

Background: Both p38 and c-Met are implicated in the tumorigenesis of cholangiocarcinoma.
Results: The inhibition of p38 inhibits human cholangiocarcinoma cell proliferation and invasion through reducing the basal activity of c-Met.
Conclusion: c-Met contributes to the pro-tumorigenic action of p38 in human cholangiocarcinoma cells.
Significance: We identify a novel molecular mechanism that contributes to understanding the pro-tumorigenic activity of p38 in human cholangiocarcinoma cells.
Keywords: Cancer Biology, Cell Biology, Cell Signaling, Molecular Biology, Oncogene
Abstract
Pro-tumorigenic function of the p38 kinase plays a critical role in human cholangiocarcinogenesis. However, the underlying mechanism remains incompletely understood. Here, we report that c-Met, the tyrosine kinase receptor for hepatocyte growth factor (HGF), contributes to the pro-tumorigenic ability of p38 in human cholangiocarcinoma cells. Both p38 and c-Met promote the proliferation and invasion of human cholangiocarcinoma cells. Importantly, inhibition or knockdown of p38 decreased the basal activation of c-Met. Tyrosine phosphatase inhibitor studies revealed that p38 promotes the activity of c-Met, at least in part, by inhibiting dephosphorylation of the receptor. Moreover, density enhanced phosphatase-1 (DEP-1) is involved in p38-mediated inhibiting dephosphorylation of c-Met. Furthermore, p38 inhibits the degradation of c-Met. Taken together, these data provide a potential mechanism to explain how p38 promotes human cholangiocarcinoma cell proliferation and invasion. We propose that the link between p38 and c-Met is implicated in the progression of human cholangiocarcinoma.
Introduction
Cholangiocarcinoma is highly aggressive malignancy of the bile ducts with poor prognosis (1, 2). Cholangiocarcinoma often arises from background conditions that cause long term inflammation, injury, and reparative biliary epithelial cell proliferation (3–7). Despite advances in therapy, the prognosis of cholangiocarcinoma remains very poor. It is well known that inhibition the proliferation and invasion of malignant biliary epithelial cells would be a potential strategy for the treatment of cholangiocarcinoma. However, little is known about the molecular mechanism responsible for proliferation and invasion of cholangiocarcinoma cells. Elucidation of the intracellular signaling events mediating proliferation and invasion of cholangiocarcinoma cells will contribute to the development of potential therapeutic targets for cholangiocarcinoma treatment.
Recent evidence suggests that the tyrosine kinase c-Met, the receptor for hepatocyte growth factor (HGF),3 is important in cholangiocarcinoma (6, 8–10). Overexpression of c-Met has been reported as a poor prognostic factor in cholangiocarcinoma patients (8, 11). Upon HGF binding, Tyr-1234 and Tyr-1235 residues in the catalytic domain of c-Met are phosphorylated and the receptor is activated (12). Once activated, c-Met activates multiple downstream signaling pathways, such as the PI3K/Akt and MEK/ERK pathways (13–17). Through these downstream pathways, HGF/c-Met governs numerous important cellular responses, including cellular proliferation, differentiation, and migration (18–21). In cancer cells, c-Met signaling plays an important role in initiating invasion and triggering metastasis (22). c-Met is a promising target for treatment of cholangiocarcinoma, but the mechanism for the deregulation of c-Met activity in human cholangiocarcinoma cells still remains unknown.
p38, a mitogen-activated protein kinase (MAPK), plays an important role in converting extracellular stimuli into a wide range of cellular responses, including inflammatory response, differentiation, proliferation, apoptosis, and survival (23–27). The role of p38 in tumor suppression has been proposed, and this function of p38 is mostly mediated by negative regulation of cell cycle progression and induction of apoptosis (23, 28–30). However, p38 also exerts oncogenic action through promoting proliferation, invasion, inflammation, and angiogenesis (31–33). The deregulation of p38 has been described in cholangiocarcinoma. Overexpression of p38δ has been observed in human cholangiocarcinomas, and elevated p38δ expression plays an important role in cholangiocarcinoma metastasis (34). p38 is essential for promoting the growth and maintaining the transformed cell phenotype in malignant human cholangiocytes (35–37). Moreover, activation of p38 in cultured cholangiocarcinoma cells results in decreasing CDK inhibitor p21WAF1/CIP1 expression, and promoting proliferation (36). These data suggest that p38 is a key oncogenic player in cholangiocarcinogenesis. Thus, it is necessary to uncover the detailed molecular mechanism responsible for the pro-tumorigenic function of p38 in cholangiocarcinoma.
In light of recent evidence showing that p38 is implicated in the pro-tumorigenic function of HGF/c-Met signaling (17, 38, 39), this study was designed to evaluate the link between c-Met and p38 in human cholangiocarcinoma cells. Our data reveal that both p38 and c-Met are required for proliferation and invasion in human cholangiocarcinoma cells. In human cholangiocarcinoma cells, p38 is not the downstream effector of c-Met. On the contrary, p38 is a key regulator of the activity of c-Met. p38 maintains high basal activity of c-Met, at least in part, through inhibiting dephosphorylation of the receptor. Moreover, p38 inhibits the degradation of c-Met upon protein synthesis inhibitor cycloheximide (CHX) treatment. Taken together, our findings suggest that c-Met contributes to pro-tumorigenic function of p38 in human cholangiocarcinoma cells.
MATERIALS AND METHODS
Chemicals and Antibodies
Cell counting kit-8 (CCK8) was purchased from Sigma. p38 MAPK inhibitor SB203580, PI3K inhibitor LY294002, MEK inhibitor U0126, JNK inhibitor SP600125, and the p38 agonist anisomycin were purchased from Merck. Recombinant human HGF was purchased from PeproTech. The c-Met inhibitor PF-2341066 was purchased from Selleck Chemicals. The human active DEP-1 kit was purchased from R&D Systems. DEP-1 and C-MET siRNA, BCA assay, and antibodies against DEP-1 and β-actin were purchased from Santa Cruz Biotechnology. P38 MAPK siRNA and antibodies against phospho-Akt (Ser-473), phospho-ERK (Thr-202/Tyr-204), phospho-Met (Tyr-1234/1235), phospho-p38 MAPK (Thr-180/Tyr-182), phospho-c-Jun, phospho-MAPKAPK2, Akt, ERK, c-Met, and p38 MAPK were purchased from Cell Signaling Technology. Constitutively active MKK6 plasmid was purchased from Addgene.
Cell Culture and Treatments
Human cholangiocarcinoma cell lines QBC939, RBE, and HCCC-9810 were cultured in RPMI 1640 medium, and hepatocellular carcinoma cell lines MHCC-97H and HepG2 were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in a humidified incubator containing 5% CO2 and 95% ambient air at 37 °C. The protocol used for P38, C-MET, and DEP-1 knockdown has been previously described (17).
Western Blot
Cells were lysed in Triton lysis buffer (20 mm Tris, pH 7.4, 137 mm NaCl, 10% glycerol, 1% Triton X-100, 2 mm EDTA, 1 mm PMSF, 10 mm sodium fluoride, 5 mg/ml of aprotinin, 20 mm leupeptin, and 1 mm sodium orthovanadate) and centrifuged at 12,000 × g for 15 min. Protein concentrations were measured using the BCA assay. Protein samples were denatured with 4× SDS-loading buffer (200 mm Tris, pH 6.8, 8% SDS, 400 mm DTT, 0.4% bromphenol blue, 40% glycerol) at 100 °C for 5 min and subjected to standard SDS-PAGE and Western blot analysis as previously described (17).
Cell Counting Kit-8 Assay
QBC939, RBE, and HCCC-9810 cells were trypsinized and seeded at 3 × 103 cells/well in 96-well plates. After 24 h, various doses of inhibitors were added and incubated for the indicated time periods. Then, 20 μl of CCK8 solution (5 g/liter) in phosphate-buffered saline (PBS) was added. After incubation for an additional 2 h, the absorbance value in each well was measured using a microculture plate reader (Bio-Tek, USA) at a wavelength of 490 nm.
Cell Migration and Invasion Assay
QBC939, RBE, and HCCC-9810 cells were transferred to 24-well Transwell chambers (Costar, Corning, NY). For migration assay, cells were seeded in the upper Transwell chamber at 1 × 105 cells/well. For the invasion assay, cells were seeded in the upper Matrigel-coated Transwell chamber at 2 × 105 cells/well. The inhibitors were added at the indicated concentrations used for the treatment, in both the upper and lower chambers. After incubation at 37 °C for the indicated time periods, migrated and invaded cells were fixed in 95% ethanol, stained with a solution of 2% crystal violet in 70% ethanol, and counted under an inverted microscope. Three fields were randomly chosen and the numbers of migrated and invaded cells were counted.
DEP-1 Activity Assay
DEP-1 activity was assayed in 96-well plates in a buffer containing final concentrations of 25 mm HEPES (pH 7.3), 5 mm DTT, and 10 μg/ml of bovine serum albumin (BSA) using DADEY(PO3)LIPQQ as a substrate. Activity was measured by following the increase in absorbance at 620 nm using a microculture plate reader (Bio-Tek, USA).
Statistical Analysis
Results were expressed as the mean ± S.D. Statistical analysis was performed using Student's t test. p < 0.05 was considered statistically significant.
RESULTS
p38 Promotes Proliferation and Invasion in Human Cholangiocarcinoma Cells
To investigate the role of p38 in cholangiocarcinoma, we first examined the expression of phosphorylated p38 in human cholangiocarcinoma cells. As shown in Fig. 1A, QBC939, RBE, and HCCC-9810 cells showed strong expression of phosphorylated p38. To determine the effects of p38 on proliferation and invasion on human cholangiocarcinoma cells, we treated QBC939, RBE, and HCCC-9810 cells with various doses of p38 selective inhibitor SB203580 for indicated time periods. Compared with dimethyl sulfoxide treatment, SB203580 inhibited the proliferation of human cholangiocarcinoma cells in a dose- and time-dependent manner (Fig. 1B). Furthermore, migration (Fig. 1C) and invasion (Fig. 1D) of human cholangiocarcinoma cells were significantly suppressed by SB203580 treatment. To confirm the effects of SB203580 on p38 activity inhibition, we assessed the levels of phosphorylated MAPKAPK2, a downstream effector of p38, in SB203580-treated human cholangiocarcinoma cells. The data showed that SB203580 inhibited the phosphorylation of MAPKAPK2 (Fig. 1, B–D), indicating that SB203580 blocked the activity of p38 in human cholangiocarcinoma cells. These data indicate that p38 is a potent promoter of proliferation and invasion in human cholangiocarcinoma cells.
FIGURE 1.

p38 promotes proliferation and invasion in human cholangiocarcinoma cells. A, Western blot analysis of phosphorylated p38 in human cholangiocarcinoma cell lines. B, p38 inhibition inhibits proliferation of human cholangiocarcinoma cells. QBC939, RBE, and HCCC-9810 cells were treated with various doses of SB203580 (SB) for the indicated time periods. Cell viability was determined by the CCK8 assay. The effects of SB203580 on p38 activity inhibition were measured by Western blot analysis. C and D, p38 inhibition suppresses migration and invasion of human cholangiocarcinoma cells. The migration (C) and invasion (D) of QBC939, RBE, and HCCC-9810 cells with or without SB203580 treatment were analyzed using a Transwell assay. The effects of SB203580 on p38 activity inhibition were measured by Western blot. Columns, mean of three individual experiments; bars, S.E. *, significantly different from control value.
p38 Maintains High Basal Activity of c-Met
Given that p38 is a downstream effector of HGF/c-Met signaling in some types of cancer (17, 38, 39), and HGF/c-Met has been reported to be involved in the carcinogenesis of cholangiocarcinoma (8–10), we tested the expression of phosphorylated c-Met in human cholangiocarcinoma cells. As shown in Fig. 2A, QBC939, RBE, and HCCC-9810 cells showed strong expression of phosphorylated c-Met. Then, the role of c-Met in regulating the activation of p38 was investigated. The data show that the phosphorylation of p38 is not regulated by c-Met (supplemental Fig. S1), suggesting that p38 is not the downstream effector of c-Met in human cholangiocarcinoma cells.
FIGURE 2.

p38 maintains high basal activation of c-Met. A, Western blot analysis of phosphorylated c-Met in human cholangiocarcinoma cell lines. B and C, p38 inhibition decreases phosphorylated c-Met. After treatment with various doses of SB203580 (SB) for 12 h (B) or treatment with SB203580 (SB, 10 μm) for the indicated time periods (C), QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis. D, p38 siRNA decreases phosphorylated c-Met. After transfection with p38 siRNA for 60 h, QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis. E, Western blot analysis of phosphorylated c-Met and p38 in human hepatocellular carcinoma cell lines. F, p38 inhibition decreases phosphorylated c-Met in MHCC-97H cells. After treatment with SB203580 (10 μm) for the indicated time periods, MHCC-97H cells were subjected to Western blot analysis. G, constitutively active MKK6 increases phosphorylated c-Met in HGF-treated HepG2 cells. After HGF (0.02 mg) treatment for 1 h with or without MKK6 pro-transfection for 24 h, serum-starved HepG2 cells were subjected to Western blot analysis.
We addressed whether p38 participates in regulating the activation of c-Met. The levels of phosphorylated c-Met were measured in p38 inhibitor SB203580-treated human cholangiocarcinoma cells. Fig. 2, B and C, showed that SB203580 decreased the levels of phosphorylated c-Met in human cholangiocarcinoma cells in a dose- and time-dependent manner. Moreover, suppression of p38 expression also decreased phosphorylated c-Met in human cholangiocarcinoma cells (Fig. 2D). To further confirm the role of p38 in sustaining the levels of phosphorylated c-Met, MHCC-97H and HepG2 cells were used in our study. As shown in Fig. 2E, strong expression of phosphorylated c-Met was detected in MHCC-97H cells, but not in HepG2 cells. In comparison with HepG2, MHCC-97H cells showed strong expression of phosphorylated p38 (Fig. 2E). Importantly, p38 inhibition decreased the levels of phosphorylated c-Met in MHCC-97H cells (Fig. 2F). Moreover, HGF-induced c-Met activation was increased by a constitutively active MKK6 expression vector in HepG2 cells (Fig. 2G). These data indicate that p38 plays an important role in sustaining c-Met activity.
Because p38 is essential for HGF expression and secretion, we further investigated whether basal activation of c-Met was due to p38-mediated autocrine secretion of HGF. The enzyme-linked immunosorbent assay (ELISA) analysis failed to demonstrate any HGF secreted by QBC939, RBE, and HCCC-9810 cells (supplemental ”Materials and Methods“ and data not shown). As the mutations in the C-MET gene correlate with autoactivation of the receptor in multiple different cancers (40–42), we investigated whether activation of c-Met in human cholangiocarcinoma cells was due to a mutation. Sequencing the C-MET gene demonstrated that none of the reported mutations was found in human cholangiocarcinoma cells (supplemental ”Materials and Methods“ and data not shown). Taken together, these results suggest that p38 is responsible for sustained high basal activity of c-Met independent of HGF production in human cholangiocarcinoma cells.
p38 Inhibits the Dephosphorylation of c-Met
To confirm that c-Met can be phosphorylated by p38, the p38 agonist anisomycin was used in our study. Compared with HGF treatment, anisomycin had no effects on c-Met activation in serum-starved HCCC-9810 cells (supplemental Fig. S2), indicating that c-Met cannot be phosphorylated by p38. To determine whether p38 inhibits the dephosphorylation of c-Met, we investigated the effects of the tyrosine phosphatase inhibitor sodium orthovanadate on p38 inhibition-mediated c-Met inactivation. As shown in Fig. 3A, sodium orthovanadate inhibited SB203580-mediated c-Met inactivation in human cholangiocarcinoma cells. However, sodium orthovanadate had no effect on the inhibition on the c-Met inhibitor PF-2341066-mediated c-Met dephosphorylation (Fig. 3A). Together, this data suggest that p38 promotes basal activation of c-Met through inhibiting its dephosphorylation. To further confirm the role of p38 in inhibiting c-Met dephosphorylation, the effects of p38 inhibition on HGF-induced c-Met activation were investigated. The results revealed that the p38 inhibitor SB203580 decreased phosphorylation of c-Met upon HGF engagement in serum-starved HCCC-9810 cells (Fig. 3B). More importantly, SB203580-mediated down-regulation of c-Met phosphorylation was inhibited by sodium orthovanadate in HGF-treated HCCC-9810 cells (Fig. 3C), confirming that p38 maintains high basal activation of c-Met, at least in part, through preventing c-Met dephosphorylation.
FIGURE 3.

p38 inhibits dephosphorylation of c-Met. A, sodium orthovanadate inhibits p38 inhibition-induced down-regulation of phosphorylated c-Met. After treatment with SB203580 (SB, 10 μm) or PF-2341066 (PF, 100 nm) for 10 h, QBC939, RBE, and HCCC-9810 cells were treated with sodium orthovanadate (Na3VO4, 100 μm) for another 2 h and then subjected to Western blot analysis. B, p38 inhibition decreases phosphorylated c-Met upon HGF treatment. After treatment with HGF (0.02 mg) for the indicated time periods with or without SB203580 (SB, 10 μm) preincubation for 1 h, serum-starved HCCC-9810 cells were subjected to Western blot analysis. C, sodium orthovanadate inhibits p38 inhibition-induced down-regulation of phosphorylated c-Met upon HGF treatment. After HGF (0.02 mg) treatment for 0.5 h with or without SB203580 (SB, 10 μm) preincubation for 1 h, serum-starved HCCC-9810 cells were treated with sodium orthovanadate (Na3VO4, 100 μm) for another 0.5 h and then subjected to Western blot analysis. D, P38 inhibits DEP-1 activity. After treatment with SB203580 (10 μm) for 12 h or p38 siRNA for 60 h, QBC939, RBE, and HCCC-9810 cells were subjected to DEP-1 activity analysis using human active DEP-1 kit. E, DEP-1 siRNA inhibits p38 inhibition-induced down-regulation of phosphorylated c-Met. After treatment with SB203580 (10 μm) for 12 h with or without DEP-1 siRNA pre-transfection for 60 h, QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis. Columns, mean of three individual experiments; bars, S.E. *, significantly different from control value.
Given that density-enhanced phosphatase-1 (DEP-1) plays an essential role in the dephosphorylation of c-Met (43–45), we postulated that p38 may inhibit the activity of DEP-1. To evaluate this hypothesis, we examined the potential effects of p38 on the activity of DEP-1 in human cholangiocarcinoma cells. As shown in Fig. 3D, both p38 inhibitor SB203580 and P38 siRNA increased the activity of DEP-1. Furthermore, suppression of the expression of DEP-1 inhibited c-Met dephosphorylation upon SB203580 treatment (Fig. 3E). These findings indicate that DEP-1 is involved in p38 inhibition-induced c-Met inactivation in human cholangiocarcinoma cells.
p38 Inhibits the Degradation of c-Met
It has been shown that internalization and proteasome-mediated degradation of c-Met are important in negatively regulating c-Met signaling (46–50). To test whether p38 is involved in regulating the stability of c-Met, CHX was employed. The data showed that p38 inhibition promoted the degradation of c-Met in response to CHX (Fig. 4A). The role of p38 in regulating the stability of c-Met in human cholangiocarcinoma cells was confirmed by suppression of the p38 expression (Fig. 4B). Furthermore, constitutively active MKK6 expression induced p38 activation and inhibited the degradation of c-Met upon CHX treatment (Fig. 4, C and D). Because the phosphorylation of c-Met is an important trigger for receptor degradation (17, 46), the role of the c-Met inhibitor PF-2341066 in the regulation of p38-mediated c-Met stability was investigated. It is notable that SB203580-promoted c-Met degradation was blocked by PF-2341066 pre-treatment (Fig. 4E). This data implies that p38 can positively regulate c-Met signaling by inhibiting its degradation.
FIGURE 4.

p38 inhibits the degradation of c-Met. A and B, p38 inhibition and knockdown promote the degradation of c-Met. After treatment with SB203580 (SB, 10 μm) for 1 h or P38 siRNA for 60 h, QBC939, RBE, and HCCC-9810 cells were treated with CHX (10 μm) for the indicated time periods and then subjected to Western blot analysis. C and D, constitutively active MKK6 inhibits the degradation of c-Met. After MKK6 was transfected for 24 h, HepG2 cells were treated with CHX (10 μm) for another 48 h and then subjected to Western blot analysis. E, c-Met inhibition inhibits c-Met degradation induced by p38 inhibition. After treatment with CHX (10 μm) for 48 h with or without SB203580 (10 μm) and PF-2341066 (PF, 100 nm) preincubation for 1 h, QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis.
c-Met Promotes Proliferation and Invasion of Human Cholangiocarcinoma Cells
To confirm the tumorigenic role of c-Met in human cholangiocarcinoma cells, we treated QBC939, RBE, and HCCC-9810 cells with different concentrations of the c-Met selective inhibitor PF-2341066 for the indicated time periods. Compared with dimethyl sulfoxide treatment, PF-2341066 inhibited the proliferation of human cholangiocarcinoma cells in a dose- and time-dependent manner (Fig. 5A). The role of c-Met in promoting human cholangiocarcinoma cell proliferation was confirmed by the suppression of c-Met expression (Fig. 5A). Furthermore, migration (Fig. 5B) and invasion (Fig. 5C) of human cholangiocarcinoma cells was significantly suppressed by c-Met blocking or knockdown. The effects of inhibitor and siRNA on c-Met inhibition were confirmed by Western blot (Fig. 5D). Thus, c-Met is a potent promoter of proliferation and invasion of human cholangiocarcinoma cells.
FIGURE 5.

c-Met promotes proliferation and invasion of human cholangiocarcinoma cells. A, c-Met inhibition inhibits human cholangiocarcinoma cell proliferation. QBC939, RBE, and HCCC-9810 cells were treated with PF-2341066 (PF, 50/100 nm) for the indicated time periods. Cell viability was determined by a CCK8 assay. B and C, c-Met inhibition and knockdown suppresses migration and invasion of human cholangiocarcinoma cells. The migration (C) and invasion (D) of QBC939, RBE, and HCCC-9810 cells with or without PF-2341066 (50/100 nm) and siMet treatment were analyzed using a Transwell assay. Columns, mean of three individual experiments; bars, S.E. *, significantly different from control value. D, the effects of PF-2341066 and C-MET siRNA on c-Met inhibition and knockdown were measured by Western blot.
p38 Inhibition Down-regulates c-Met Downstream Pathways
As PI3K/Akt and MEK/ERK are two typical downstream pathways of c-Met (13, 14, 17), we tested whether the phosphorylation of Akt and ERK are c-Met-dependent in human cholangiocarcinoma cells. The results showed that c-Met activity inhibition and its expression suppression obviously decreased the phosphorylation of Akt and ERK (Fig. 6, A-C), indicating that PI3K/Akt and MEK/ERK are downstream pathways of c-Met in human cholangiocarcinoma cells.
FIGURE 6.

p38 up-regulates c-Met downstream pathways. A and B, c-Met inhibition inhibits PI3K/Akt (A) and MEK/ERK (B) activation. After treated with PF-2341066 (PF, 100 nm) for the indicated time periods, QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis. C, C-MET siRNA inhibits PI3K/Akt and MEK/ERK activation. After transfection with C-MET siRNA for 60 h, QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis. D and E, p38 inhibition inhibits PI3K/Akt (D) and MEK/ERK (E) activation. After treatment with SB203580 (SB, 10 μm) for the indicated time periods, QBC939, RBE, and HCCC-9810 cells were subjected to Western blot analysis. LY294002 (LY, 20 μm) and U0126 (10 μm) were used as the positive control.
Considering the role of p38 in regulating the activity of c-Met, we investigated the effects of p38 activity blocking on the phosphorylation of c-Met downstream effectors Akt and ERK in human cholangiocarcinoma cells. As shown in Fig. 6, D and E, p38 inhibition down-regulated the levels of phosphorylated Akt and ERK, suggesting that p38 promotes the activity of Akt and ERK, at least in part, through c-Met.
The PI3K/Akt and MEK/ERK Pathways Promote Proliferation and Invasion of Human Cholangiocarcinoma Cells
To explore the role of c-Met downstream pathways PI3K/Akt and MEK/ERK in cholangiocarcinoma, we tested the effects of PI3K/Akt and MEK/ERK inhibition on proliferation and invasion of human cholangiocarcinoma cells. The results showed that both PI3K inhibitor LY294002 and MEK inhibitor U0126 inhibited the proliferation of human cholangiocarcinoma cells (Fig. 7A). Furthermore, migration and invasion of human cholangiocarcinoma cells were significantly suppressed by blocking the activity of PI3K and MEK (Fig. 7, B and C). These data indicate that c-Met downstream pathways PI3K/Akt and MEK/ERK are involved in the tumorigenesis of cholangiocarcinoma.
FIGURE 7.

c-Met downstream pathways promote proliferation and invasion of human cholangiocarcinoma cells. A, PI3K/Akt and MEK/ERK inhibition inhibit human cholangiocarcinoma cell proliferation. QBC939, RBE, and HCCC-9810 cells were treated with LY294002 (LY, 20 μm) and U0126 (10 μm) for the indicated time periods. Cell viability was determined by a CCK8 assay. B and C, PI3K/Akt and MEK/ERK inhibition suppress migration and invasion of human cholangiocarcinoma cells. The migration (C) and invasion (D) of QBC939, RBE, and HCCC-9810 cells with or without LY294002 (LY, 20 μm) and U0126 (10 μm) treatment were analyzed using a Transwell assay. Columns, mean of three individual experiments; bars, S.E. *, significantly different from control value.
c-Met Is Involved in the Pro-tumorigenic Function of P38
Because c-Met is a downstream pathway of p38, we investigated whether the protumorigenic action of p38 is dependent on c-Met signaling in human cholangiocarcinoma cells. The results showed that C-MET knockdown and p38 blocking had no synergy effect on the inhibition of RBE cell invasion (Fig. 8A), implying that c-Met is involved in the oncogenic function of p38. To further confirm the role of c-Met in the pro-tumorigenic activity of p38, the constitutively active MKK6 expression vector was used to activate p38 in RBE cells. The results showed that both RBE cell invasion and Akt or ERK activation promoted by MKK6/p38 were inhibited by c-Met blocking (Fig. 8, B and C). Moreover, c-Met blocking-mediated inhibition of RBE cell invasion and inactivation of Akt and ERK were attenuated by the activation of MKK6/p38 (Fig. 8, B and C), indicating that p38 promotes carcinogenesis of cholangiocarcinoma is partially, but not totally, dependent on c-Met. Taken together, these data suggest that p38 promotes the development of cholangiocarcinoma, at least in part, through sustaining the activity of c-Met.
FIGURE 8.

c-Met is involved in the protumorigenic function of p38. A, C-MET knockdown and p38 blocking had no synergy effect on the inhibition of RBE cell invasion. After C-MET siRNA treatment for 60 h, RBE cells were treated with SB203580 (SB, 10 μm) for another 20 h. Then, the invasion of RBE cells were analyzed using Transwell assay. Columns, mean of three individual experiments; bars, S.E. *, significantly different from control value. B, c-Met inhibition inhibits the invasion of RBE cells induced by MKK6/p38. After constitutively active MKK6 treatment for 24 h, RBE cells were treated with PF-2341066 (PF, 100 nm) for another 20 h. Then, the invasion of RBE cells were analyzed using a Transwell assay. Columns, mean of three individual experiments; bars, S.E. *, significantly different from control value. C, c-Met inhibition inhibits the activation of Akt and ERK induced by MKK6/p38. After constitutively active MKK6 treatment for 24 h, RBE cells were treated with PF-2341066 (PF, 100 nm) for another 6 h. Then, RBE cells were subjected to Western blot analysis.
DISCUSSION
Although p38 is implicated in the carcinogenesis and progression of cholangiocarcinoma, it is unclear how p38 exerts its pro-tumorigenic action. The present work reveals that p38 promotes proliferation and invasion of human cholangiocarcinoma cells, at least in part, through sustaining high basal activation of c-Met.
c-Met has been reported to be associated with a wide variety of human malignancies, including colon, gastric, lung, breast, prostate, liver, and bile duct cancer (51–53). Based on the data that blocking c-Met by PF-2341066 inhibited proliferation and invasion of human cholangiocarcinoma cells, we suggest c-Met can exert its oncogenic potential both by stimulating cell proliferation and promoting invasion. In human cholangiocarcinoma cells, the PI3K/Akt and MEK/ERK pathways were partially regulated by c-Met, indicating the PI3K/Akt and MEK/ERK pathways are involved in the oncogenic activity of c-Met. c-Met signaling plays an essential role in the development of cholangiocarcinoma, but the detailed mechanism of the aberrant activation of c-Met remains unknown. The aberrant activation of c-Met is strongly associated with overexpression of HGF/c-Met, and C-MET gene mutation, rearrangement, or amplification (39). As the phosphorylation of c-Met is independent of HGF engagement and C-MET gene mutation in human cholangiocarcinoma cells, it is necessary to address the mechanism how human cholangiocarcinoma cells sustain high basal activity of c-Met.
It has been reported that the p38 pathway, which is involved in the development of cholangiocarcinoma, is one of the c-Met downstream pathways (39). Thus, we tested the potential link between p38 and c-Met. As our data showed that p38 is not regulated by c-Met, we suggest that p38 is not the effector of c-Met in the tumorigenesis of human cholangiocarcinoma. Interestingly, both blocking the activity and suppression of the expression of p38 decreased basal c-Met phosphorylation in human cholangiocarcinoma cells. These data suggest that c-Met is the downstream pathway, but not upstream pathway, of p38 in human cholangiocarcinoma cells. The role of p38 in sustaining the activity of c-Met was confirmed by our data that MKK6/p38 activation increased the activation of c-Met upon HGF treatment. Considering that both the PI3K/Akt and MEK/ERK pathways were involved in the oncogenic role of c-Met and p38 in human cholangiocarcinoma cells, it is reasonable to suggest that p38 can exert pro-tumorigenic effects through c-Met/Akt and c-Met/ERK signaling. Because inhibition of c-Met tyrosine kinase activity by PF-2341066 attenuated Akt or ERK activation as well as the promotion of cell invasion induced by MKK6/p38, it is reasonable to draw the conclusion that p38 promotes the carcinogenesis of cholangiocarcinoma, at least in part, through sustaining the activity of c-Met.
An important question now before us is how p38 sustains high basal activation of c-Met. Based on our findings, we propose that p38 cannot phosphorylate c-Met. As protein-tyrosine phosphatase has been implicated in negatively regulating HGF/c-Met signaling by dephosphorylating the phosphotyrosine residues of c-Met, we suspected that p38 might inhibit protein-tyrosine phosphatase-mediated dephosphorylation of c-Met. This hypothesis was supported by the data, which demonstrated that the tyrosine phosphatase inhibitor sodium orthovanadate inhibited the inactivation of c-Met induced by p38 inhibition. The role of p38 in inhibiting c-Met dephosphorylation was further supported by the data that inhibition of p38 up-regulated the activity of c-Met phosphatase DEP-1. Moreover, DEP-1 knockdown suppressed c-Met dephosphorylation induced by p38 inhibition, confirming that p38 can sustain high basal activity of c-Met through inhibiting its phosphatase. Furthermore, inhibition of other MAPK family members MEK and JNK did not affect the phosphorylation levels of c-Met (supplemental Fig. S3), indicating the effects of p38 in c-Met activity regulating is specific.
In addition to dephosphorylation, the degradation of c-Met also plays an essential role in negatively regulating HGF/c-Met signaling (46). Thus, the role of p38 in regulating the stability of c-Met was tested. It is notable that neither p38 inhibition nor p38 activation had effects on the protein level of c-Met. However, p38 inhibited the degradation of c-Met in CHX-treated human cholangiocarcinoma cells. Furthermore, p38 inhibition-promoted c-Met degradation was abolished by c-Met inhibitor pretreatment. Further studies are needed to investigate the mechanism of this effect of c-Met inhibition on c-Met degradation upon p38 blocking. Thus, we suggest that inhibiting the degradation c-Met also is involved in the role of p38 in positively regulating HGF/c-Met signaling.
In brief, the present work reveals that c-Met contributes to the protumorigenic function of p38 in human cholangiocarcinoma cells. High levels of phosphorylated p38 and c-Met are found in human cholangiocarcinoma cells. Inhibition of c-Met dephosphorylation by p38 is required for maintaining the sustained c-Met activation. Sustained c-Met activation results in constitutive activation of c-Met downstream pathways, which leads to proliferation and invasion of human cholangiocarcinoma cells (Fig. 9). More detailed studies on the mechanism of p38 and c-Met aberrant activation and their role in cholangiocarcinoma will contribute to the understanding of molecular mechanisms of cholangiocarcinogenesis and the development of new therapeutic strategies against cholangiocarcinoma.
FIGURE 9.
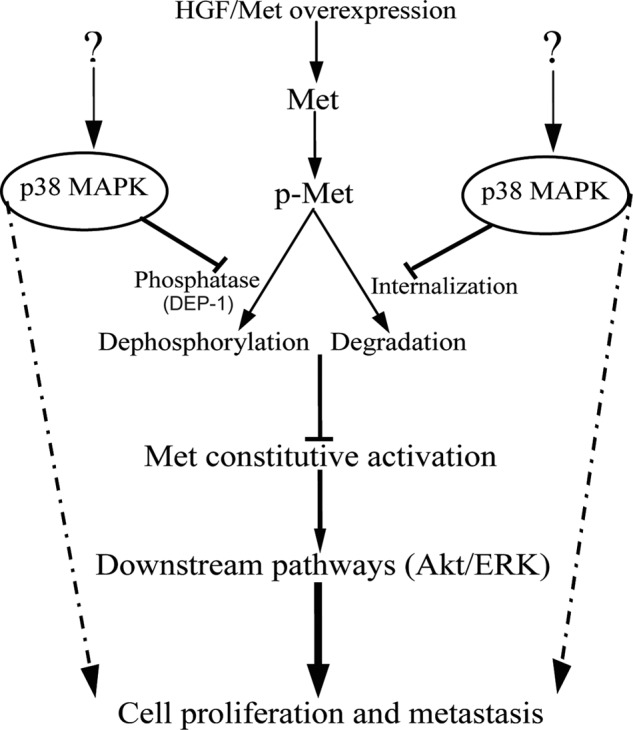
Proposed mechanism for p38-mediated proliferation and invasion of human cholangiocarcinoma cells. The activation of c-Met signaling is controlled by phosphorylation, dephosphorylation, and receptor degradation. Deregulated p38 activity maintains high basal c-Met activation, at least in part, through inhibiting its dephosphorylation and degradation. p38-mediated c-Met sustained activation results in constitutive activation of c-Met downstream pathways, which leads to proliferation and invasion of human cholangiocarcinoma cells.
Supplementary Material
This work was supported by National Natural Science Foundation of China Grants 81000886, 81170377, 81101714, 30921006, and 91029732, State Key Project for Infectious Diseases Grant 2012ZX10002-009 and -011, New Century Excellent Talents in University Grant NCET-11-1058, and State Key Laboratory of Oncogenes and Related Genes Grants 91-10-02, 90-10-03, and 10JC1418500.

This article contains supplemental ”Materials and Methods“ and Figs. S1–S3.
- HGF
- hepatocyte growth factor
- CCK8
- cell counting kit-8
- DMSO
- dimethyl sulfoxide
- DEP-1
- density-enhanced phosphatase-1
- CHX
- cycloheximide.
REFERENCES
- 1. Figueras J., Valls C., Jaurrieta E. (2000) Biliary tract cancers. N. Engl. J. Med. 342, 663–664 [DOI] [PubMed] [Google Scholar]
- 2. Olnes M. J., Erlich R. (2004) A review and update on cholangiocarcinoma. Oncology 66, 167–179 [DOI] [PubMed] [Google Scholar]
- 3. Han C., Wu T. (2005) Cyclooxygenase-2-derived prostaglandin E2 promotes human cholangiocarcinoma cell growth and invasion through EP1 receptor-mediated activation of the epidermal growth factor receptor and Akt. J. Biol. Chem. 280, 24053–24063 [DOI] [PubMed] [Google Scholar]
- 4. Patel T. (2006) Cholangiocarcinoma. Nat. Clin. Pract. Gastroenterol. Hepatol. 3, 33–42 [DOI] [PubMed] [Google Scholar]
- 5. Wehbe H., Henson R., Meng F., Mize-Berge J., Patel T. (2006) Interleukin-6 contributes to growth in cholangiocarcinoma cells by aberrant promoter methylation and gene expression. Cancer Res. 66, 10517–10524 [DOI] [PubMed] [Google Scholar]
- 6. Farazi P. A., Zeisberg M., Glickman J., Zhang Y., Kalluri R., DePinho R. A. (2006) Chronic bile duct injury associated with fibrotic matrix microenvironment provokes cholangiocarcinoma in p53-deficient mice. Cancer Res. 66, 6622–6627 [DOI] [PubMed] [Google Scholar]
- 7. Xu L., Han C., Wu T. (2006) A novel positive feedback loop between peroxisome proliferator-activated receptor-δ and prostaglandin E2 signaling pathways for human cholangiocarcinoma cell growth. J. Biol. Chem. 281, 33982–33996 [DOI] [PubMed] [Google Scholar]
- 8. Terada T., Nakanuma Y., Sirica A. E. (1998) Immunohistochemical demonstration of MET overexpression in human intrahepatic cholangiocarcinoma and in hepatolithiasis. Hum. Pathol. 29, 175–180 [DOI] [PubMed] [Google Scholar]
- 9. Leelawat K., Leelawat S., Tepaksorn P., Rattanasinganchan P., Leungchaweng A., Tohtong R., Sobhon P. (2006) Involvement of c-Met/hepatocyte growth factor pathway in cholangiocarcinoma cell invasion and its therapeutic inhibition with small interfering RNA specific for c-Met. J. Surg. Res. 136, 78–84 [DOI] [PubMed] [Google Scholar]
- 10. Sirica A. E. (2005) Cholangiocarcinoma. Molecular targeting strategies for chemoprevention and therapy. Hepatology 41, 5–15 [DOI] [PubMed] [Google Scholar]
- 11. Miyamoto M., Ojima H., Iwasaki M., Shimizu H., Kokubu A., Hiraoka N., Kosuge T., Yoshikawa D., Kono T., Furukawa H., Shibata T. (2011) Prognostic significance of overexpression of c-Met oncoprotein in cholangiocarcinoma. Br. J. Cancer 105, 131–138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ferracini R., Longati P., Naldini L., Vigna E., Comoglio P. M. (1991) Identification of the major autophosphorylation site of the Met/hepatocyte growth factor receptor tyrosine kinase. J. Biol. Chem. 266, 19558–19564 [PubMed] [Google Scholar]
- 13. Boccaccio C., Comoglio P. M. (2006) Invasive growth. A MET-driven genetic programme for cancer and stem cells. Nat. Rev. Cancer 6, 637–645 [DOI] [PubMed] [Google Scholar]
- 14. You H., Ding W., Dang H., Jiang Y., Rountree C. B. (2011) c-Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 54, 879–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Graziani A., Gramaglia D., Cantley L. C., Comoglio P. M. (1991) The tyrosine-phosphorylated hepatocyte growth factor/scatter factor receptor associates with phosphatidylinositol 3-kinase. J. Biol. Chem. 266, 22087–22090 [PubMed] [Google Scholar]
- 16. Ponzetto C., Bardelli A., Zhen Z., Maina F., dalla Zonca P., Giordano S., Graziani A., Panayotou G., Comoglio P. M. (1994) A multifunctional docking site mediates signaling and transformation by the hepatocyte growth factor/scatter factor receptor family. Cell 77, 261–271 [DOI] [PubMed] [Google Scholar]
- 17. Dai R., Li J., Fu J., Chen Y., Yu L., Zhao X., Qian Y., Zhang H., Chen H., Ren Y., Su B., Luo T., Zhu J., Wang H. (2012) Disturbance of Ca2+ homeostasis converts Pro-Met into non-canonical tyrosine kinase p190MetNC in response to endoplasmic reticulum stress in MHCC97 cells. J. Biol. Chem. 287, 14586–14597 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ma P. C., Maulik G., Christensen J., Salgia R. (2003) c-Met. Structure, functions and potential for therapeutic inhibition. Cancer Metastasis Rev. 22, 309–325 [DOI] [PubMed] [Google Scholar]
- 19. Forte G., Minieri M., Cossa P., Antenucci D., Sala M., Gnocchi V., Fiaccavento R., Carotenuto F., De Vito P., Baldini P. M., Prat M., Di Nardo P. (2006) Hepatocyte growth factor effects on mesenchymal stem cells. Proliferation, migration, and differentiation. Stem Cells 24, 23–33 [DOI] [PubMed] [Google Scholar]
- 20. Gay B., Suarez S., Weber C., Rahuel J., Fabbro D., Furet P., Caravatti G., Schoepfer J. (1999) Effect of potent and selective inhibitors of the Grb2 SH2 domain on cell motility. J. Biol. Chem. 274, 23311–23315 [DOI] [PubMed] [Google Scholar]
- 21. Tajima H., Matsumoto K., Nakamura T. (1992) Regulation of cell growth and motility by hepatocyte growth factor and receptor expression in various cell species. Exp. Cell Res. 202, 423–431 [DOI] [PubMed] [Google Scholar]
- 22. Zhang Y. W., Su Y., Volpert O. V., Vande Woude G. F. (2003) Hepatocyte growth factor/scatter factor mediates angiogenesis through positive VEGF and negative thrombospondin 1 regulation. Proc. Natl. Acad. Sci. U.S.A. 100, 12718–12723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wagner E. F., Nebreda A. R. (2009) Signal integration by JNK and p38 MAPK pathways in cancer development. Nat. Rev. Cancer 9, 537–549 [DOI] [PubMed] [Google Scholar]
- 24. Maher P. (1999) p38 mitogen-activated protein kinase activation is required for fibroblast growth factor-2-stimulated cell proliferation but not differentiation. J. Biol. Chem. 274, 17491–17498 [DOI] [PubMed] [Google Scholar]
- 25. Enslen H., Raingeaud J., Davis R. J. (1998) Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. J. Biol. Chem. 273, 1741–1748 [DOI] [PubMed] [Google Scholar]
- 26. Matsumoto T., Yokote K., Tamura K., Takemoto M., Ueno H., Saito Y., Mori S. (1999) Platelet-derived growth factor activates p38 mitogen-activated protein kinase through a Ras-dependent pathway that is important for actin reorganization and cell migration. J. Biol. Chem. 274, 13954–13960 [DOI] [PubMed] [Google Scholar]
- 27. Ono K., Han J. (2000) The p38 signal transduction pathway. Activation and function. Cell. Signal. 12, 1–13 [DOI] [PubMed] [Google Scholar]
- 28. Bulavin D. V., Fornace A. J., Jr. (2004) p38 MAP kinase's emerging role as a tumor suppressor. Adv. Cancer Res. 92, 95–118 [DOI] [PubMed] [Google Scholar]
- 29. Hui L., Bakiri L., Stepniak E., Wagner E. F. (2007) p38α. A suppressor of cell proliferation and tumorigenesis. Cell Cycle 6, 2429–2433 [DOI] [PubMed] [Google Scholar]
- 30. Kogai T., Liu Y. Y., Mody K., Shamsian D. V., Brent G. A. (2012) Regulation of sodium iodide symporter gene expression by Rac1/p38β mitogen-activated protein kinase signaling pathway in MCF-7 breast cancer cells. J. Biol. Chem. 287, 3292–3300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Platanias L. C. (2003) MAP kinase signaling pathways and hematologic malignancies. Blood 101, 4667–4679 [DOI] [PubMed] [Google Scholar]
- 32. Lee R. J., Albanese C., Stenger R. J., Watanabe G., Inghirami G., Haines G. K., 3rd, Webster M., Muller W. J., Brugge J. S., Davis R. J., Pestell R. G. (1999) pp60v-src induction of cyclin D1 requires collaborative interactions between the extracellular signal-regulated kinase, p38, and Jun kinase pathways. A role for cAMP response element-binding protein and activating transcription factor-2 in pp60v-src signaling in breast cancer cells. J. Biol. Chem. 274, 7341–7350 [DOI] [PubMed] [Google Scholar]
- 33. Neve R. M., Holbro T., Hynes N. E. (2002) Distinct roles for phosphoinositide 3-kinase, mitogen-activated protein kinase and p38 MAPK in mediating cell cycle progression of breast cancer cells. Oncogene 21, 4567–4576 [DOI] [PubMed] [Google Scholar]
- 34. Tan F. L., Ooi A., Huang D., Wong J. C., Qian C. N., Chao C., Ooi L., Tan Y. M., Chung A., Cheow P. C., Zhang Z., Petillo D., Yang X. J., Teh B. T. (2010) p38δ/MAPK13 as a diagnostic marker for cholangiocarcinoma and its involvement in cell motility and invasion. Int. J. Cancer 126, 2353–2361 [DOI] [PubMed] [Google Scholar]
- 35. Park J., Tadlock L., Gores G. J., Patel T. (1999) Inhibition of interleukin 6-mediated mitogen-activated protein kinase activation attenuates growth of a cholangiocarcinoma cell line. Hepatology 30, 1128–1133 [DOI] [PubMed] [Google Scholar]
- 36. Tadlock L., Patel T. (2001) Involvement of p38 mitogen-activated protein kinase signaling in transformed growth of a cholangiocarcinoma cell line. Hepatology 33, 43–51 [DOI] [PubMed] [Google Scholar]
- 37. Yamagiwa Y., Marienfeld C., Tadlock L., Patel T. (2003) Translational regulation by p38 mitogen-activated protein kinase signaling during human cholangiocarcinoma growth. Hepatology 38, 158–166 [DOI] [PubMed] [Google Scholar]
- 38. Recio J. A., Merlino G. (2002) Hepatocyte growth factor/scatter factor activates proliferation in melanoma cells through p38 MAPK, ATF-2, and cyclin D1. Oncogene 21, 1000–1008 [DOI] [PubMed] [Google Scholar]
- 39. Trusolino L., Bertotti A., Comoglio P. M. (2010) MET signalling. Principles and functions in development, organ regeneration and cancer. Nat. Rev. Mol. Cell Biol. 11, 834–848 [DOI] [PubMed] [Google Scholar]
- 40. Jeffers M., Fiscella M., Webb C. P., Anver M., Koochekpour S., Vande Woude G. F. (1998) The mutationally activated Met receptor mediates motility and metastasis. Proc. Natl. Acad. Sci. U.S.A. 95, 14417–14422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmidt L., Junker K., Nakaigawa N., Kinjerski T., Weirich G., Miller M., Lubensky I., Neumann H. P., Brauch H., Decker J., Vocke C., Brown J. A., Jenkins R., Richard S., Bergerheim U., Gerrard B., Dean M., Linehan W. M., Zbar B. (1999) Novel mutations of the MET proto-oncogene in papillary renal carcinomas. Oncogene 18, 2343–2350 [DOI] [PubMed] [Google Scholar]
- 42. Jeffers M., Schmidt L., Nakaigawa N., Webb C. P., Weirich G., Kishida T., Zbar B., Vande Woude G. F. (1997) Activating mutations for the Met tyrosine kinase receptor in human cancer. Proc. Natl. Acad. Sci. U.S.A. 94, 11445–11450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Palka H. L., Park M., Tonks N. K. (2003) Hepatocyte growth factor receptor tyrosine kinase met is a substrate of the receptor protein-tyrosine phosphatase DEP-1. J. Biol. Chem. 278, 5728–5735 [DOI] [PubMed] [Google Scholar]
- 44. Wang Z., Wang M., Carr B. I. (2010) Involvement of receptor tyrosine phosphatase DEP-1 mediated PI3K-cofilin signaling pathway in sorafenib-induced cytoskeletal rearrangement in hepatoma cells. J. Cell. Physiol. 224, 559–565 [DOI] [PubMed] [Google Scholar]
- 45. Arora D., Stopp S., Böhmer S. A., Schons J., Godfrey R., Masson K., Razumovskaya E., Rönnstrand L., Tänzer S., Bauer R., Böhmer F. D., Müller J. P. (2011) Protein-tyrosine phosphatase DEP-1 controls receptor tyrosine kinase FLT3 signaling. J. Biol. Chem. 286, 10918–10929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kermorgant S., Zicha D., Parker P. J. (2003) Protein kinase C controls microtubule-based traffic but not proteasomal degradation of c-Met. J. Biol. Chem. 278, 28921–28929 [DOI] [PubMed] [Google Scholar]
- 47. Jeffers M., Taylor G. A., Weidner K. M., Omura S., Vande Woude G. F. (1997) Degradation of the Met tyrosine kinase receptor by the ubiquitin-proteasome pathway. Mol. Cell. Biol. 17, 799–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Peschard P., Fournier T. M., Lamorte L., Naujokas M. A., Band H., Langdon W. Y., Park M. (2001) Mutation of the c-Cbl TKB domain binding site on the Met receptor tyrosine kinase converts it into a transforming protein. Mol. Cell 8, 995–1004 [DOI] [PubMed] [Google Scholar]
- 49. Carter S., Urbé S., Clague M. J. (2004) The met receptor degradation pathway. Requirement for Lys48-linked polyubiquitin independent of proteasome activity. J. Biol. Chem. 279, 52835–52839 [DOI] [PubMed] [Google Scholar]
- 50. Li N., Hill K. S., Elferink L. A. (2008) Analysis of receptor tyrosine kinase internalization using flow cytometry. Methods Mol. Biol. 457, 305–317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Birchmeier C., Birchmeier W., Gherardi E., Vande Woude G. F. (2003) Met, metastasis, motility and more. Nat. Rev. Mol. Cell Biol. 4, 915–925 [DOI] [PubMed] [Google Scholar]
- 52. Corso S., Comoglio P. M., Giordano S. (2005) Cancer therapy. Can the challenge be MET? Trends Mol. Med. 11, 284–292 [DOI] [PubMed] [Google Scholar]
- 53. Christensen J. G., Burrows J., Salgia R. (2005) c-Met as a target for human cancer and characterization of inhibitors for therapeutic intervention. Cancer Lett. 225, 1–26 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.