

Background: TF and TFPI, two primary components of the coagulation cascade, play important roles in nonhomeostatic and pathological events.
Results: PAK1 stimulates and represses the expression of TF and TFPI, respectively, and promotes TF procoagulant activity.
Conclusion: PAK1 is identified as an important player in the coagulation processes.
Significance: Targeting PAK1 might lead to a novel therapeutic strategy in the control of coagulation.
Keywords: Chromatin Immunoprecipitation (ChIP), Coagulation Factors, Gene Regulation, Tissue Factor, Transcription Factors, Cell Signaling, PAK1, Tissue Factor Pathway Inhibitor, Transcription
Abstract
Tissue factor (TF) is a cell-surface glycoprotein responsible for initiating the coagulation cascade. Besides its role in homeostasis, studies have shown the implication of TF in embryonic development, cancer-related events, and inflammation via coagulation-dependent and -independent (signaling) mechanisms. Tissue factor pathway inhibitor (TFPI) plays an important role in regulating TF-initiated blood coagulation. Therefore, transcriptional regulation of TF expression and its physiological inhibitor TFPI would allow us to understand the critical step that controls many different processes. From a gene profiling study aimed at identifying differentially regulated genes between wild-type (WT) and p21-activated kinase 1-null (PAK1-KO) mouse embryonic fibroblasts (MEFs), we found TF and TFPI are differentially expressed in the PAK1-KO MEFs in comparison with wild-type MEFs. Based on these findings, we further investigated in this study the transcriptional regulation of TF and TFPI by PAK1, a serine/threonine kinase. We found that the PAK1·c-Jun complex stimulates the transcription of TF and consequently its procoagulant activity. Moreover, PAK1 negatively regulates the expression of TFPI and additionally contributes to increased TF activity. For the first time, this study implicates PAK1 in coagulation processes, through its dual transcriptional regulation of TF and its inhibitor.
Introduction
Tissue factor (TF)2 (also known as F3, coagulation factor III, or thromboplastin), a 47-kDa transmembrane glycoprotein and the primary initiator of the coagulation cascade (1), plays a critical role in homeostasis and thrombosis (2). It functions as a high affinity receptor for the serine proteases factors VII and VIIa. The resulting TF FVIIa complex activates both factors IX and X (FIX and FX) leading to thrombin generation and fibrin formation (1). TF-induced coagulation is regulated by a specific inhibitor termed tissue factor pathway inhibitor (TFPI). The mechanism of inhibition by TFPI involves its binding first to factor Xa (FXa) and then to the FVIIa TF complex (1).
In the last few years, it has become evident that TF is involved in various other (patho-) physiological processes apart from homeostasis, such as embryonic development, transmission of signals, promotion of cell migration, adhesion, tumor initiation, growth, and inflammation (3). Concomitantly, TFPI, the physiological inhibitor of TF, has also been widely implicated in the regulation of these nonhomeostatic functions of TF (4). TF is essential for the normal embryonic development, and its absence could lead to a defective vessel development and embryonic death in mice (5). The expression of TF is deregulated in many cancers, and this up-regulation is often linked with aggressive malignancies (6) and increased tumor growth (7). Moreover, expression of TF amplifies the inflammatory reaction in patients with sepsis (8, 9).
TF contributes to pathologies by activation of the coagulation cascade (proteolysis-dependent signaling) as well as by coagulation-independent (proteolysis-independent) signaling events via the cytoplasmic domain of TF (3, 10). Thus, TF might not only function as the initiator of coagulation but also as a transmembrane signaling receptor that regulates angiogenesis, tumor growth, metastasis, and inflammation. Therefore, understanding the transcriptional regulation of TF expression and its inhibitor, TFPI, would appear to be a critical step in the control of many different processes.
The PAKs are serine/threonine kinases that were originally identified as binding partners and downstream effectors of Cdc42 and Rac1 in actin reorganization and cell migration (11). PAK1, the best characterized member of the PAK family, is activated by the p21ras-related proteins Cdc42 and Rac1 (11) and also by a wide variety of extracellular signals (12) that promote PAK1's auto-phosphorylation and stimulation of its kinase activity (13). PAK1 kinase activity has been implicated in a wide variety of cellular processes, including cell motility, survival and proliferation, morphogenesis, cytoskeleton remodeling, and transcription (14). In addition to its well characterized kinase activity, the PAK1 pathway also affects nuclear events in a profound manner (15–17) and also regulates the transcription of target gene chromatin (18–20).
During a previous gene profiling study that includes wild-type (WT) and knock-out PAK1 (PAK1-KO) mouse embryonic fibroblasts (MEFs), we identified TF and TFPI to be down- and up-regulated, respectively, in the PAK1-KO MEFs in comparison with its wild-type controls,3 suggesting that PAK1 might regulate the expression of TF and TFPI. Given the involvement of TF and TFPI in several pathological and physiological events, this study was undertaken to investigate the genomic regulation of TF and TFPI by PAK1 signaling. To date, nothing is known about the regulation of TF expression by PAK1 in cancer cells. We discovered, for the first time, that PAK1 signaling stimulates and inhibits the transcription of TF and TFPI, respectively, and modulates the coagulation process.
EXPERIMENTAL PROCEDURES
Cell Culture, Antibodies, and Reagents
HeLa, HEK 293T, and MDA-MB-231 cells were obtained from the American Type Culture collection (ATCC, and cultured in Dulbecco's modified Eagle's medium/F-12 supplemented with 10% fetal bovine serum and 1× antibiotic/antimycotic solution in humidified 5% CO2 at 37 °C. PAK1 WT and KO MEFs have been described earlier (21). Sources of antibodies were as follows: mouse anti-phospho-PAK1 (Thr-212) (Sigma); rabbit anti-PAK1 (Bethyl Laboratories, Montgomery, TX); mouse anti-tissue factor (10H10) (Novus Biologicals, Littleton, CO); mouse anti-vinculin (Sigma); rabbit anti-c-Jun (Santa Cruz Biotechnology); mouse anti-Myc Tag (9B11) (Cell Signaling Technology, Danvers, MA), and mouse anti-FLAG M2 (Sigma). Normal mouse IgG and rabbit IgG were from Sigma. All primary antibodies were used according to the manufacturer's instructions. Horseradish peroxidase-conjugated secondary antibodies were from GE Healthcare, and enhanced chemiluminescence (ECL) reagents were from Amersham Biosciences. The blocking antibody for human tissue factor TF8-5G9 was a generous gift of Dr. James H. Morrissey (University of Illinois).
Expression Vectors, Recombinant Proteins, siRNAs, and Transfections
Expression vectors encoding PAK1-myc, PAK1 T423E, and PAK1 K229R were generously provided by Jonathan Chernoff (Fox Chase Cancer Center, Philadelphia, PA) and have been described previously (22), pCDNA3.1-PAK1 and GST-PAK1 have been described previously (23). pcDNA-c-Jun-FLAG (c-Jun-FLAG) and GST-c-Jun have been used previously (24). The dominant-negative mutant of c-Jun was a generous gift from Dr. Powell H. Brown (NCI, National Institutes of Health, Rockville, MD) and has already been described (25). All GST recombinant proteins were expressed in Escherichia coli strain BL21 (DE3) (Stratagene, La Jolla, CA) and subsequently purified using the glutathione-Sepharose 4B batch method (GE Healthcare). In vitro translation was performed using the TnT® QuickCoupled transcription/translation systems (Promega, Madison, WI). Plasmid transfections were carried out using FuGENE HD transfection reagent (Roche Applied Science) according to the manufacturer's instructions. When needed, after 24 h of transfection, cells were serum-starved for another 24 h before serum stimulation for the indicated times. Specific siRNAs targeting human PAK1 or control siRNAs were obtained from Cell Signaling Technology (Danvers, MA). The transfection of siRNA was performed twice at 24-h intervals with OligofectamineTM reagent (Invitrogen) according to the manufacturer's protocol. 24 h after the second round of transfection, cells were serum-starved for 24 h before serum stimulation for the indicated times.
Microarray Gene Expression Assays and Analysis
Microarray gene expression assays and data analysis have been described previously (26). The datasets of differentially expressed genes between PAK1 WT and KO MEFs were analyzed using Ingenuity Pathway Analysis to discover relationships between the genes as well as their association with various processes and disorders.4 The Fisher's exact test was used to identify significant functions and pathways represented within the respective datasets.
Quantitative Real Time PCR (qPCR)
Total RNA was isolated from cultured cells using TRIzol reagent (Invitrogen) according to the manufacturer's protocol, and 2 μg of extracted RNA was converted to cDNA using the SuperScriptTM III First-strand Synthesis System for RT-PCR (Invitrogen). The resultant cDNA was subjected to qPCR by using the iQTM SYBR® Green Supermix (Bio-Rad) on an iCycler iQTM real time PCR detection system (Bio-Rad). The values for specific genes were normalized to human actin or mouse 18 S housekeeping controls. Mean values are displayed ± S.D. The primers used were as follows: hACTIN_F, 5′-TCC CTG GAG AAG AGC TAC GA-3′, and hACTIN_R, 5′-GTA CTT GCG CTC AGG AGG AG-3′; hTF_F, 5′-GCC CGG TAG AGT GTA TGG GCC AG-3′, and hTF_R, 5′-GCT CTG CCC CAC TCC TGC CTT-3′; hTFPI_F, 5′-AGT ATG GTG GAT GCC TGG GCA ATA-3′, and hTFPI_R, 5′-ACC TGG AAA CCA TTC GGA CCA TCT-3′; m18 S_F, 5′-CCG GAG CTA GGA ATA ATG GA-3′, and m18 S_R, 5′-CCC TCT TAA TCA TGG CCT CA-3′; mTF_F, 5′-AGA ACA CCC CGT CGC GCT TG-3′, and mTF_R, GCT CTC CGC AAC AGT GCC GT-3′; mTFPI_F, 5′-TGT TGC TTA GCC TTG TTC CCG AGT-3′, and mTFPI_R, TGC TTT GCA TGG ACC ATC ATC TGC-3′. All qPCR primers were synthesized in Sigma.
Western Blot and Immunoprecipitation
Protein extracts were prepared by lysing the cells in RIPA buffer containing 50 mm Tris-HCl, pH 7.4, 1% Nonidet P-40, 0.25% sodium deoxycholate, 150 mm NaCl, 1 mm EDTA, 1× protease inhibitor mixture (Roche Applied Science), and 1× phosphatase inhibitor mixture I and II (Sigma), and protein concentrations were determined using Bio-Rad DC protein assay reagents (Bio-Rad). Cell extracts were then resolved by SDS-PAGE, transferred to nitrocellulose membranes, and incubated with the indicated antibodies. Detections were performed using the ECL reagents. For immunoprecipitation analysis, a total of 1 mg of protein material was incubated with 1 μg of primary antibody overnight at 4 °C on a rocket platform, followed by incubation with total 50 μl of protein A/G PLUS-agarose (Santa Cruz Biotechnology) or Trueblot immunoprecipitation beads (eBioscience, San Diego) for 2 h at 4 °C. The immunoprecipitates were collected by centrifugation in a microcentrifuge at 6000 rpm for 5 min. The supernatant was discarded, whereupon the pellet was washed with Nonidet P-40 buffer (Nonidet P-40 lysis buffer) (50 mm Tris-HCl, pH 8.0, 0.5% Nonidet P-40, 10% glycerol, 150 mm NaCl, 2 mm MgCl2, and 1 mm EDTA), with protease inhibitors for three to five times, and then dissolved in a sample buffer for SDS-PAGE.
GST Pulldown Assays
The GST pulldown assays were performed by incubating equal amounts of GST or GST fusion protein immobilized to glutathione-Sepharose beads with in vitro translated 35S-labeled protein, in a 400-μl reaction volume. The mixtures were incubated for 2 h at 4 °C and washed three to six times with Nonidet P-40 lysis buffer. Bound proteins were eluted with 2× SDS buffer, separated by SDS-PAGE, and visualized by autoradiography. The transferred protein on the blot was visualized using Ponceau S stain.
Chromatin Immunoprecipitation (ChIP) and Re-ChIP Assays
The ChIP assays were performed as described previously (27). Briefly, ∼106 cells were treated with 1% formaldehyde (final concentration) for 10 min at 37 °C to cross-link histones to DNA and washed twice with phosphate-buffered saline containing protease inhibitor mixture. Cells were lysed by sonication and immunoprecipitated with specific antibodies. The immunoprecipitates were washed; DNA was eluted off the beads, and purified DNA (phenol/chloroform extraction) was subjected to PCR. PCR primers for ChIP assays for TF and TFPI promoters are provided in supplemental Tables 1 and 2, respectively. For re-ChIP assays, the first ChIP using a PAK1 antibody (Bethyl Laboratories) was followed by a second ChIP using a c-Jun antibody (Santa Cruz Biotechnology). Quantification of the bands amplified by PCR was done using ImageJ software.
Electrophoretic Mobility Shift Assay
Nuclear extracts were prepared using a Nonidet P-40 lysis method (28). EMSA for the binding of different transcription factors was performed using the annealed and [γ-32P]ATP end-labeled oligonucleotides in a 20-μl reaction mixture for 15 min at 20 °C. Samples were run on a nondenaturing 5% polyacrylamide gel and imaged by autoradiography. Supershift EMSAs were performed by incubating the nuclear extracts with 2 μg of either the PAK1 (Bethyl Laboratories, Montgomery, TX) or the c-Jun (Santa Cruz Biotechnology) antibody for 1 h before the addition of the radiolabeled oligonucleotides. Oligonucleotides used are listed in supplemental Table 3.
Tissue Factor Procoagulant Activity Assay
TF activity, measured as the amount of FXa generated, was determined using the Assay Sense human TF chromogenic activity assay kit (AssayPro) with minor modifications, according to Drake et al. (29). Cells were detached by brief exposure to trypsin/EDTA, washed three times with DMEM/F-12, and resuspended in DMEM/F-12 at 4 × 105 cells/ml. For blocking cell-surface expressed TF, anti-TF mAb TF8–5G9 (10 μg/ml) was incubated with 1 ml of the corresponding intact cell suspensions for 15 min at 37 °C, washed three times with DMEM/F-12, and divided into two equal portions. One portion was resuspended in Hepes/saline buffer (25 mm Hepes, 0.85% NaCl, pH 7.4) and kept at 4 °C before assay of intact cell surface TF activity. The other portion was lysed by freezing the cell suspension at −70 °C for 15 min and solubilizing with 15 mm octyl-β-d-glucopyranoside at 37 °C for 15 min for assay of total TF procoagulant activity. Identical aliquots of intact (cell surface TF procoagulant activity) and lysed cells (total TF procoagulant activity) were assayed, following the manufacturer's recommendations, and the amount of FXa generated was determined by reading the absorbance at 405 nm every 2 min for 20 min, using a kinetic 96-well microplate reader (Synergy-H1 Hybrid Reader, Biotek).
RESULTS AND DISCUSSION
PAK1 Up-regulates the Expression of Tissue Factor
A microarray gene profiling study that includes wild-type (WT) and PAK1-knock-out (KO) MEFs was performed to identify the new genes or pathways that are influenced by PAK1.2 When the statistically significant differentially expressed genes were studied using Ingenuity Pathway Analysis, a total of 16 genes assigned to thrombosis were differentially expressed in between wild-type and PAK1-KO MEFs (Table 1). These findings raise a new interesting line of research and for the first time introduce PAK1 as a possible new regulatory player in thrombosis.
TABLE 1.
List of the down-regulated or up-regulated genes, involved in thrombosis, in the PAK1 knock-out MEFs compared to its wild-type controls
| Refseq | Gene symbol | Fold change | Regulation | Gene description |
|---|---|---|---|---|
| NM_009378 | Thbd | 7.86 | Down | Thrombomodulin |
| NM_011058 NM_001083316 | Pdgfra | 5.04 | Down | Platelet-derived growth factor receptor, α-polypeptide |
| NM_009255 | Serpine2 | 3.27 | Down | Serine (or cysteine) peptidase inhibitor, clade E, member 2 |
| NM_010171 | F3 | 2.53 | Down | Coagulation factor III |
| NM_008610 | Mmp2 | 2.51 | Down | Matrix metallopeptidase 2 |
| NM_031168 | Il6 | 2.42 | Down | Interleukin 6 |
| NM_009506 | Vegfc | 2.37 | Down | Vascular endothelial growth factor C |
| NM_001110504 NM_007600 | Capn1 | 4.88 | Up | Calpain 1 |
| NM_011576 | Tfpi | 3.46 | Up | Tissue factor pathway inhibitor |
| NM_011198 | Ptgs2 | 3.26 | Up | Prostaglandin-endoperoxide synthase 2 |
| NM_010217 | Ctgf | 2.70 | Up | Connective tissue growth factor |
| NM_008873 | Plau | 2.25 | Up | Plasminogen activator, urokinase |
| NM_009931 | Col4a1 | 2.08 | Up | Collagen, type IV, α1 |
| NM_019521 | Gas6 | 2.02 | Up | Growth arrest specific 6 |
| NM_011057 | Pdgfb | 2.01 | Up | Platelet-derived growth factor, B polypeptide |
| NM_008969 | Ptgs1 | 2.01 | Up | Prostaglandin-endoperoxide synthase 1 |
In the microarray analysis, TF, the primary initiator of the coagulation cascade, was remarkably found to be differentially expressed between both cell lines. The expression of TF was 2.53-fold down-regulated in the PAK1-KO compared with wild-type MEFs (Table 1). Consistent with the above data, RT-quantitative PCR confirmed that TF expression was down-regulated in the PAK1-KO MEFs as compared with its wild-type counterparts (Fig. 1A), positioning TF as a potential downstream target of PAK1. Through its impact on the coagulation cascade and cellular signaling, TF acts as an important regulator of vascular development, several cancer-related processes, such as hypercoagulability, angiogenesis, and metastasis, and inflammation (3). Therefore, the knowledge of its transcriptional regulation will be of great scientific interest.
FIGURE 1.
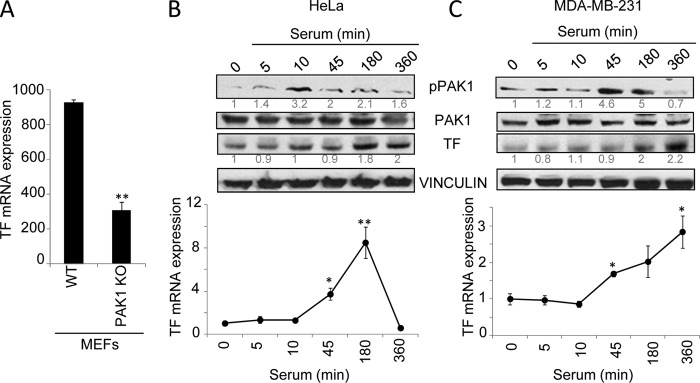
PAK1 up-regulates tissue factor expression. A, microarray RT-qPCR validation of TF mRNA expression in the WT and PAK1-KO MEFs. The relative mRNA expression levels are represented; mouse 18 S was used to normalize the values. **, p < 0.001 versus WT MEFs. B and C, expression of tissue factor protein (upper panel) and mRNA (lower panel) levels was analyzed after serum induction for the indicated times (minutes) in HeLa (B) and MDA-MB-231 cells (C). B and C, upper panel, Western blot using phospho-PAK1 (pPAK1), PAK1, and TF antibodies. The expression of vinculin was used as a loading control. The numbers below the panels (in gray) indicate the densitometric values corresponding to the quantification of the bands detected by autoradiography. A representative Western blot, from a total of three independent experiments, is shown. B and C, lower panel, total mRNA from HeLa (B) or MDA-MB-231 (C) cells induced with serum for the indicated times was analyzed for the expression of TF mRNA by RT-qPCR. The relative mRNA expression levels are represented; actin expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments. *, p < 0.005; **, p < 0.001 in serum-stimulated cells compared with nonstimulated cells. min, minutes.
A previous study already implicated PAK1 in the regulation of TF expression in pulmonary artery smooth muscle cells (30). These authors observed that PAK1 is involved in thrombin signaling in pulmonary artery smooth muscle cells and regulates the expression of TF by activating p38MAPK, PDK1, and PKB, and thus, PAK1 could play a role in vascular remodeling. Given the established connection between malignancy and hypercoagulant states (31) and PAK1's role in malignant processes (32), as well as the linkage of TF with cancer-related processes (6, 33), the possible role of PAK1 as a modulator of thrombosis in cancer cells was viewed of high interest for a more in-depth study. To date, nothing is known about the regulation of TF by PAK1 in cancer cells. Further studies in human epithelial cancer cell lines, such as HeLa and MDA-MB-231, demonstrated that the serum induction of PAK1 activity induced up-regulation of TF protein as well as mRNA levels (Fig. 1, B and C), suggesting a role of PAK1 in the regulation of TF expression.
PAK1 Is Required for TF Up-regulation by Serum
Since the induction of TF by serum has been well described (34), to further demonstrate the implication of PAK1 in the regulation of TF expression by serum, we examined the effect of PAK1 down-regulation conditions on TF expression. As shown in Fig. 2A, PAK1 down-regulation by siRNA transfection in HeLa cells compromised the ability of serum to stimulate TF protein as compared with cells transfected with a control siRNA. The induction of TF mRNA expression was also analyzed in wild-type and PAK1-KO MEFs (Fig. 2B). The results show that the serum induction of TF mRNA observed in the wild-type MEFs was almost completely abolished in PAK1-KO MEFs, highlighting the significance of PAK1 in the regulation of TF expression.
FIGURE 2.

PAK1 is required for the up-regulation of tissue factor by serum. A, Western blot analysis of phospho-PAK1, PAK1 and TF in HeLa cells transiently transfected with an siRNA against PAK1 (siPAK1) or an siRNA control (siC) induced with serum for the indicated times (in minutes). The expression of vinculin was used as a loading control. The numbers below the panels (in gray) indicate the densitometric values corresponding to the quantification of the bands detected by autoradiography. A representative Western blot, from a total of three independent experiments, is shown. B, RT-qPCR analysis of TF mRNA in WT and PAK1-KO MEFs cells induced with serum for the indicated times (in minutes). The relative mRNA expression levels are represented; mouse 18 S expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments.*, p < 0.005; **, p < 0.001 versus nonstimulated WT MEFs. #, p < 0.005; ##, p < 0.001 versus serum-stimulated WT MEFs. C, Western blot (upper panel) and RT-qPCR analysis (lower panel) in HeLa cells transiently transfected with the catalytically active (T423E), kinase-dead (K229R) PAK1 mutants, or with the empty vector (CTRL) induced with serum for the indicated times. C, upper panel, cell lysates were blotted with p-PAK1, PAK1, and TF antibodies. The expression of vinculin was used as a loading control. The numbers below the panels (in gray) indicate the densitometric values corresponding to the quantification of the bands detected by autoradiography. A representative Western blot, from a total of three independent experiments, is shown. C, lower panel, total mRNA was analyzed for the expression of TF mRNA by RT-qPCR. The relative mRNA expression levels are represented; actin expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments. *, p < 0.005; **, p < 0.001 versus nonstimulated control (CTRL) cells. #, p < 0.005 versus serum-stimulated control (CTRL) cells. min, minutes.
We next examined whether the kinase activity of PAK1 is essential for the regulation of TF expression. To test this, we analyzed the effect of a dominant-negative K229R and catalytically active T423E PAK1 mutants (22, 35) on the expression of TF by serum (Fig. 2C). Transient transfection of HeLa cells with the catalytically active T423E PAK1 mutant resulted in a superior stimulation of TF protein and mRNA by serum as compared with the control cells (Fig. 2C). However, cells expressing the kinase-dead K229R PAK1 partially suppressed the ability of serum to stimulate TF protein and mRNA expression levels (Fig. 2C). These results demonstrate the significance of PAK1 expression and activity in the induction of TF expression by serum.
PAK1 Recruitment onto TF Promoter
The previously observed nuclear localization of PAK1 (18), as well as its already reported modulatory effect in the transcription of some genes, such as nuclear factor of activated T-cell (NFAT1) and the phosphofructokinase-muscle isoform (PFK-M) (18), prompted us to investigate the direct genomic regulation of TF transcription by PAK1 in cancer cells.
Therefore, we examined the recruitment of PAK1 in the promoter of TF using a ChIP-based promoter walk assay. To this end, cross-linked chromatin from HeLa cells was immunoprecipitated with an anti-PAK1 antibody or IgG and then PCR-amplified using primers designed against about every 400-bp region of the TF promoter (Fig. 3A). Results from Fig. 3B show that PAK1 was recruited to the TF promoter at a region encompassing −666 to −1529 bp from the transcription start site. In an attempt to narrow down the region of the TF promoter in which PAK1 was recruited, regions 3 and 4, which are positive for PAK1 recruitment, were subdivided further into two smaller regions, of about 200 bp each. New primers encompassing these regions were designed, and the same purified eluted DNA was PCR-amplified using these primers (Fig. 3C). We discovered that PAK1 was indeed recruited to the regions 3.2 to 4.2 but not to the region 3.1 (Fig. 3C). Our results indicate, for the first time, that PAK1 is able to specifically bind to the TF promoter region, demonstrating that TF is a target of PAK1.
FIGURE 3.

PAK1 is recruited to the promoter of tissue factor. A, schematic representation of the promoter of tissue factor, showing the regions (R) analyzed. B and C, ChIP analysis of the recruitment of PAK1 on the human TF promoter in HeLa cells. Negative (Neg) controls, without DNA, are shown for each region amplified. Input represents around 1–10% of the total immunoprecipitated DNA. The histogram on the right show the densitometric analysis of the bands amplified by PCR. The intensity of the bands amplified in the samples immunoprecipitated with IgG was used as the reference and assigned the value 1. Ab, antibody; TSS, transcription start site.
Involvement of c-Jun Transcription Factor in PAK1-mediated TF Up-regulation
Since PAK1 lacks transcriptional activity but is able to induce TF expression, we next hypothesized that PAK1 might be co-recruited to the TF promoter along with a transcription factor, which could be responsible for the direct stimulation of TF transcription. For this reason, we next performed a sequence analysis of the TF promoter-PAK1 interacting regions (regions 3–4, see Fig. 3B) using the Promo 3.0 software (36) to identify the putative binding sites for transcription factors that might be responsible for the PAK1-mediated stimulation of TF transcription. The complete list of the resulting transcription factor binding sites is presented in Table 2. We found that TF promoter harbors binding sites for some transcription factors already involved in cell growth, proliferation, and cancer-related signaling events. These included TCF-4E, GATA-1, TFII-I, PXR-1:RXR-α, c-ETS-1, or AP-1/c-Jun (37–42). Similarly, the involvement of both PAK1 and TF in such processes has been well established (13, 33). Therefore the above mentioned transcription factors appear to be good candidates for PAK1-mediated TF transcriptional up-regulation.
TABLE 2.
Complete list of the transcription factor-binding sites predicted in the tissue factor promoter-PAK1 interacting region using Promo 3.0 software analysis
The transcription factors selected for EMSA are highlighted.
| Transcription factor | No. of sites | Activator/repressor | Function |
|---|---|---|---|
| TCF-4E (T02878) | 2 | Activator | Associated with reduced survival in estrogen receptor-α breast cancer cells |
| Positive regulation of epithelial cell proliferation | |||
| GR (T05076) | 1 | Activator/repressor | Regulation of genes controlling development, metabolism, and immune response |
| GR-β (T01920) | 14 | Activator/repressor | |
| GR-α (T00337) | 7 | Activator/repressor | |
| XBP-1 (T00902) | 1 | Activator | Immune system and cellular stress response |
| AP-2αA (T00035) | 4 | Activator/repressor | Differentiation and development of neural crestal cells |
| C/EBPβ(T00581) | 19 | Repressor | Inflammatory and immune responses |
| HOXD9 (T01424) | 2 | Activator | Morphogenesis |
| HOXD10 (T01425) | 2 | Activator | Differentiation and limb development |
| TFIID (T00820) | 1 | Activator | Basal transcription and transcription initiation |
| FOXP3 (T04280) | 2 | Repressor | Immune system responses |
| YY1 (T00915) | 2 | Activator/repressor | Proposed core component of chromatin remodeling complexes |
| GATA-1 (T00306) | 2 | Activator | Platelet aggregation and formation |
| Cell growth and cancer | |||
| TFII-I (T00824) | 2 | Activator/repressor | Growth factor signaling. Implications in cancer |
| HNF-1A (T00368) | 1 | Activator | Liver-specific genes. Glucose homeostasis |
| PXR-1:RXR-α (T05671) | 1 | Activator/repressor | Assembly of functional peroxisomes (PXR-1) |
| Mediator of retinoids (RXR-α) | |||
| c-Ets-1 (T00112) | 1 | Activator/repressor | Stem cell development, cell senescence and death, and tumorigenesis |
| STAT4 (T01577) | 1 | Activator | Mediation of cellular responses to IL-12 in T-lymphocytes |
| Signaling molecule for TH1 differentiation | |||
| AP-1 (T00029) | 1 | Activator | Differentiation, proliferation, and apoptosis |
| c-Jun (T00133) | 1 | ||
| LEF-1 (T02905) | 1 | Activator | Wnt signaling |
To assess the above possibility, we examined the direct recruitment of these transcription factors to the TF promoter by electrophoretic mobility shift assay (EMSA) (Fig. 4A). To this end, we designed double-stranded oligonucleotides, containing the binding site(s) for each transcription factor(s), that were radiolabeled, and the binding of each transcription factor was analyzed using nuclear extracts from control and serum-stimulated HeLa cells. After electrophoresis and autoradiography, we detected the formation of DNA·protein complexes with the oligonucleotides containing binding sites for AP-1/c-Jun as well as TFII-I binding site b among the tested transcription factors (Fig. 4A). In addition, the observed complex was induced in serum-stimulated HeLa cells compared with nonstimulated cells, only when using the specific oligonucleotides for AP-1/c-Jun-binding site (Fig. 4A), suggesting that PAK1-serum stimulated signaling uses AP-1/c-Jun to induce TF transcription. Accordingly, results from Fig. 4B, using nuclear extracts from control and serum-stimulated HeLa (left panel) or MDA-MB-231 cells (right panel), revealed that the formation of AP-1/c-Jun·DNA complex(s) at the TF promoter was completely abolished using an oligonucleotide containing the putative binding site mutated, confirming the presence of AP-1/c-Jun. In addition, the observed complex could be highly inhibited, especially under serum-induced conditions, in the presence of an anti-c-Jun antibody, as well as in the presence of an anti-PAK1 antibody, but not with a control immunoglobulin IgG antibody, suggesting the co-existence of PAK1·DNA and c-Jun·DNA complexes. However, the presence of an anti-c-Jun antibody failed to exhibit the appearance of a super-shifted band, probably due to inefficient blocking of the recognition site for c-Jun in the DNA sequence and/or antibody-specific reasons.
FIGURE 4.

c-Jun is involved in the PAK1-mediated regulation of tissue factor expression. A, EMSA analysis of the binding of GATA-1, PXR-1:RXR-α, TCF-4E, AP1/c-Jun, c-ETS, and TFII-I transcription factors to the human TF promoter using radiolabeled oligonucleotides encompassing the predicted consensus binding sites and nuclear extracts from HeLa cells under serum starvation/induction conditions. a and b denote different binding sites predicted for the annotated transcription factor. c indicates control lane without nuclear lysate. +/− indicates nuclear lysate from HeLa cells induced or not with serum, respectively. B, EMSA analysis of c-Jun and PAK1 binding to the human TF promoter using the wild-type (WT) and mutant (MUT) oligonucleotides encompassing the c-Jun consensus binding sites using HeLa (left panel) or MDA-MB-231 (right panel) nuclear lysates prepared from cells stimulated or not with serum. c indicates the control lane without nuclear lysate. +/− indicates nuclear lysate from HeLa cells induced or not with serum, respectively. Ab, antibody. C, ChIP analysis of the recruitment of c-Jun onto the human TF promoter in HeLa cells. R, region. D, double ChIP analysis of the recruitment of PAK1·c-Jun complex onto the TF chromatin region 1 (R1) and 3 (R3) in HeLa cells. The first ChIP was carried out with an anti-PAK1 antibody followed by second ChIP with anti-c-Jun antibody. Negative (Neg) controls, without DNA, are shown for each region amplified by ChIP. Input represents around 1–10% of the immunoprecipitated DNA. E, RT-qPCR analysis of TF mRNA expression in HeLa cells transiently overexpressing the catalytically active mutant of PAK1 (T423E), and the dominant-negative mutant of c-Jun (TAM 67) after serum stimulation for the indicated times (in minutes). The relative mRNA expression levels are represented; actin expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments. *, p < 0.005; **, p < 0.001, versus nonstimulated T423E PAK1 mutant (T423E) cells. #, p < 0.005; ##, p < 0.001, versus serum- stimulated T423E PAK1 mutant (T423E) cells. min, minutes.
To gain a deeper insight into the molecular mechanism involved in c-Jun regulation of TF expression, we next analyzed the in vivo recruitment of c-Jun to the TF promoter by ChIP, using cross-linked chromatin from HeLa cells. We found the recruitment of c-Jun onto region 3 of the TF promoter (Fig. 4C). This is in agreement with the Promo 3.0 sequence analysis of the TF promoter-PAK1 interacting DNA region, which indicates this region includes the binding site for c-Jun.
The regulation of TF gene expression by c-Jun has been demonstrated in human monocytic and endothelial cells (43, 44), as well as in cultured fibroblasts (45). However, the binding region of c-Jun reported in these studies (located at a region around −200 to −220 bp, relative positions to the transcription start site (43–45)) is different from the one detected in this study (−1450 bp, relative to the transcription start site). However, a previous report located elements required for serum and phorbol myristate acetate induction of TF promoter in HeLa cells in a region between −96 and +121 bp, from a total region analyzed for 300 bp relative to the transcription start site (46). This region lacks any AP1/c-Jun-binding site, but it is possible, however, that c-Jun could account for the induction of TF gene transcription by serum in HeLa cells in a different region than that analyzed in a previous study (46) as compared with results obtained in this study. The apparent controversy between different results could be explained by the fact that the TF gene is expressed in a cell type-specific manner (29). Moreover, our results suggest that not only the expression of TF appears to be mediated by distinct regions of the TF promoter in different cell types but also the induction in response of different stimuli can be mediated by distinct regions of the promoter in the same cell type.
Results from Fig. 4B, as well as the fact that c-Jun is recruited to the TF promoter region 3 (Fig. 4C), suggest that both PAK1 and c-Jun could be co-recruited at the same region onto the TF promoter. To determine whether the PAK1·c-Jun complex associates to the same region on the TF promoter, a sequential double-ChIP analysis using the indicated antibodies was performed. We found that indeed PAK1 and c-Jun are co-recruited onto the c-Jun-TF promoter-interacting region 3 (Fig. 4D), demonstrating the co-recruitment of both proteins onto the TF promoter.
To further demonstrate the importance of c-Jun in PAK1-mediated TF up-regulation, HeLa cells were co-transfected with the catalytically active mutant of PAK1 (T423E) and with a dominant-negative mutant of c-Jun (TAM 67) that can repress its transcriptional activity (25). The cells were induced with serum for the indicated times, prior to the analysis of TF expression by qPCR. The presence of a dominant-negative mutant of c-Jun partially suppressed the ability of the catalytically active mutant of PAK1 to stimulate TF mRNA expression after serum induction (Fig. 4E), confirming the involvement of c-Jun in the regulation of TF expression by PAK1.
PAK1 Physically Interacts with c-Jun
It has been previously reported that PAK1 activates JNK, resulting in the phosphorylation and activation of c-Jun (47). However, our findings that PAK1 and c-Jun are co-recruited onto the TF promoter raise the possibility of physical interaction between them. To test this notion, HEK293T cells were co-transfected with expression vectors encoding FLAG-tagged c-Jun and Myc-tagged PAK1, and protein extracts were subjected to the sequential immunoprecipitation/Western blot analysis.
Results from Fig. 5A show that indeed exogenously expressed FLAG-c-Jun but not control IgG could be coimmunoprecipitated with Myc-PAK1, suggesting that the two proteins interact in vivo. In addition, similar interaction was observed between PAK1 and c-Jun at the endogenous protein level in HeLa cells (Fig. 5B). To determine whether PAK1 directly binds to c-Jun, we next performed in vitro GST pulldown assays using 35S-labeled in vitro translated c-Jun protein and full-length GST-PAK1 fusion protein, as well as 35S-labeled in vitro translated PAK1 protein and full-length GST-c-Jun fusion protein, and we found that 35S-labeled c-Jun and 35S-labeled PAK1 strongly bound to GST-PAK1 and GST-c-Jun proteins, respectively (Fig. 5, C and D). These findings demonstrate that PAK1 physically associates with c-Jun both in vivo and in vitro. It is noteworthy that PAK2 is also shown to bind and phosphorylate c-Jun modulating its activity (48). Given the similarity in the phosphorylation recognition motifs of PAK1 and PAK2, these results suggest that PAK1 interaction with c-Jun promotes its transcriptional activation and leads to induction of TF expression.
FIGURE 5.

PAK1 interacts physically with c-Jun. A, HEK 293T cells were co-transfected with expression vectors encoding FLAG-c-Jun and Myc-PAK1. Protein extracts were immunoprecipitated (IP) with control IgG or an anti-Myc antibody and were immunoblotted (WB) with the indicated antibodies. The input represents around 10% of the total amount of protein used for the immunoprecipitation. B, nuclear protein extracts from HeLa cells were immunoprecipitated with an anti-PAK1 or anti-c-Jun antibodies, followed by Western blotting using the indicated antibodies. C, in vitro transcribed-translated c-Jun protein (35S-labeled) was incubated with GST or GST-PAK1 constructs. Bound proteins were separated and analyzed by SDS-PAGE and autoradiography. D, in vitro transcribed-translated PAK1 protein (35S-labeled) was incubated with GST or GST-c-Jun constructs. Bound proteins were separated and analyzed by SDS-PAGE and autoradiography. The lower panels in C and D are Ponceau S-stained blots showing either GST-PAK1 or GST-c-Jun and GST. * denotes the presence of the GST-PAK1 or GST-c-Jun fusion proteins.
PAK1 Stimulation Regulates the Procoagulant Activity of Tissue Factor
To investigate the possibility of PAK1 involvement in the coagulation cascade triggered by TF, we analyzed the TF activity as means of its ability to activate Factor X (FX) to FXa. The amount of FXa generated was quantitated using a highly specific FXa substrate releasing a yellow para-nitroaniline (pNA) chromophore. The change in absorbance of the pNA at 405 nm is directly proportional to the TF enzymatic activity. HeLa cells transiently overexpressing the dominant-negative K229R or catalytically active T423E PAK1 mutants or control cells (empty vector) stimulated or not with serum for 3 h were used for these experiments (Fig. 6).
FIGURE 6.

PAK1 activation regulates the procoagulant activity of tissue factor. A and B, monitorization of TF activity in cell lysates from control HeLa cells (CTRL) or cells transiently overexpressing the catalytically active (T423E) (A) and kinase-deficient (K229R) (B) PAK1 mutants under serum starvation/induction conditions. The change in absorbance at 405 nm of the released pNA chromophore, which is directly proportional to the TF enzymatic activity, was recorded each 2 min for 20 min. The average and standard deviation from three independent experiments is shown. Arrows indicate the time points in which the amount of active TF was calculated in C. C, amount of active endogenous TF expressed in lysates from control HeLa cells (CTRL) or cells transiently overexpressing the catalytically active (T423E) and kinase-deficient (K229R) mutants of PAK1 under serum starvation/induction conditions. Values were extrapolated from a standard curve generated with different known concentrations of a recombinant human TF preparation and expressed as pm of TF per 105 cells. The average and standard deviation from triplicates is shown. *, p < 0.005; **, p < 0.001 versus nonstimulated cells. #, p < 0.005 in T423E PAK1 mutant cells (T423E) compared with control HeLa cells (CTRL) under serum stimulation conditions. min, minutes.
The corresponding intact cell suspensions were incubated with either control or anti-TF antibody for 15 min at 37 °C and then washed to remove unbound antibody before cells were either lysed (for assay of total TF activity) or collected (for assay of cell surface TF activity), and the TF activity was measured. As reported previously (49), intact HeLa cells expressed very little cell surface TF activity compared with the total TF activity in disrupted cells (data not shown). In addition, the presence of the TF blocking antibody was able to reduce <5% of the total activity (data not shown), thus indicating that the majority of TF in HeLa cells was expressed intracellularly, which is in agreement with previous reports (49). The presence of an intracellular pool may be particularly important because it is likely to be protected from extracellular inhibitors of TF. Furthermore, previous studies have found that a substantial portion of TF is not active on the surface of unperturbed cells, and studies have suggested that much of the TF is encrypted on the cell surface (50). Therefore, to further verify the implication of TF in the generation of FXa, the experiment was performed in the absence of FVII. This approach completely blocked all the activity (data not shown), thus demonstrating that the results obtained were entirely due to TF activity.
To gain insight into the ability of PAK1 to induce TF procoagulant activity, we monitored the ability of the endogenously expressed TF to enhance the specific proteolytic activation of FX to FXa during the whole assay (Fig. 6, A and B). This is achieved through the constant annotation of the change in absorbance of the released chromophore pNA at 405 nm. A clear increase in the TF procoagulant activity during the whole analysis was observed after serum treatment of the control and T423E PAK1-expressing cells, and this induction was more notable in cells expressing the catalytically active form of PAK1, T423E (Fig. 6A). On the contrary, serum was unable to further induce any TF activity in cells expressing the dominant-negative PAK1 mutant (K229R) (Fig. 6B).
Active TF was determined from the standard curves generated, at two different time points (10 and 20 min), using known concentrations of a recombinant human TF preparation, and expressed as pm of TF per number of cells (Fig. 6C). We found that control cells as well as catalytically active mutant PAK1 (T423E) cells showed a higher level of active TF after serum stimulation. Moreover, cells expressing the catalytically active PAK1 (T423E) showed an even higher level of active TF in serum stimulation conditions, as compared with control cells. However, the levels of active TF remained without any change in cells transiently transfected with the kinase-dead mutant of PAK1 (K229R) (Fig. 6C), thus highlighting the implication of PAK1 activation in the control of TF coagulation activity. Together, these results implicate PAK1 signaling acts as a modulator of the coagulation protease cascade by playing a role in the modulation of the ability of TF to activate FX.
PAK1 Recruitment onto TFPI Promoter and Modulation of Its Expression
The above results demonstrate the significance of PAK1 signaling in the coagulation cascade triggered by TF. Interestingly, the natural inhibitor of the TF-mediated coagulation, TFPI, was also found to be differentially expressed between wild-type and PAK1-KO MEFs (Table 1). PAK1-KO MEFs showed a 3.46-fold up-regulation of TFPI compared with wild-type MEFs, suggesting a possible negative regulation of TFPI expression by PAK1.
RT-quantitative PCR analysis confirmed the up-regulation of TFPI in the PAK1-KO cells as compared with its wild-type counterparts (Fig. 7A). Furthermore, serum induction of PAK1 in HeLa and MDA-MB-231 human cell lines led to down-regulation of TFPI mRNA expression (Fig. 7, B and C, respectively), suggesting that PAK1 can negatively modulate the expression of TFPI. Similar to TF, the expression of TFPI has also been reported to be modulated by serum (51). Hence, further studies were designed to demonstrate the significance of PAK1 signaling in the down-regulation of TFPI expression. To this end, we analyzed the expression of TFPI mRNA in PAK1-KO MEFs or wild-type MEFs under serum stimulation conditions. We found a down-regulation of TFPI mRNA levels by serum stimulation in the wild-type MEFs, and this down-regulation was not only compromised in the serum-stimulated PAK1-KO MEFs, but we observed instead a transient up-regulation of TFPI mRNA expression levels in these cells, suggesting that PAK1 may be essentially involved in the repression of TFPI expression.
FIGURE 7.

PAK1 down-regulates TFPI mRNA expression by its direct recruitment to the TFPI promoter. A, RT-qPCR validation of TFPI mRNA expression in the WT and PAK1-KO MEFs. The relative mRNA expression levels are represented; mouse 18 S was used to normalize the values. **, p < 0.001 versus WT MEFs. B and C, RT-qPCR analysis of TFPI mRNA in HeLa (B) and MDA-MB-231 (C) cells after serum induction for the indicated times (in minutes). The relative mRNA expression levels are represented; actin expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments. min, minutes. *, p < 0.005; **, p < 0.001 in serum-stimulated cells compared with nonstimulated cells. D, RT-qPCR analysis of TFPI mRNA in WT and PAK1-KO MEFs cells induced with serum for the indicated times. The relative mRNA expression levels are represented; mouse 18 S expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments. *, p < 0.005; **, p < 0.001 in serum-stimulated cells compared with nonstimulated cells. min, minutes. E, RT-qPCR analysis of TFPI mRNA in HeLa cells transiently transfected with the catalytically active (T423E), kinase-dead (K229R) PAK1 mutants, or with the empty vector (CTRL) induced with serum for the indicated times. The relative mRNA expression levels are represented; actin expression was used to normalize the values. Each value represents the mean ± S.E. of three independent experiments. *, p < 0.005; **, p < 0.001 versus nonstimulated control cells. #, p < 0.005; ##, p < 0.001 versus serum-stimulated control cells. min, minutes. F, schematic representation of the promoter of TF pathway inhibitor, showing the regions (R) analyzed by ChIP. TSS, transcription start site. G, ChIP analysis of the recruitment of PAK1 on the human (h) TFPI promoter in HeLa cells. Negative (Neg) controls, without DNA, are shown for each region amplified. Input represents around 1–10% of the total immunoprecipitated DNA. Ab, antibody. H, schematic representation of TF and TFPI regulation by PAK1. Serum stimulation of PAK1 results in the up-regulation of TF, by the co-recruitment of PAK1 and c-Jun onto the human (h) TF promoter. The up-regulation of TF expression by PAK1 is followed by an induction of the procoagulant activity of TF. At the same time, serum stimulation of PAK1 results in the down-regulation of TFPI expression by the co-recruitment of PAK1 and a yet unknown transcription factor onto the TFPI promoter, thereby releasing the TF-FVIIa coagulation·initiation complex from its inhibition and possibly contributing to a hypercoagulant state.
We next investigated the importance of PAK1 kinase activity in the regulation of TFPI expression. We analyzed the effect of the dominant-negative K229R and catalytically active T423E PAK1 mutants (22, 35) on TFPI mRNA expression after serum induction (Fig. 7E). As expected, serum induction of cells overexpressing a kinase-dead PAK1 mutant (K229R) resulted in a progressive up-regulation of TFPI mRNA expression, but no stimulation of TFPI mRNA levels was observed in those cells expressing the catalytically active PAK1 mutant (T423E). Results from Fig. 7, D and E, suggest that PAK1 expression and activation negatively regulate TFPI expression at the transcriptional level. To further strengthen this hypothesis, we next explored the recruitment of PAK1 to the promoter of TFPI using a ChIP-based promoter walk assay in cross-linked chromatin from HeLa cells immunoprecipitated with an anti-PAK1 antibody or IgG control antibody. A region of the human TFPI promoter of ∼1700 bp (+25- to −1653-bp relative positions from the transcription start site) was analyzed by PCR using primers designed about each 400 bp (Fig. 7F). As shown in Fig. 7G, ChIP results showed the recruitment of PAK1 onto the TFPI promoter to a region encompassing −798 to −1198 bp from the transcription start site, named region 3. In summary, results from Fig. 7 demonstrate for the first time that PAK1 is recruited to the TFPI promoter and that PAK1 acts as a negative modulator of TFPI expression.
Balanced levels of TF and TFPI are essential to maintain normal homeostasis. In this regard, the ability of PAK1 to up-regulate TF expression and down-regulate TFPI expression places PAK1 as a new key molecule involved in the deregulation of normal homeostasis. In agreement with the results presented here, previous work from Diebold et al. (52) demonstrates the expression of PAK1 in the media of remodeled pulmonary vessels from patients with pulmonary vasculopathy and its implication in vascular remodeling by thrombin signaling in pulmonary artery smooth muscle cells. Future studies, with the use of smooth muscle cells, are guaranteed to better elucidate the role of PAK1 in thrombosis. From another point of view, given the role of PAK1 as a central signaling transducer and the emerging functions of TF, as well as of TFPI in different signaling events apart from homeostasis (53, 54), our findings open a new transcriptional regulatory axis that could be highly relevant not only in coagulation, physiological signaling pathways, but also in several malignant processes.
In summary, we provide here a novel regulatory mechanism of TF expression and procoagulant activity in epithelial cancer cells by the PAK1·c-Jun complex. Furthermore, PAK1 is able to bind to the TFPI promoter and negatively regulate its expression, presumably releasing the inhibition of the TF·FVIIa complex from TFPI and, in turn, contributing to a hypercoagulant state (Fig. 7H). Our working model provides a novel function for PAK1 in the control of coagulation processes, suggesting that the control of PAK1 activity may act as a new and exciting potential therapeutic strategy.
Supplementary Material
Acknowledgments
We thank the members of our laboratory for insightful discussions.
This work was supported, in whole or in part, by National Institutes of Health Grant CA90970 (to R. K.). This work was also supported by the McCormick Proteomic and Genomic Center.
This article was selected as a Paper of the Week.

This article contains supplemental Tables S1–S3.
R. Kumar, unpublished data.
M. Motwani, D.-Q. Li, A. Horvath, and R. Kumar, unpublished data.
- TF
- tissue factor
- TFPI
- tissue factor pathway inhibitor
- MEF
- mouse embryonic fibroblast
- qPCR
- quantitative PCR
- pNA
- para-nitroaniline
- F
- factor.
REFERENCES
- 1. McVey J. H. (1994) Tissue factor pathway. Bailliere's Clin. Haematol. 7, 469–484 [DOI] [PubMed] [Google Scholar]
- 2. Nemerson Y. (1988) Tissue factor and hemostasis. Blood 71, 1–8 [PubMed] [Google Scholar]
- 3. Versteeg H. H., Ruf W. (2006) Emerging insights in tissue factor-dependent signaling events. Semin. Thromb. Hemostasis 32, 24–32 [DOI] [PubMed] [Google Scholar]
- 4. Bajaj M. S., Birktoft J. J., Steer S. A., Bajaj S. P. (2001) Structure and biology of tissue factor pathway inhibitor. Thromb. Haemost. 86, 959–972 [PubMed] [Google Scholar]
- 5. Bugge T. H., Xiao Q., Kombrinck K. W., Flick M. J., Holmbäck K., Danton M. J., Colbert M. C., Witte D. P., Fujikawa K., Davie E. W., Degen J. L. (1996) Fatal embryonic bleeding events in mice lacking tissue factor, the cell-associated initiator of blood coagulation. Proc. Natl. Acad. Sci. U.S.A. 93, 6258–6263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Contrino J., Hair G., Kreutzer D. L., Rickles F. R. (1996) In situ detection of tissue factor in vascular endothelial cells. Correlation with the malignant phenotype of human breast disease. Nat. Med. 2, 209–215 [DOI] [PubMed] [Google Scholar]
- 7. Ruf W. (2007) Tissue factor and PAR signaling in tumor progression. Thromb. Res. 120, S7–S12 [DOI] [PubMed] [Google Scholar]
- 8. Levi M., ten Cate H., Bauer K. A., van der Poll T., Edgington T. S., Büller H. R., van Deventer S. J., Hack C. E., ten Cate J. W., Rosenberg R. D. (1994) Inhibition of endotoxin-induced activation of coagulation and fibrinolysis by pentoxifylline or by a monoclonal anti-tissue factor antibody in chimpanzees. J. Clin. Invest. 93, 114–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Poll T., Büller H. R., ten Cate H., Wortel C. H., Bauer K. A., van Deventer S. J., Hack C. E., Sauerwein H. P., Rosenberg R. D., ten Cate J. W. (1990) Activation of coagulation after administration of tumor necrosis factor to normal subjects. N. Engl. J. Med. 322, 1622–1627 [DOI] [PubMed] [Google Scholar]
- 10. Pendurthi U. R., Rao L. V. (2002) Factor VIIa/tissue factor-induced signaling. A link between clotting and disease. Vitam. Horm. 64, 323–355 [DOI] [PubMed] [Google Scholar]
- 11. Manser E., Leung T., Salihuddin H., Zhao Z. S., Lim L. (1994) A brain serine/threonine protein kinase activated by Cdc42 and Rac1. Nature 367, 40–46 [DOI] [PubMed] [Google Scholar]
- 12. Tsakiridis T., Taha C., Grinstein S., Klip A. (1996) Insulin activates a p21-activated kinase in muscle cells via phosphatidylinositol 3-kinase. J. Biol. Chem. 271, 19664–19667 [DOI] [PubMed] [Google Scholar]
- 13. Molli P. R., Li D. Q., Murray B. W., Rayala S. K., Kumar R. (2009) PAK signaling in oncogenesis. Oncogene 28, 2545–2555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vadlamudi R. K., Kumar R. (2003) p21-activated kinases in human cancer. Cancer Metastasis Rev. 22, 385–393 [DOI] [PubMed] [Google Scholar]
- 15. Barnes C. J., Vadlamudi R. K., Mishra S. K., Jacobson R. H., Li F., Kumar R. (2003) Functional inactivation of a transcriptional corepressor by a signaling kinase. Nat. Struct. Biol. 10, 622–628 [DOI] [PubMed] [Google Scholar]
- 16. Bagrodia S., Dérijard B., Davis R. J., Cerione R. A. (1995) Cdc42 and PAK-mediated signaling leads to Jun kinase and p38 mitogen-activated protein kinase activation. J. Biol. Chem. 270, 27995–27998 [DOI] [PubMed] [Google Scholar]
- 17. Brown J. L., Stowers L., Baer M., Trejo J., Coughlin S., Chant J. (1996) Human Ste20 homologue hPAK1 links GTPases to the JNK MAP kinase pathway. Curr. Biol. 6, 598–605 [DOI] [PubMed] [Google Scholar]
- 18. Singh R. R., Song C., Yang Z., Kumar R. (2005) Nuclear localization and chromatin targets of p21-activated kinase 1. J. Biol. Chem. 280, 18130–18137 [DOI] [PubMed] [Google Scholar]
- 19. Li F., Adam L., Vadlamudi R. K., Zhou H., Sen S., Chernoff J., Mandal M., Kumar R. (2002) p21-activated kinase 1 interacts with and phosphorylates histone H3 in breast cancer cells. EMBO Rep. 3, 767–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Balasenthil S., Sahin A. A., Barnes C. J., Wang R. A., Pestell R. G., Vadlamudi R. K., Kumar R. (2004) p21-activated kinase-1 signaling mediates cyclin D1 expression in mammary epithelial and cancer cells. J. Biol. Chem. 279, 1422–1428 [DOI] [PubMed] [Google Scholar]
- 21. Molli P. R., Li D. Q., Bagheri-Yarmand R., Pakala S. B., Katayama H., Sen S., Iyer J., Chernoff J., Tsai M. Y., Nair S. S., Kumar R. (2010) Arpc1b, a centrosomal protein, is both an activator and substrate of Aurora A. J. Cell Biol. 190, 101–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sells M. A., Knaus U. G., Bagrodia S., Ambrose D. M., Bokoch G. M., Chernoff J. (1997) Human p21-activated kinase (Pak1) regulates actin organization in mammalian cells. Curr. Biol. 7, 202–210 [DOI] [PubMed] [Google Scholar]
- 23. Vadlamudi R. K., Li F., Adam L., Nguyen D., Ohta Y., Stossel T. P., Kumar R. (2002) Filamin is essential in actin cytoskeletal assembly mediated by p21-activated kinase 1. Nat. Cell Biol. 4, 681–690 [DOI] [PubMed] [Google Scholar]
- 24. Li D. Q., Pakala S. B., Reddy S. D., Ohshiro K., Zhang J. X., Wang L., Zhang Y., Moreno de Alborán I., Pillai M. R., Eswaran J., Kumar R. (2011) Bidirectional autoregulatory mechanism of metastasis-associated protein 1-alternative reading frame pathway in oncogenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 8791–8796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Brown P. H., Chen T. K., Birrer M. J. (1994) Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene 9, 791–799 [PubMed] [Google Scholar]
- 26. Li D. Q., Pakala S. B., Reddy S. D., Ohshiro K., Peng S. H., Lian Y., Fu S. W., Kumar R. (2010) Revelation of p53-independent function of MTA1 in DNA damage response via modulation of the p21 WAF1-proliferating cell nuclear antigen pathway. J. Biol. Chem. 285, 10044–10052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kumar R., Wang R. A., Mazumdar A., Talukder A. H., Mandal M., Yang Z., Bagheri-Yarmand R., Sahin A., Hortobagyi G., Adam L., Barnes C. J., Vadlamudi R. K. (2002) A naturally occurring MTA1 variant sequesters oestrogen receptor-α in the cytoplasm. Nature 418, 654–657 [DOI] [PubMed] [Google Scholar]
- 28. Schreiber E., Matthias P., Müller M. M., Schaffner W. (1989) Rapid detection of octamer binding proteins with “mini-extracts,” prepared from a small number of cells. Nucleic Acids Res. 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Drake T. A., Morrissey J. H., Edgington T. S. (1989) Selective cellular expression of tissue factor in human tissues. Implications for disorders of hemostasis and thrombosis. Am. J. Pathol. 134, 1087–1097 [PMC free article] [PubMed] [Google Scholar]
- 30. Görlach A., BelAiba R. S., Hess J., Kietzmann T. (2005) Thrombin activates the p21-activated kinase in pulmonary artery smooth muscle cells. Role in tissue factor expression. Thromb. Haemost. 93, 1168–1175 [DOI] [PubMed] [Google Scholar]
- 31. Rickles F. R., Levine M. N. (2001) Epidemiology of thrombosis in cancer. Acta Haematol. 106, 6–12 [DOI] [PubMed] [Google Scholar]
- 32. Kumar R., Gururaj A. E., Barnes C. J. (2006) p21-activated kinases in cancer. Nat. Rev. Cancer 6, 459–471 [DOI] [PubMed] [Google Scholar]
- 33. Ruf W., Disse J., Carneiro-Lobo T. C., Yokota N., Schaffner F. (2011) Tissue factor and cell signaling in cancer progression and thrombosis. J. Thromb. Haemost. 9, Suppl. 1, 306–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mackman N., Fowler B. J., Edgington T. S., Morrissey J. H. (1990) Functional analysis of the human tissue factor promoter and induction by serum. Proc. Natl. Acad. Sci. U.S.A. 87, 2254–2258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Chong C., Tan L., Lim L., Manser E. (2001) The mechanism of PAK activation. Autophosphorylation events in both regulatory and kinase domains control activity. J. Biol. Chem. 276, 17347–17353 [DOI] [PubMed] [Google Scholar]
- 36. Messeguer X., Escudero R., Farré D., Núñez O., Martínez J., Albà M. M. (2002) PROMO. Detection of known transcription regulatory elements using species-tailored searches. Bioinformatics 18, 333–334 [DOI] [PubMed] [Google Scholar]
- 37. Angus-Hill M. L., Elbert K. M., Hidalgo J., Capecchi M. R. (2011) T-cell factor 4 functions as a tumor suppressor whose disruption modulates colon cell proliferation and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 108, 4914–4919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Zheng R., Blobel G. A. (2010) GATA transcription factors and cancer. Genes Cancer 1, 1178–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tanikawa M., Wada-Hiraike O., Nakagawa S., Shirane A., Hiraike H., Koyama S., Miyamoto Y., Sone K., Tsuruga T., Nagasaka K., Matsumoto Y., Ikeda Y., Shoji K., Oda K., Fukuhara H., Nakagawa K., Kato S., Yano T., Taketani Y. (2011) Multifunctional transcription factor TFII-I is an activator of BRCA1 function. Br. J. Cancer 104, 1349–1355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Soprano D. R., Qin P., Soprano K. J. (2004) Retinoic acid receptors and cancers. Annu. Rev. Nutr. 24, 201–221 [DOI] [PubMed] [Google Scholar]
- 41. Khanna A., Okkeri J., Bilgen T., Tiirikka T., Vihinen M., Visakorpi T., Westermarck J. (2011) ETS1 mediates MEK1/2-dependent overexpression of cancerous inhibitor of protein phosphatase 2A (CIP2A) in human cancer cells. PloS ONE 6, e17979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Shen Q., Uray I. P., Li Y., Krisko T. I., Strecker T. E., Kim H. T., Brown P. H. (2008) The AP-1 transcription factor regulates breast cancer cell growth via cyclins and E2F factors. Oncogene 27, 366–377 [DOI] [PubMed] [Google Scholar]
- 43. Mackman N. (1996) Regulation of tissue factor gene expression in human monocytic and endothelial cells. Haemostasis 26, 17–19 [DOI] [PubMed] [Google Scholar]
- 44. Bierhaus A., Zhang Y., Deng Y., Mackman N., Quehenberger P., Haase M., Luther T., Müller M., Böhrer H., Greten J., et al. (1995) Mechanism of the tumor necrosis factor α-mediated induction of endothelial tissue factor. J. Biol. Chem. 270, 26419–26432 [DOI] [PubMed] [Google Scholar]
- 45. Felts S. J., Stoflet E. S., Eggers C. T., Getz M. J. (1995) Tissue factor gene transcription in serum-stimulated fibroblasts is mediated by recruitment of c-Fos into specific AP-1 DNA-binding complexes. Biochemistry 34, 12355–12362 [DOI] [PubMed] [Google Scholar]
- 46. Cui M. Z., Parry G. C., Edgington T. S., Mackman N. (1994) Regulation of tissue factor gene expression in epithelial cells. Induction by serum and phorbol 12-myristate 13-acetate. Arterioscler. Thromb. 14, 807–814 [DOI] [PubMed] [Google Scholar]
- 47. Adam L., Vadlamudi R., Mandal M., Chernoff J., Kumar R. (2000) Regulation of microfilament reorganization and invasiveness of breast cancer cells by kinase dead p21-activated kinase-1. J. Biol. Chem. 275, 12041–12050 [DOI] [PubMed] [Google Scholar]
- 48. Li T., Zhang J., Zhu F., Wen W., Zykova T., Li X., Liu K., Peng C., Ma W., Shi G., Dong Z., Bode A. M. (2011) p21-activated protein kinase (PAK2)-mediated c-Jun phosphorylation at five threonine sites promotes cell transformation. Carcinogenesis 32, 659–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Carson S. D., Archer P. G. (1986) Tissue factor activity in HeLa cells measured with a continuous chromogenic assay and ELISA reader. Thromb. Res. 41, 185–195 [DOI] [PubMed] [Google Scholar]
- 50. Carson S. D. (1996) Manifestation of cryptic fibroblast tissue factor occurs at detergent concentrations that dissolve the plasma membrane. Blood Coagul. Fibrinolysis 7, 303–313 [DOI] [PubMed] [Google Scholar]
- 51. Bajaj M. S., Steer S., Kuppuswamy M. N., Kisiel W., Bajaj S. P. (1999) Synthesis and expression of tissue factor pathway inhibitor by serum-stimulated fibroblasts, vascular smooth muscle cells, and cardiac myocytes. Thromb. Haemost. 82, 1663–1672 [PubMed] [Google Scholar]
- 52. Diebold I., Petry A., Djordjevic T., Belaiba R. S., Fineman J., Black S., Schreiber C., Fratz S., Hess J., Kietzmann T., Görlach A. (2010) Reciprocal regulation of Rac1 and PAK-1 by HIF-1α: a positive-feedback loop promoting pulmonary vascular remodeling. Antioxid. Redox Signal. 13, 399–412 [DOI] [PubMed] [Google Scholar]
- 53. Amirkhosravi A., Meyer T., Amaya M., Davila M., Mousa S. A., Robson T., Francis J. L. (2007) The role of tissue factor pathway inhibitor in tumor growth and metastasis. Semin. Thromb. Hemost. 33, 643–652 [DOI] [PubMed] [Google Scholar]
- 54. Hembrough T. A., Ruiz J. F., Papathanassiu A. E., Green S. J., Strickland D. K. (2001) Tissue factor pathway inhibitor inhibits endothelial cell proliferation via association with the very low density lipoprotein receptor. J. Biol. Chem. 276, 12241–12248 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.