16 and S-17, indicating that mutation of the two primary cleavage sites had greatly reduced their suitability as EP-B substrates, whereas the introduction of L-15 created a new primary cleavage site. In a second mutant C hordein (P-22 → S), there was no effect on the cleavage pattern with EP-B (Fig. 4).
Figure 4.

N-terminal amino acid sequence of mutant C hordeins deduced from their nucleotide sequences. Cleavage motifs are underlined, and the scissile bonds are indicated by ↓. Residues in bold indicate mutated amino acids and show that substitution of L by S or P at P2 destroys primary cleavage sites, whereas the substitution of Q by L can create a new primary cleavage site.
Determination of EP Specificity by Using Synthetic Substrates
Several synthetic, internally quenched fluorogenic substrates were synthesized, the sequence of which was based on the determined major cleavage sites of C hordeins, i.e. LQ↓SP, LQ↓QP, VQ↓QP, FQ↓QP, and FR↓QQ, with the general sequence Abz-P2-P1-P1′-P2′-Tyr(NO2)-D. In addition to these, longer substrates based on the C hordein sequence, with residues extending to P6 and P5′, were also synthesized to investigate the importance of these residues in determining substrate kinetics. Each peptide substrate was assayed at three different concentrations under pseudo-first-order conditions (≪Km) with EP-A, EP-B, and papain, which had been titrated with E-64 to determine the concentration of the active enzyme. The second-order kinetic constant kcat/Km was calculated for each substrate (Table I). It can be seen that substrates with Phe at P2 had the highest values for kcat/Km, followed by Val and Leu. Extension of the substrate by the addition of residues from P3 to P6 and P3′ to P5′ usually decreased the kcat/Km. The cleavage of the same substrates by papain was measured and in several cases, in contrast to EP-A and EP-B, increasing substrate length gave increasing kcat/Km values (Table I).
Table I.
Second-order kinetic constants kcat/Km for plant cysteine endoproteases
| Substrate | EP-A | EP-B | Papain |
|---|---|---|---|
| mm−1 s−1 | |||
| XFQ ↓QPZDa | 609 ± 29 | 2650 ± 132 | 166 ± 6 |
| XQQIIFQ ↓QPQQZD | 149 ± 3 | 45 ± 8 | 98 ± 5 |
| XQIIFQ ↓QPQQSZPPD | 63 ± 2 | 150 ± 10 | 333 ± 9 |
| XFR ↓QQZD | 4400 ± 290 | 8110 ± 573 | 319 ± 9 |
| XQPFR ↓QQZD | 230 ± 4 | 365 ± 16 | 219 ± 13 |
| XQQPFR ↓QQAELZPPD | 180 ± 11 | 160 ± 5 | 312 ± 11 |
| XVQ ↓QPZD | 108 ± 3 | 495 ± 25 | 23 ± 1 |
| XFPVQ ↓QPFZD | 78 ± 4 | 680 ± 21 | 317 ± 8 |
| XLQ ↓QPZD | 960 ± 61 | 6500 ± 700 | 53 ± 2 |
| XQSYLQ ↓QPYPQZPPD | 300 ± 8 | 1340 ± 17 | 125 ± 5 |
| XLQ ↓SPZD | 930 ± 19 | 7130 ± 460 | 182 ± 10 |
| XSQELQ ↓SPQQSZPPD | 84 ± 1 | 540 ± 36 | 208 ± 3 |
| XLQ PQZD | ndb | nd | 0.32 ± 0.007 |
| XFP QQZD | nd | nd | 0.15 ± 0.004 |
| XQPQ QPZD | nd | 2.8 | 1.10 ± 0.11 |
| XSQ QPZD | nd | 2.5 | 2.88 ± 0.08 |
X, 2-Aminobenzoyl; Z, 3-nitrotyrosine.
nd, Not detected; rate too low.
DISCUSSION
Whereas Cys EPs such as EP-A and EP-B are known to contribute to storage-protein degradation in the germinating barley grain, the cleavage site specificity on native substrates has not, to our knowledge, previously been studied. A C hordein polypeptide, expressed as a recombinant protein, was selected as a substrate for EP-B, because the fidelity of its refolding to the native conformation has been demonstrated (Tamas et al., 1994). C hordein proteolysis was initiated by cleavage at a limited number of sites, followed by cleavage to smaller peptides consisting of 2 to 15 residues. The initial cleavage sites characteristically had hydrophobic residues (F, L, V, and I) at P2. To study the kinetics of cleavage at these sites, we synthesized synthetic, internally quenched, fluorogenic substrates based on their sequence. EP-A and EP-B belong to the papain-type group of Cys EPs (each have 51% sequence identity to papain), and the same substrates were used with papain for comparison. The most important residue for papain-type Cys EP specificity is that at P2, which is usually large and hydrophobic (Berger and Schechter, 1970).
For substrates of the type Abz-XaaQQP-Tyr(NO2)D (in which Xaa = F, L, V, P, or S), the order of decreasing activity for EP-B and EP-A was L > F > V ≫ P, S, whereas for papain, the order was F > L > V ≫ S > P (Table I). Substrates with P or S at P2 were particularly poor substrates, which was confirmed by site-directed mutagenesis of the C hordein polypeptide, where cleavage at LQ↓SP and LQ↓QP was greatly reduced after mutation to SQ↓SP and PQ↓QP, respectively (Fig. 4). This indicates that cleavage of C hordein by EP-B is mainly determined by the primary sequence at the cleavage site. These results are in broad agreement with those reported for EP-B cleavage of hordothionins, with cleavage at sites containing L or V at P2 (Jones and Poulle, 1990). Cleavage of other polypeptides by a presumptive isoform of EP-B yielded similar results with W, F, L, I, V, Y, and A at P2 (Zhang and Jones, 1996). Most of the B1 hordein-cleavage sites predicted by these authors have Pro at P1, which would make them extremely poor EP-B substrates.
The effect of a possible secondary structure on kcat/Km was investigated by increasing the length of the substrate based on the C hordein sequence around the primary cleavage sites. In most cases, kcat/Km values decreased with increased substrate length for EP-A and EP-B (Table I). This was unexpected, since the papain substrate-binding cleft is reported to consist of up to seven subsites (S4, S3, S2, S1, S1′, S2′, and S3′) (Berger and Schechter, 1970), and one might expect EP-A and EP-B to have the same number of subsites as papain. Similar results with substrate elongation have been reported for subtilisin and were attributed to adverse effects of secondary structure (Meldal and Breddam, 1991). This does not appear to be the case here, because longer substrates were often better substrates for papain, with kcat/Km values increasing from 2 to 14 times (LQ↓QP versus QSYLQ↓QPYPQ, VQ↓QP versus FPVQ↓QPF, and FQ↓QP versus QIIFQ↓QPQQS), in contrast to EP-A and EP-B. It should be noted that in two of these series, Pro was placed at P3, which may have been undesirable for EP-A and EP-B.
An examination of the secondary EP-B cleavage sites in C hordein after total digestion revealed that PQ↓QP was cleaved 14 times (Table II), although this sequence is not a good substrate (Table I). The residues found at the four different positions (P2–P2′) are listed in decreasing order of frequency. Between 9 to 12 amino acids were found at each position, with the total number probably being limited by the amino acid composition and sequence of C hordein (Fig. 2). It is striking, however, that Pro appears at P2 and P2′, but not at P1 or P1′ (Table II). A list of potential EP-B cleavage sites in C hordein (i.e. those with F, V, or L at P2) that are not cleaved shows that they all contain Pro at P1 or P1′ (Table II). The substrates Abz-LQPQ-Tyr(NO2)D and Abz-FPQQ-Tyr(NO2)D were made to measure kcat/Km values of substrates with Pro at these positions. It was not possible to detect cleavage with EP-A and EP-B, but high concentrations of papain yielded values that were 500 times less for Pro at P1′ (LQ↓PQ versus LQ↓SP) and 2000 times less for Pro at P1 (FP↓QQ versus FR↓QQ). Comparable ratios probably exist for EP-A and EP-B.
It is clear from Table I that kcat/Km values are higher for EP-A and EP-B than for papain. We therefore determined kcat and Km values for all three enzymes using the substrate CBZ-Phe-Arg-AMC (Table III). Values obtained for papain of 79 μm (Km) and 45 s−1 (kcat) are close to published values (Gauthier et al., 1993). The high kcat/Km values for EP-A and EP-B were primarily due to their much lower Km values, indicating a higher affinity of these enzymes for the substrate. Better papain substrates are known, such as Cap-Leu-Arg-AMC (Km = 6 μm, kcat = 55 s−1, and kcat/Km = 9166 mm−1 s−1) (Alves et al., 1996), in which the presence of ε-aminocaproic acid (Cap) at P3 may cause a drastic reduction in Km. However, Abz-FRQQ-Tyr(NO2)D and CBZ-FR-AMC appear to be equally good substrates for all three enzymes, even though the residue at P1′ and particularly P2′ can enhance substrate suitability considerably (García-Echeverría and Rich, 1992a, 1992b; Lalmanach et al., 1995).
Table III.
Kinetic constants for plant cysteine endoproteases with CBZ-Phe-Arg-AMC
| Enzyme | Km | kcat | kcat/Km |
|---|---|---|---|
| μm | s−1 | mm−1 s−1 | |
| EP-B | 3.6 ± 0.23 | 24.4 ± 1.6 | 6780 ± 233 |
| EP-A | 1.0 ± 0.13 | 4.1 ± 0.2 | 2750 ± 83 |
| Papain | 79 ± 11 | 45 ± 2.6 | 577 ± 9 |
This work has shown that, although EP-A and EP-B are similar proteases (66% amino acid sequence identity), they do exhibit differences in substrate specificity (e.g. LQ↓QP). This may be because EP-B can tolerate Pro at P2′, whereas EP-A cannot because there is a relatively smaller difference between EP-A and EP-B with FR↓QQ. A detailed study of substrate requirements at P1 and P2 and the importance of the primary cleavage sites for subsequent attack at secondary cleavage sites in C hordein will be the subject of future investigations.
ACKNOWLEDGMENTS
We thank Professor Peter Shewry for the gift of the C hordein expression vector, Dr. Finn Lok for providing the EP-A cDNA clone, and Suksawad Vongvisuttikun for C hordein DNA sequencing. We are grateful to Dr. Phaedria St. Hilaire, Bodil Corneliussen, and Lone Sørensen for assistance with amino acid sequencing and MALDI-TOF MS, and to Hanne Christiansen and Kirsten Lilja for help and advice with peptide synthesis and HPLC. Ann-Sofi Steinholz and Nina Rasmussen are acknowledged for making Figure 1.
Abbreviations:
- Abz
2-aminobenzoyl
- AMC
7-amido-4-methylcoumarin
- CBZ
benzyloxycarbonyl
- E-64
trans-epoxysuccinyl-l-leucylamido-(4-guanido)butane
- EP
endoprotease
- Fmoc
fluoren-9-ylmethyloxycarbonyl
- MALDI-TOF
matrix-assisted laser desorption ionization-time-of-flight
- Tyr(NO2)
3-nitrotyrosine
- Xaa
unspecified amino acid
Footnotes
This work was supported by the Danish Academy of Technical Sciences (A.D. received a studentship). This is Adapting Barley for Industrial Needs publication no. 159.
The accession number for EP-A is Z97023.
LITERATURE CITED
- Alves LC, Almeida PC, Franzoni L, Juliano L, Juliano MA. Synthesis of Nα-protected aminoacyl 7-amino-4-methyl-coumarin amide by phosphorus oxychloride and preparation of specific fluorogenic substrates for papain. Peptide Res. 1996;9:92–96. [PubMed] [Google Scholar]
- Barrett AJ, Kirschke H. Cathepsin B, cathepsin H, and cathepsin L. Methods Enzymol C. 1981;80:535–561. doi: 10.1016/s0076-6879(81)80043-2. [DOI] [PubMed] [Google Scholar]
- Berger A, Schechter I. Mapping the active site of papain with the aid of peptide substrates and inhibitors. Philos Trans R Soc Lond B. 1970;257:249–264. doi: 10.1098/rstb.1970.0024. [DOI] [PubMed] [Google Scholar]
- Entwistle J. Primary structure of a C-hordein gene from barley. Carlsberg Res Commun. 1988;53:247–258. doi: 10.1007/BF02907181. [DOI] [PubMed] [Google Scholar]
- Fling SP, Gregerson DS. Peptide and protein molecular weight determination by electrophoresis using a high-molarity Tris buffer system without urea. Anal Biochem. 1986;155:83–88. doi: 10.1016/0003-2697(86)90228-9. [DOI] [PubMed] [Google Scholar]
- García-Echeverría C, Rich DH. New intramolecularly quenched fluorogenic peptide substrates for the study of the kinetic specificity of papain. FEBS Lett. 1992a;297:100–102. doi: 10.1016/0014-5793(92)80336-f. [DOI] [PubMed] [Google Scholar]
- García-Echeverría C, Rich DH. Effect of P2′ substituents on kinetic constants for hydrolysis by cysteine proteinases. Biochem Biophys Res Commun. 1992b;187:615–619. doi: 10.1016/0006-291x(92)91239-m. [DOI] [PubMed] [Google Scholar]
- Gauthier F, Moreau T, Lalmanach G, Brillard-Bourdet M, Ferrer-Di Martino M, Juliano L. A new, sensitive fluorogenic substrate for papain based on the sequence of the cystatin inhibitory site. Arch Biochem Biophys. 1993;306:304–308. doi: 10.1006/abbi.1993.1516. [DOI] [PubMed] [Google Scholar]
- Guerin JR, Lance RCM, Wallace W. Release and activation of barley beta-amylase by malt endopeptidases. J Cereal Sci. 1992;15:5–14. [Google Scholar]
- Halford NG, Tatham AS, Sui E, Daroda L, Dreyer T, Shewry PR. Identification of a novel beta-turn-rich repeat motif in the D hordeins of barley. Biochim Biophys Acta. 1992;1122:118–122. doi: 10.1016/0167-4838(92)90313-3. [DOI] [PubMed] [Google Scholar]
- I'Anson KJ, Morris VJ, Shewry PR, Tatham AS. Small-angle x-ray-scattering studies of the C hordeins of barley (Hordeum vulgare) Biochem J. 1992;287:183–185. doi: 10.1042/bj2870183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones BL, Poulle M. A proteinase from germinated barley. II. Hydrolytic specificity of a 30 kilodalton cysteine proteinase from green malt. Plant Physiol. 1990;94:1062–1070. doi: 10.1104/pp.94.3.1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler SM, Ho T-HD. Purification and characterization of gibberellic acid-induced cysteine endoproteases in barley aleurone layers. Plant Physiol. 1988;87:95–103. doi: 10.1104/pp.87.1.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler SM, Ho T-HD. A major gibberellic acid-induced barley aleurone cysteine proteinase which digests hordein. Plant Physiol. 1990a;94:251–258. doi: 10.1104/pp.94.1.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehler SM, Ho T-HD. Hormonal regulation, processing, and secretion of cysteine proteinases in barley aleurone layers. Plant Cell. 1990b;2:769–783. doi: 10.1105/tpc.2.8.769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lalmanach G, Serveau C, Brillard-Bourdet M, Chagas JR, Mayer R, Juliana L, Gauthier F. Conserved cystatin segments as models for designing specific substrates and inhibitors of cysteine proteinases. J Protein Chem. 1995;14:645–653. doi: 10.1007/BF01886903. [DOI] [PubMed] [Google Scholar]
- Marttila S, Jones BL, Mikkonen A. Differential localization of two acid proteinases in germinating barley (Hordeum vulgare) seed. Physiol Plant. 1995;93:317–327. [Google Scholar]
- Meldal M, Breddam K. Anthranilamide and nitrotyrosine as a donor-acceptor pair in internally quenched fluorescent substrates for endopeptidases: multicolumn peptide synthesis of enzyme substrates for subtilisin Carlsberg and pepsin. Anal Biochem. 1991;195:141–147. doi: 10.1016/0003-2697(91)90309-h. [DOI] [PubMed] [Google Scholar]
- Ménard R, Khouri HE, Plouffe C, Dupras R, Ripoll D, Vernet T, Tessier DC, Lalberte F, Thomas DY, Storer AC. A protein engineering study of the role of aspartate 158 in the catalytic mechanism of papain. Biochemistry. 1990;29:6706–6713. doi: 10.1021/bi00480a021. [DOI] [PubMed] [Google Scholar]
- Phillips HA, Wallace W. A cysteine endopeptidase from barley malt which degrades hordein. Phytochemistry. 1989;28:3285–3290. [Google Scholar]
- Poulle M, Jones BL. A proteinase from germinated barley. I. Purification and some physical properties of a 30-kD cysteine endoproteinase from green malt. Plant Physiol. 1988;88:1454–1460. doi: 10.1104/pp.88.4.1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sissons MJ. Studies on the activation and release of bound limit dextrinase in malted barley. J Am Soc Brew Chem. 1996;54:19–25. [Google Scholar]
- Tamas L, Greenfield J, Halford NG, Tatham AS, Shewry PR. Protein Exp Purif. 1994;5:357–363. doi: 10.1006/prep.1994.1052. [DOI] [PubMed] [Google Scholar]
- Zhang N, Jones BL. Purification and partial characterization of a 31-kDa cysteine endopeptidase from germinated barley. Planta. 1996;199:565–572. doi: 10.1007/BF00195188. [DOI] [PubMed] [Google Scholar]