

Background: The role of MEN1 gene in development of lung cancer is poorly understood.
Results: K-Ras inhibits menin expression via increasing DNA methylation, whereas menin inhibits Ras-mediated signaling via suppressing activation of Ras.
Conclusion: The interplay between K-Ras and menin plays an important role in regulating the development of lung cancer.
Significance: These results have unraveled a novel mechanism underlying menin-mediated repression of lung cancer.
Keywords: DNA Methylation, DNA Methyltransferase, Epigenetics, Lung Cancer, Ras, DNA Methylation, K-Ras, MEN1, Menin
Abstract
MEN1, which encodes the nuclear protein menin, acts as a tumor suppressor in lung cancer and is often inactivated in human primary lung adenocarcinoma. Here, we show that the inactivation of MEN1 is associated with increased DNA methylation at the MEN1 promoter by K-Ras. On one hand, the activated K-Ras up-regulates the expression of DNA methyltransferases and enhances the binding of DNA methyltransferase 1 to the MEN1 promoter, leading to increased DNA methylation at the MEN1 gene in lung cancer cells; on the other hand, menin reduces the level of active Ras-GTP at least partly by preventing GRB2 and SOS1 from binding to Ras, without affecting the expression of GRB2 and SOS1. In human lung adenocarcinoma samples, we further demonstrate that reduced menin expression is associated with the enhanced expression of Ras (p < 0.05). Finally, excision of the Men1 gene markedly accelerates the K-RasG12D-induced tumor formation in the Men1f/f;K-RasG12D/+;Cre ER mouse model. Together, these findings uncover a previously unknown link between activated K-Ras and menin, an important interplay governing tumor activation and suppression in the development of lung cancer.
Introduction
Lung cancer is the leading cause of cancer death in males and the 5-year survival rate remains poor, because of the difficulty in early diagnosis and lack of effective chemotherapy/radiotherapy in advanced cancer (1). Mutations of proto-oncogenes such as Ras, epidermal growth factor receptor (EGFR),3 anaplastic lymphoma receptor kinase, and tumor suppressor gene p53 are important molecular events in the development of lung cancer (2–4). Ras proteins are widely expressed membrane-anchored small GTPases that have active GTP-bound states and inactive GDP-bound states (5). Mutation or activation of proto-oncogene Ras is involved in a variety of tumors including lung cancer, especially adenocarcinomas (6). The Ras activation and cycling is under the tight control of Ras GTPase-activating proteins (GAPs) and guanine nucleotide exchange factors (GEFs). Ras GAPs facilitate the conversion of Ras-GTP to Ras-GDP, whereas GEFs switch the inactive Ras-GDP to the active Ras-GTP state (5). SOS (son of sevenless) proteins are ubiquitously expressed GEFs in mammals (5). The adapter GRB2, following binding of a growth factor to its cell surface receptor, recruits SOS to the membrane to activate membrane-anchored Ras by converting inactive Ras-GDP to active Ras-GTP (5). More than 90% of Ras mutations occur in the K-Ras gene in lung adenocarcinoma (7, 8). Different mutation sites have different mechanisms to activate Ras. The mutations at codons 12 and 61 in Ras coding sequences can inhibit the hydrolysis of GTP that is normally enhanced by GAP, resulting in the increased level of active Ras-GTP and the activation of multiple downstream pro-proliferative and survival-promoting signaling pathways (5, 6, 9).
MEN1, a bona fide tumor suppressor gene, encodes a nuclear protein menin whose mutation is causative for the development of MEN1 (multiple endocrine neoplasia type 1) syndrome (10). The observation of loss of heterozygocity of the MEN1 allele in MEN1 tumors indicates the tumor-suppressing function of menin. Recently emerging evidence suggests that menin plays a vital tumor-promoting role in mixed lineage leukemia (MLL). In this regard, menin interacts with MLL proteins and increases MLL-mediated H3K4 methylation and expression of multiple Hox genes, which are required for hematopoiesis and leukemogenesis (11–13). There is now considerable and increasing evidence showing an aberrant or compromised activity of menin in human lung cancers. For example, MEN1 gene mutations were detected in sporadic lung carcinoid tumors of lung in patients (14). In addition, the probability of developing non-small cell lung cancer (NSCLC) was significantly increased in Men1+/− or Men1+/−;P18−/− mice (15). Phosphorylation of Rb protein by either CDK2 or CDK4/6 sites results in activation of the activity of pro-proliferative transcription factor E2Fs, and the phosphorylation of Rb is significantly increased in bronchial epithelia and tumor cells derived from p18−/−;Men1+/− mice (15). These findings suggest that menin is important for repressing the development of lung cancer, but the molecular mechanisms remain unclear. In our previous report, we have found that ectopic expression of menin markedly reduced lung cancer cell proliferation and migration through epigenetic repressing pleiotrophin signaling (16, 17). We also found that menin expression was markedly reduced in certain human primary lung adenocarcinoma (16), suggesting a potential tumor-suppressing role for menin in NSCLC progression. It remains unclear how menin is inactivated or compromised in NSCLC. Moreover, the molecular details of repression of lung cancer cell phenotype by menin remain elusive. The potential link between menin and NSCLC offers a fresh opportunity to uncover novel mechanisms underlying menin-mediated suppression of tumorigenesis in lung epithelial cells.
Our studies reveal a novel molecular basis controlling the development or progression of lung adenocarcinoma that is governed by the interplay between K-Ras and menin. Menin expression is reduced in certain human lung adenocarcinoma, at least partly by K-Ras-induced DNA methylation at the MEN1 promoter. Moreover, menin appears to reduce the Ras-GTP levels by inhibiting the binding of GRB2 and SOS1 to K-Ras, an important step in activation of Ras by increasing conversion of the inactive Ras-GDP to the active Ras-GTP state. Excision of the Men1 gene can accelerate the tumor formation induced by temporally controlled induction K-RasG12D in Men1f/f;K-RasG12D/+;Cre ER mouse model. Together, these findings unravel a previously unknown link between the oncogene K-Ras and tumor suppressor menin, whose intertwining may play a crucial role in regulating the development of lung cancer.
EXPERIMENTAL PROCEDURES
Cell Culture and Gene Transfection
Cell culture and transfection of human lung adenocarcinoma cell line A549 were described previously (16). The cells were treated with or without 5 μmol/liter 5-aza-2′-deoxycytidine (5-aza-dc; Sigma) for 4 days to determine the impact of DNA demethylation on the lung cancer cells.
Bisulfite Modification, Methylation-specific PCR, and DNA Cloning
CpG islands in the promoter regions of MEN1 were identified by using CpG island searcher. The primers for methylation-specific PCR were designed by using the online program MethPrimer (supplemental Table S1). The bisulfite modification of genomic DNA, which was extracted from clinical lung adenocarcinoma samples, was performed using the CpGenome DNA modification kit (Chemicon International). PCR products were cloned into the pMD18-T plasmid and sequenced for analysis by using ABI-PRISM 7300.
Ras Activity Assay
A549 cells were grown to 70% confluence in a 100-mm dish, starved overnight in serum-free DMEM, and recovered in 10% FBS DMEM for 20 min. The activity of Ras was determined by Ras activation assay kits (Upstate), based on the manufacturer's instructions.
Real Time qRT-PCR and ChIP
qRT-PCRs were performed as described previously using an ABI-PRISM 7300 detection system (16) with the primers shown in supplemental Table S2. ChIP assays were performed essentially as previously described (18). Antibodies used for ChIP assays were: anti-DNMT1 (Abcam), anti-trimethylated histone H3 Lys-4 (Abcam), anti-trimethyl-histone H3 Lys-27 (Millipore), and control IgG (Santa Cruz). We performed PCR using primers specific for MEN1 and p18 promoter sequences (supplemental Table S3).
Immunoprecipitation Assay
IP assays were performed as previously described (17, 18). Antibodies used for IP assays were: anti-SOS1 (Santa Cruz), GRB2 (Santa Cruz), Ras (BD Biosciences), and menin (Bethyl Laboratory).
Mouse Breeding
All of the mouse experiments were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania and were performed according to institutional and national guidelines. The K-RasG12D/+ mice (C57BL/6 and 129/Sv mixed background) were obtained from the group of Dr. Tyler Jacks and crossed with Men1f/f mice to generate Men1f/f;K-RasG12D/+ mice. They were then crossed with Ubc9 promoter driven Cre-ER mice (C57B6 background) as previously described (19). Men1f/f and Men1f/f;Cre-ER mice were as described previously (20). Animal genotyping was done by PCR according to published methods using DNA extracted from tails.
Activation of K-Ras by Tamoxifen
K-RasG12D/+;Ubc9 Cre-ER and K-RasG12D/+;Men1f/f Ubc9 Cre-ER were fed tamoxifen at 200 mg/kg of body weight/day by oral gavage for two consecutive days (20). The animals were sacrificed on day 9, and the lungs were removed for further analysis.
RESULTS
Expression Level of Menin Is Inversely Correlated with K-Ras Expression in Lung Adenocarcinoma
Our previous studies indicate that menin expression is reduced in certain primary human lung cancer, but the underlying mechanism is unknown (16). MEN1 missense mutations usually reduce the stability of menin and thus its expression level in MEN1 tumors (21). To determine whether the reduced menin expression is correlated with MEN1 mutations in the lung cancer samples, we extracted the genomic DNA from six paraffin-embedded lung adenocarcinoma samples in which menin expression was reduced. Through the whole exon PCR amplification and sequencing of the MEN1 gene, we only found one single nucleotide polymorphism (C to T change at codon 7), with no change in amino acid sequence (date not shown).
We then turned our attention to the possibility that reduced menin expression might be affected by alteration of other oncogenes in lung cancer. Ras is the most frequently mutated gene in lung cancer (6) and is a driving force for the development for lung adenocarcinoma (6). The mutated K-Ras becomes constitutively active because of the locked state of Ras-GTP (5). Thus, we examined the expression of menin and Ras in 34 lung adenocarcinoma samples, along with adjacent normal tissues. Immunohistochemistry (IHC) analysis revealed that menin expression was high in the nucleus of the normal bronchiole epithelial cells and alveoli (Fig. 1A) but much weaker or even undetectable in certain tumors (Fig. 1B), consistent with our previous finding (16). The expression of menin was markedly reduced in eight adenocarcinoma samples, compared with its adjacent normal tissue, accounting for 23.5% of the tumors we examined (supplemental Table S4). In contrast, Ras staining in the cytoplasm and cell membrane was noticeably increased in 16 of 34 (47.1%) adenocarcinomas (Fig. 1D), compared with the adjacent normal tissue (Fig. 1C). In eight cases with reduced menin, Ras expression was significantly up-regulated in six cases (supplemental Table S4). It is noteworthy that the attenuated menin expression was associated with the enhanced expression of Ras, analyzed by chi-squared test (p < 0.05). These results suggest that menin expression was correlated with a high level of Ras expression in human lung adenocarcinomas.
FIGURE 1.

Down-regulated menin expression was correlated with up-regulated Ras. A–D, sections from paraffin-embedded adjacent normal tissues and cancer tissues were stained with antibodies against menin (A and B) and Ras (C and D) for IHC staining, respectively, Original magnification, ×200. E and F, the expression of menin, detected by real time qRT-PCR (E) and Western blot (F), was increased in A549 cells that were transfected with shRNA vector against K-Ras compared with luciferase. G, menin expression, detected by Western blot, was reduced in K-Ras overexpression A549 cells.
K-Ras Inhibits Menin Expression in A549 Lung Cancer Cells
The Ras subfamily consists of H-Ras, K-Ras, and N-Ras. Noticeably, 90% of Ras mutations in lung adenocarcinomas occurred in K-Ras, and ∼97% of K-Ras mutations in NSCLC involve codons 12 or 13 (9). Thus, we focus on the relationship of K-Ras and menin in lung cancer cells. A549 cell line harbors a mutation in codon 12 of K-Ras (e.g., G12 to C), thereby maintaining constitutive activation of K-Ras. We transduced A549 cells with either control luciferase (Luc) shRNA or a K-Ras shRNA. Notably, real time qRT-PCR showed that K-Ras knockdown (KD) led to an increase in MEN1 expression, as compared with the control Luc shRNA (Fig. 1E). Likewise, compared with controls, the menin protein level was elevated in K-Ras KD A549 cells, as shown by Western blot (Fig. 1F). Next, we examined whether overexpression of K-Ras affects menin expression. The full-length K-Ras cDNA was cloned from A549 cells, and sequencing results confirmed that G12C mutation was detectable in A549 cells (date not shown). The empty plasmid and pcDNA3.1(+)-K-RasG12C were transfected into A549 cells, which were selected by G418. The Western blot analysis indicated that the menin level was reduced when ectopic K-RasG12C was overexpressed in A549 cells (Fig. 1G). These observations collectively indicate that K-Ras down-regulates menin expression in A549 human lung adenocarcinoma cells.
Reduced MEN1 Expression Was Correlated with DNA Methylation at Its Promoter
Genetic and epigenetic processes, such as DNA methylation at the CpG island of the promoter region, play an important role in gene silencing in cancer (22). To determine whether menin reduction is associated with DNA methylation, we examined the MEN1 promoter methylation in lung adenocarcinoma samples. We designed DNA methylation-specific PCR (MS-PCR) primers spanning the CpG islands of the MEN1 promoter (Fig. 2A). MS-PCR amplification was used to verify the DNA methylation of the six paired samples of primary human lung adenocarcinoma and the adjacent tissue obtained from surgery. Consistent with less MEN1 mRNA in the cancer, DNA methylation level at the MEN1 promoter was considerably increased in lung cancer, as compared with the adjacent tissue (Fig. 2B; n = 6, p = 0.001). To investigate the status of each CpG site, we cloned the MS-PCR amplification products from two pairs of cancer and the adjacent tissues to pMD18-T plasmids and sequenced 10 clones. Bisulfite sequencing analysis showed the representative CpG sites, which were methylated in cancer and unmethylated in the adjacent normal tissues (Fig. 2C). The −1051-bp site (ATG defined as 0 bp) had a higher level of DNA methylation, compared with the adjacent tissue, i.e., 60% (12/20) versus 25% (5/20), respectively, and the CpG methylation probability of the −1013bp site increased from 5% (1 of 20) in normal tissues to 45% (9 of 20) in the cancer (Fig. 2D). The collective results suggest that cancer tissue contains far more methylated MEN1 CpG sites than the adjacent normal tissue. To determine whether genomic DNA demethylation in cells affects the expression of the MEN1 gene, we treated A549 cells with DNA methylation inhibitor, 5-aza-dc (5 μm, for 3 days). Real time RT-PCR analysis revealed that treatment with 5-aza-dc elevated the MEN1 mRNA level (Fig. 2E). Collectively, these findings indicate that increased DNA methylation at the MEN1 promoter is an important means to suppress menin expression in lung adenocarcinoma.
FIGURE 2.
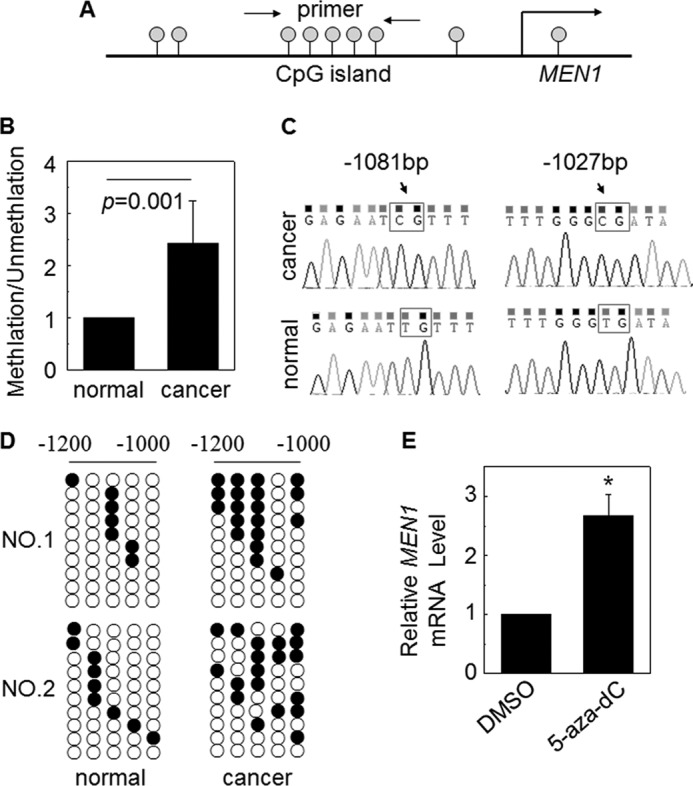
Down-regulated menin expression was partly associated with promoter DNA methylation. A, the scheme shows the CpG islands of MEN1 promoter that is the aim of the MS-PCR primer. B, MS-PCR analysis of the MEN1 promoter methylation of lung cancer tissue and adjacent normal tissue, obtained from the surgeries of six patients. The methylation/unmethylation of normal samples was set to 1 (n = 6). C, the representative bisulfite sequencing chromatograms of cancer and normal tissues in the two MEN1 promoter CpG sites. D, DNA methylation analysis by bisulfite cloning and sequencing. The analysis was carried out for two paired cancer and normal tissues bisulfite sequencing. Each circle in each row represents a single CpG site. Each line represents an individually sequenced clone, and circles represent CpG residues. White and black circles represent unmethylated and methylated CpG sites, respectively. E, A549 cells were either treated or not treated with DNA methylation inhibitor 5-aza-dc (5 μm). Real time qRT-PCR was performed to detect the MEN1 mRNA expression. *, p < 0.05 versus control. DMSO, dimethyl sulfoxide.
K-Ras Affects DNA Methylation at the MEN1 Promoter by Promoting the Binding of DNMT1 to the MEN1 Locus
To assess whether there is an intrinsic relationship between K-Ras and DNA methylation in regulating menin expression, we examined the methylation profile of the MEN1 promoter DNA in K-Ras KD A549 cells. Interestingly, MS-PCR result revealed that the DNA methylation of the MEN1 promoter was reduced, which was only 0.18-fold as compared with the control (Fig. 3A). Bisulfite sequencing also showed that the methylated CpG sites were reduced in K-Ras KD A549 cells (Fig. 3B), in agreement with reduced methylation of the MEN1 promoter. These results suggest that K-Ras increases the DNA methylation of the MEN1 promoter. Mammalian DNA methylation was controlled by DNA methyltransferases (DNMTs). DNMT1 is overexpressed in lung cancer samples (23), and the tobacco-specific carcinogen 4-(methylnitrosamino)-l-(3-pyridyl)-1-butanone induces DNMT1 expression, which leads to hypermethylation of certain tumor suppressor genes (24). We examined the protein expression of three main DNMTs: DNMT1, DNMT3A, and DNMT3B in K-Ras KD A549 cells. Western blot analysis showed that the diminished K-Ras expression in A549 led to reduction of DNMT1 and DNMT3B, but not DNMT3A (Fig. 3C). Also the increased DNMT1 protein expression by K-Ras was shown in supplemental Fig. S1A. Next, we performed ChIP assays with vector and K-RasG12C overexpressed A549 cells. ChIP assays clearly showed that DNMT1 bound to the MEN1 promoter locus, marked with primer pair PP1 and PP2, and the binding in K-Ras overexpressing A549 cells was more than that in the control cells (Fig. 3D, lanes 3 and 6). These results indicate that K-Ras down-regulates MEN1 expression at least partly by promoting the binding of DNMT1 to the MEN1 locus, leading to the DNA methylation at the MEN1 promoter.
FIGURE 3.

K-Ras increases the MEN1 promoter DNA methylation via DNMTs. A, quantitative MS-PCR was used to analyze the methylation level of MEN1 promoter in K-Ras knockdown A549 cells (K-Ras shRNA), compared with Luc shRNA control. *, p < 0.05 versus control. B, bisulfite sequence analysis of each clone from K-Ras shRNA or Luc shRNA A549 cells (10 clones/sample). C, Western bolt detection of DNMTs and EZH2 in K-Ras diminished A549 cells. D, a schematic representation of the MEN1 gene loci and amplicons used for ChIP assay. ChIP assay was performed using the antibody against DNMT1 in vector and K-Ras-transfected A549 cells. PCR was carried out using primers for each amplicon.
A previous report has shown that histone methylation is mechanistically linked to DNA methylation in mammals (25). EZH2 is a member of PcG family and acts as a gene silencer by catalyzing histone H3 lysine 27 trimethylation (H3K27me3), which can compress chromatin structure (26). However, the EZH2 expression in K-Ras KD A549 cells was indistinguishable from that of control (Fig. 3C). Furthermore, ChIP assays showed that down-regulated K-Ras expression did not affect H3K27me3 level at MEN1 locus (supplemental Fig. S1B). On the other hand, histone H3 lysine 4 trimethylation (H3K4me3) is correlated with positive gene transcription (11). However, we failed to detect the difference between H3K4me3 at the MEN1 locus in control and K-Ras knocked down A549 cells by ChIP assays (supplemental Fig. S1C). Together, these results suggest that K-Ras represses menin expression mainly through increasing DNMT1 binding to the MEN1 locus.
Menin Inhibits Ras Activity by Repressing Its Binding to SOS1
Next, we studied whether menin also reciprocally affects expression of Ras. To this end, we examined the K-Ras mRNA level in vector and menin-overexpressing A549 cells using two sets of primers and found that menin had no effect on the mRNA level of K-Ras (supplemental Fig. S2A). The Western blot assay showed that ectopic expression of menin barely affected the protein level of total Ras (supplemental Fig. S2B). Interestingly, the Ras-GTP level, detected by Ras activity assay, showed that the quantity of Ras-GTP was noticeably reduced in menin-overexpressing cells (supplemental Fig. S2B). These findings suggest that menin mainly reduces the level of the active Ras-GTP form in human lung adenocarcinoma cancer cells, suggesting the regulation at the post-translational level.
Ras activity is regulated by GAP and GEFs (5, 6). GAP reduces Ras activity through hydrolysis of GTP to GDP in complex with Ras. To assess how menin regulates the active Ras-GTP, we examined GAP expression in stably menin expressed A549 cells. However, Western blotting results indicate that ectopic expression of menin did not affect the expression of GAP (Fig. 4A). There are numerous members of GEF, which may be responsible for activation of different substrates. SOS1 is a Ras-specific GEF (27, 28). Formation of GRB2, SOS1, and Ras complex after ligand binding to the growth factor receptor leads to Ras activation (increased level of Ras-GTP) (29, 30). We found that menin did not affect the expression of SOS1 and GRB2 (Fig. 4A). Because binding to SOS1 is necessary for Ras-GDP to get activated, we performed co-IP and found that ectopic expression of menin reduced the binding of GRB2 and SOS1 to Ras but did not affect the interaction between SOS1 and GRB2 (Fig. 4, B–D). We next sought to determine whether menin associates with Ras complex. Fig. 4E showed that menin was co-immunoprecipitated with GRB2, SOS1, and Ras. Consistent with these observations, the reverse co-IP also showed that endogenous GRB2, SOS1, and Ras were co-immunoprecipitated with endogenous menin (Fig. 4F).
FIGURE 4.

Menin suppresses Ras GTP activity. A, Western blotting detected the impact of menin overexpression on SOS1, GAP, GRB2, and Ras expression in A549 cells. B–E, menin-overexpressed and control A549 cells lysates were immunoprecipitated for Ras, GRB2, or SOS1, and the immunoprecipitates were analyzed by Western blotting (WB) for the presence of SOS1, Ras, GRB2, and menin. F, total protein lysates of A549 cells were used for endogenous immunoprecipitation with antibody to menin and IgG as a control, followed by Western blotting for SOS1, GRB2, and Ras. V, vector; M, MEN1.
EGF binds its cell surface receptor and induces the receptor phosphorylation, leading to sequential recruitment of GRB2, SOS1, and Ras to the cell membrane, thereby increasing Ras-GTP (31, 32). The Ras-GTP level was significantly increased 5 min after treatment with EGF (supplemental Fig. S3A). However, there was no significant change in the EGF and EGF2 mRNA levels upon overexpression of menin (supplemental Fig. S3, B and C). Furthermore, the EGFR expression and its various phosphorylation forms were not affected by menin (supplemental Fig. S3D), and menin also did not inhibit EGF-mediated induction of EGFR phosphorylation; in contrast, the phosphorylation was only slightly increased (supplemental Fig. S3E). Together, these findings suggest that menin could bind to either GRB2 and SOS1 or Ras to block their interaction and thus inhibit Ras activation, and it is independent of EGF and its receptor. The precise biochemical model of menin interaction with the Ras complex needs further investigation.
Menin Excision Promotes Lung Epithelial Cell Proliferation
Menin represses proliferation of human endocrine tumor cells, insulinoma cells, intestinal epithelial cells, and Ras-transformed NIH3T3 cells via several distinct mechanisms (33, 34). Previously, we found that menin suppresses proliferation of lung cancer cells and migration partly through polycomb gene (PcG)-dependent repression of pleitrophin expression in vitro (16). In this study, we used Men1 conditional KO (20) and Men1 loss of heterozygosity mice (35) to investigate the proliferation of lung epithelial cells by detecting BrdU uptake proliferating cells. As expected, IHC detection confirmed that menin expression in Men1l/l;Cre-ER mice, in which Men1 gene was excised when treated with tamoxifen, was markedly reduced in lung epithelial cells and bronchial epithelial cells, as compared with control Men1l/l mice (Fig. 5A). With the reduction of menin, the number of proliferating cells were increased as assessed by the IHC detection of BrdU+ cells (Fig. 5A). We quantified the number of BrdU+ cells in each 400 magnification field. Statistical analysis showed that the numbers of BrdU + cells/field in Men1l/l and Men1l/l;Cre-ER mice were 6.5 and 10.7, respectively (Fig. 5C; n = 6), similar to conditional Men1 KO mice. The BrdU+ lung epithelial cells were also significantly increased, whereas menin expression was decreased in Men1+/− mice, as detected by IHC (Fig. 5B). The statistical analysis indicates that there was an average of 3.7 BrdU + cells/field in Men1+/+ mice, whereas the value was 11.2 in Men1+/− mice (Fig. 5D; n = 6, p < 0.05). These experiments confirmed that Men1 excision leads to enhanced proliferation of lung epithelial cells. Moreover, 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assays revealed that the proliferation was much slower in menin cDNA-transfected A549 cells (supplemental Fig. S4, A and B) but accelerated in MEN1 KD A549 cells, as compared with the control (supplemental Fig. S4, C and D).
FIGURE 5.

Men1 KO promotes lung epithelial cell proliferation. A, control Men1l/l and Men1l/l;Cre-ER mice (n = 8 mice) were fed tamoxifen at the age of 12 weeks at 200 mg/kg of body weight/day for 2 consecutive days. Four weeks after the last dose of tamoxifen feeding, the mice were sacrificed. The lung tissue sections from Men1l/l and Men1l/l;Cre-ER mice were stained with antibody of menin, BrdU, and p18. B, sections from paraffin-embedded Men+/+ and Men1+/− mice lung (3 months old) samples were also immunostained with menin, BrdU, and p18 antibody (-200; insets, ×400). C and D, quantification of BrdU+ lung cells from the groups of Men1l/l (control) versus tamoxifen-fed Men1l/l;Cre-ER mice and Men +/+ (control) versus Men1+/− mice. *, p < 0.05 versus control. E, real time qRT-PCR shows p18 mRNA expression in vector- or menin-expressing A549 cells. F, Western blot analysis the expression of P18, CDK4, and CDK6 in vector and menin overexpression A549 cells. G, a schematic representation of the p18 gene loci and amplicons used for ChIP assay. ChIP assay using the antibody against H3K4me3 in vector and K-Ras transfected A549 cells.
Menin regulates cell proliferation via many different ways. It not only regulates cell cycle related proteins, such as cyclin D, CDK4, p18INK4C (p18), and p27Kip1 (p27), but also affects DNA replication proteins such as Cdc7/ASK (34). We investigated the possible role of menin in regulation the proliferation of lung epithelial cells and lung cancer cells. IHC results indicate that p18 was highly expressed in the nucleus of bronchial epithelial cells in Men1l/l and Men1+/+ mice but hardly detectable in Men1l/l;Cre-ER and Men1+/− mice (Fig. 5, A and B). To test whether menin regulates lung cancer proliferation through the p18 signaling pathway, we measured the p18 mRNA level in MEN1-transformed A549 cells. qRT-PCR analysis revealed that p18 mRNA level was increased 5.6 times when A549 cells were transformed with the menin cDNA (Fig. 5E). Consistent with these findings, Western blot analysis indicates that the overexpressed menin level in A549 cells was accompanied by the increased p18 protein level and the decreased levels of CDK4 and CDK6 (Fig. 5F), but not Cdc7 (supplemental Fig. S4E). It is well known that p18 inhibits CDK4 and CDK6 to arrest cell cycle (36, 37). It is likely that menin can facilitate the transcription of p18 and then repress the activity of CDK4, preventing the cell cycle progression and inhibiting proliferation of lung cancer cells. Further ChIP assay results indicate that ectopic expression of K-Ras represses menin protein level and the H3K4me3 in p18 promoter loci (Fig. 5G, lanes 3 and 6). This is consistent with the previous report that menin promotes methylation of H3K4 at the p18 promoter loci and up-regulates p18 transcription in islet cells (38). To test the biological significance of the K-Ras knockdown, we measured proliferation of A549 cells that were transfected with either control Luc shRNA or K-Ras shRNAs. Correlated with K-Ras knockdown, the proliferation of A549 cells was significantly reduced, as compared with the control shRNA (supplemental Fig. S5; n = 8, p < 0.05).
To test whether menin affects the development of K-Ras induced lung cancer, we bred mice with a conditionally inducible knock-in K-RasG12D with mice harboring the floxed Men1;Cre-ER (see “Experimental Procedures”). This mouse model (knock-in K-RasG12D) is widely used to investigate the development/maintenance of the lung cancer (39). We found that all three K-Ras mutant mice (K-RasG12D/+;Cre ER) show hyperplasia and some small tumors in lungs as seen in hematoxylin- and eosin-stained tissues (Fig. 6A). We calculated the number of tumors and measured the area of each tumor nodule. Notably, we observed that not only the number but also the volume of the lung tumors was significantly increased when the Men1 gene was knocked out in K-RasG12D mice (Men1 f/f;K-RasG12D/+;Cre ER mice), as compared with K-RasG12D/+;Cre ER mice (Fig. 6, B and C). These observations collectively demonstrated that the excision of the Men1 gene can promote the tumor formation induced by K-Ras mutation.
FIGURE 6.

Menin excision promotes the development of K-Ras-induced lung cancer. A, the lung tissue hematoxylin and eosin staining of K-RasG12D/+ mice (C57BL/6 and 129/Sv mixed background) and Men1f/f;K-RasG12D/+ mice (K-RasG12D/+ mice crossed with Men1f/f mice) fed tamoxifen. B and C, the quantitative analysis of the number and the size of tumors in K-RasG12D/+;Cre-ER mice and Men1f/f;K-RasG12D/+;Cre-ER mice, respectively. *, p < 0.05 versus control.
DISCUSSION
Previous studies have identified that multiple genes, including TP53, CDKN2A, EGFR, and K-Ras were involved in the development of lung cancer (1–3). Our previous studies have shown reduced expression of menin in lung adenocarcinoma, but the mechanism remains elusive (16). In the present study, we found that although menin expression was lower in lung adenocarcinoma samples, no mutation was identified in the MEN1 gene. As a high percentage of NSCLCs contain activating mutation in the K-Ras gene (7, 8). We examined whether menin was repressed by K-Ras expression in lung cancer. The histological study of clinical lung adenocarcinoma samples revealed that menin reduction was associated with increased Ras expression in cancer tissue. Further cellular study verified that K-Ras down-regulates expression of MEN1 mRNA and protein. The animal model confirmed that excision of the Men1 gene markedly promotes the tumor formation that is triggered by K-Ras mutation (G12D), consistent with a previous report that menin can repress Ras-mediated tumor transformation in NIH-3T3 cells (33). Intriguingly, we also found that menin significantly represses the active Ras-GTP form while slightly reducing the amount of the total Ras in A549 cells. Our findings offer the first biochemical evidence that menin expression reduces the active form of K-Ras-GTP. This further supports the correlation between reduced menin expression and increased Ras expression in the lung tumor samples.
DNA methylation plays an important role in gene silencing, and a number of methylated tumor suppressors were identified in lung cancer, such as p16, DAPK1, RASSF1A, and HOX genes (40). Our results have shown that DNA methylation of MEN1 promoter was increased in lung cancer tissues as compared with the adjacent tissue, and the MEN1 mRNA level can be up-regulated by DNA methylation inhibitor in A549 cells. In turn, methylation of MEN1 promoter was decreased in K-Ras KD A549 cells. These observations are consistent with the notion that K-Ras may repress menin expression via increasing MEN1 methylation. Supporting this notion, we demonstrated that the expression of DNMT1 and DNMT3B was substantially reduced in K-Ras KD A549 cells, but DNMT3A expression was not changed. DNMT1 and DNMT3B may cooperate to silence the MEN1 gene in A549 as previously reported for methylation of other genes in colorectal cancer cell line (41). ChIP assay results provided direct evidence that K-Ras can promote DNMT1 binding to the MEN1 promoter. This is similar to the finding that increased binding of DNMT3B to E-cadherin promoter by K-Ras causes promoter hypermethylation for reduced expression of E-cadherin in human prostate cancer cells (42).
Intriguingly, we also found that menin reduced the active Ras-GTP form in A549 cells. In investigating the mechanism whereby menin inhibits Ras activity, we found that ectopic expression of menin in A549 cells decreased binding of SOS1 and GRB2 to Ras, without affecting the expression of GAP, SOS1, and GRB2. Menin bound to endogenous GRB2 and SOS1. Thus, it is likely that menin binds to the GRB2-SOS1 complex to reduce the binding of SOS1 and GRB2 to Ras, resulting in the decreased Ras-GTP. Although Ras mutations at positions 12, 13, and 61 impair the intrinsic GTPase activity, causing the accumulation of Ras-GTP (9), menin-mediated repression of SOS1 binding to K-Ras may still help to reduce K-Ras-GFP levels. In our previous study, we found that menin represses phosphorylation of ERK1/2 and AKT in lung cancer cells (17). Recently, Wang et al. (43) demonstrated that menin is an important negative regulator of AKT kinase activity in nonendocrine and endocrine cells. Consistent with these observations, our results uncover a new mechanism whereby menin represses the K-Ras, through inhibiting the formation of GRB2-SOS1-Ras complex, thereby inhibited the Ras-RAF-MEK-MAPK pathway in the formation of lung cancer.
We also found that the Ras-GTP level was increased in A549 cells by EGF stimulation, but menin expression did not affect phosphorylation of EGFR. These results suggest that menin regulates the Ras-GTP level likely downstream of EGFR, consistent with the impact of menin on repressing interaction between SOS1 and Ras.
In summary, we have unraveled a novel feedback loop of Ras and tumor suppressor gene MEN1 in lung adenocarcinoma. K-Ras inhibits menin expression by promoting DNMT1 binding to the MEN1 promoter and increased DNA methylation, whereas menin inhibits Ras-mediated signaling at least partly via suppressing SOS1-mediated activation of Ras by blocking GRB2-SOS1 from binding to Ras. The mouse model suggests that the excision of the Men1 gene can promote the tumor formation caused by K-Ras mutation. The imbalance of MEN1 and Ras may play a crucial role to promote the lung cancer development. These results have unraveled a novel mechanism underlying menin-mediated repression of lung cancer and provide a novel potential target for treating menin-negative and Ras active lung adenocarcinoma via inhibiting DNMTs.
The latter part of our study showed that menin excision can promote lung epithelial cell proliferation via p18 regulation, but the mechanism is unclear. One study revealed that menin plays a critical role in the MLL-trithorax Histone methyltransferases complex by recruiting MLL to the p18 and p27 promoters (44). Function loss of either menin or MLL results in down-regulation of p18 and p27 expression and deregulating cell proliferation (44). Another study showed that the menin-MLL complex can up-regulate p18 and p27 transcription by increasing H3K4 methylation at their loci in both cultured and murine pancreatic islet (45). Further research is necessary to determine whether the loss of menin expression will diminish MLL-Histone methyltransferases activity and epigenetic regulation of p18, p27, and other cancer-related genes in K-Ras involved lung cancer. Further experiments remains on determining the H3K4 methylation of p18 in A549 cells or K-Ras-mediated pulmonary tumors.
Supplementary Material
Acknowledgment
We thank Dr. Tyler Jacks (Massachusetts Institute of Technology) for providing the K-RasG12D/+ mice.
This work was supported by Natural Science Foundation of China Grants 81071926 and 81272719 (to G. H. J.), 81101763 (to S. B. G.), and 81101924 (to S. L.); Natural Science Foundation of Fujian Province Grant 2011J06016 (to G. H. J.); Natural Science Foundation of Xiamen Grant 3502Z20104001 (to G. H. J.); Fundamental Research Grant 2010121106 for the central universities, Xiamen University (to G. H. J.). This work was also supported by National Institutes of Health Grant R01 DK085121 (to X. H.) and a Lung Cancer Research Foundation Grant (to X. H.).

This article contains supplemental text, Tables S1–S4, and Figs. S1–S5.
- EGFR
- epidermal growth factor receptor
- GAP
- GTPase-activating protein
- GEF
- guanine nucleotide exchange factor
- MLL
- mixed lineage leukemia
- NSCLC
- non-small cell lung cancer
- 5-aza-dc
- 5-aza-2′-deoxycytidine
- qRT-PCR
- quantitative RT-PCR
- IP
- immunoprecipitation
- IHC
- immunohistochemistry
- Luc
- luciferase
- KD
- knockdown
- MS-PCR
- methylation-specific PCR.
REFERENCES
- 1. Jemal A., Bray F., Center M. M., Ferlay J., Ward E., Forman D. (2011) Global Cancer Statistics. CA-Cancer J. Clin. 61, 69–90 [DOI] [PubMed] [Google Scholar]
- 2. Jemal A., Thomas A., Murray T., Thun M. (2002) Cancer statistics, 2002. CA-Cancer J. Clin. 52, 23–47 [DOI] [PubMed] [Google Scholar]
- 3. Herbst R. S., Heymach J. V., Lippman S. M. (2008) Lung cancer. N. Engl. J. Med. 359, 1367–1380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Soda M., Choi Y. L., Enomoto M., Takada S., Yamashita Y., Ishikawa S., Fujiwara S., Watanabe H., Kurashina K., Hatanaka H., Bando M., Ohno S., Ishikawa Y., Aburatani H., Niki T., Sohara Y., Sugiyama Y., Mano H. (2007) Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 448, 561–566 [DOI] [PubMed] [Google Scholar]
- 5. Vigil D., Cherfils J., Rossman K. L., Der C. J. (2010) Ras superfamily GEFs and GAPs: validated and tractable targets for cancer therapy? Nat. Rev. Cancer 10, 842–857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bos J. L. (1989) ras oncogenes in human cancer. A review. Cancer Res. 49, 4682–4689 [PubMed] [Google Scholar]
- 7. Mitsudomi T., Viallet J., Mulshine J. L., Linnoila R. I., Minna J. D., Gazdar A. F. (1991) Mutations of ras genes distinguish a subset of non-small-cell lung cancer cell lines from small-cell lung cancer cell lines. Oncogene 6, 1353–1362 [PubMed] [Google Scholar]
- 8. Rodenhuis S., Slebos R. J. (1992) Clinical significance of ras oncogene activation in human lung cancer. Cancer Res. 52, 2665s–2669s [PubMed] [Google Scholar]
- 9. Schubbert S., Shannon K., Bollag G. (2007) Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7, 295–308 [DOI] [PubMed] [Google Scholar]
- 10. Chandrasekharappa S. C., Guru S. C., Manickam P., Olufemi S. E., Collins F. S., Emmert-Buck M. R., Debelenko L. V., Zhuang Z., Lubensky I. A., Liotta L. A., Crabtree J. S., Wang Y., Roe B. A., Weisemann J., Boguski M. S., Agarwal S. K., Kester M. B., Kim Y. S., Heppner C., Dong Q., Spiegel A. M., Burns A. L., Marx S. J. (1997) Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 276, 404–407 [DOI] [PubMed] [Google Scholar]
- 11. Yokoyama A., Somervaille T. C., Smith K. S., Rozenblatt-Rosen O., Meyerson M., Cleary M. L. (2005) The menin tumor suppressor protein is an essential oncogenic cofactor for MLL-associated leukemogenesis. Cell 123, 207–218 [DOI] [PubMed] [Google Scholar]
- 12. Jin S., Zhao H., Yi Y., Nakata Y., Kalota A., Gewirtz A. M. (2010) c-Myb binds MLL through menin in human leukemia cells and is an important driver of MLL-associated leukemogenesis. J. Clin. Invest. 120, 593–606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Thiel A. T., Blessington P., Zou T., Feather D., Wu X., Yan J., Zhang H., Liu Z., Ernst P., Koretzky G. A., Hua X. (2010) MLL-AF9-induced leukemogenesis requires coexpression of the wild-type Mll allele. Cancer Cell 17, 148–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Debelenko L. V., Brambilla E., Agarwal S. K., Swalwell J. I., Kester M. B., Lubensky I. A., Zhuang Z., Guru S. C., Manickam P., Olufemi S. E., Chandrasekharappa S. C., Crabtree J. S., Kim Y. S., Heppner C., Burns A. L., Spiegel A. M., Marx S. J., Liotta L. A., Collins F. S., Travis W. D., Emmert-Buck M. R. (1997) Identification of MEN1 gene mutations in sporadic carcinoid tumors of the lung. Hum. Mol. Genet. 6, 2285–2290 [DOI] [PubMed] [Google Scholar]
- 15. Pei X. H., Bai F., Smith M. D., Xiong Y. (2007) p18Ink4c collaborates with Men1 to constrain lung stem cell expansion and suppress non-small-cell lung cancers. Cancer Res. 67, 3162–3170 [DOI] [PubMed] [Google Scholar]
- 16. Gao S. B., Feng Z. J., Xu B., Wu Y., Yin P., Yang Y., Hua X., Jin G. H. (2009) Suppression of lung adenocarcinoma through menin and polycomb gene-mediated repression of growth factor pleiotrophin. Oncogene 28, 4095–4104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Feng Z. J., Gao S. B., Wu Y., Xu X. F., Hua X., Jin G. H. (2010) Lung cancer cell migration is regulated via repressing growth factor PTN/RPTP β/ζ signaling by menin. Oncogene 29, 5416–5426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Xu B., Zeng D. Q., Wu Y., Zheng R., Gu L., Lin X., Hua X., Jin G. H. (2011) Tumor suppressor menin represses paired box gene 2 expression via Wilms tumor suppressor protein-polycomb group complex. J. Biol. Chem. 286, 13937–13944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schnepp R. W., Chen Y. X., Wang H., Cash T., Silva A., Diehl J. A., Brown E., Hua X. (2006) Mutation of tumor suppressor gene Men1 acutely enhances proliferation of pancreatic islet cells. Cancer Res. 66, 5707–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yang Y., Gurung B., Wu T., Wang H., Stoffers D. A., Hua X. (2010) Reversal of preexisting hyperglycemia in diabetic mice by acute deletion of the Men1 gene. Proc. Natl. Acad. Sci. U.S.A. 107, 20358–20363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yaguchi H., Ohkura N., Takahashi M., Nagamura Y., Kitabayashi I., Tsukada T. (2004) Menin missense mutants associated with multiple endocrine neoplasia type 1 are rapidly degraded via the ubiquitin-proteasome pathway. Mol. Cell Biol. 24, 6569–6580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jones P. A., Baylin S. B. (2007) The epigenomics of cancer. Cell 128, 683–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin R. K., Hsu H. S., Chang J. W., Chen C. Y., Chen J. T., Wang Y. C. (2007) Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer 55, 205–213 [DOI] [PubMed] [Google Scholar]
- 24. Lin R. K., Hsieh Y. S., Lin P., Hsu H. S., Chen C. Y., Tang Y. A., Lee C. F., Wang Y. C. (2010) The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. J. Clin. Invest. 120, 521–532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henckel A., Nakabayashi K., Sanz L. A., Feil R., Hata K., Arnaud P. (2009) The tobacco-specific carcinogen NNK induces DNA methyltransferase 1 accumulation and tumor suppressor gene hypermethylation in mice and lung cancer patients. Hum. Mol. Genet. 18, 3375–338319515852 [Google Scholar]
- 26. Cao R., Wang L., Wang H., Xia L., Erdjument-Bromage H., Tempst P., Jones R. S., Zhang Y. (2002) Role of histone H3 lysine 27 methylation in polycomb-group silencing. Science 298, 1039–1043 [DOI] [PubMed] [Google Scholar]
- 27. Chardin P., Camonis J. H., Gale N. W., van Aelst L., Schlessinger J., Wigler M. H., Bar-Sagi D. (1993) Human Sos1. A guanine nucleotide exchange factor for Ras that binds to GRB2. Science 260, 1338–1343 [DOI] [PubMed] [Google Scholar]
- 28. Egan S. E., Giddings B. W., Brooks M. W., Buday L., Sizeland A. M., Weinberg R. A. (1993) Association of Sos Ras exchange protein with Grb2 is implicated in tyrosine kinase signal transduction and transformation. Nature 363, 45–51 [DOI] [PubMed] [Google Scholar]
- 29. Simon M. A., Dodson G. S., Rubin G. M. (1993) An SH3-SH2-SH3 protein is required for p21Ras1 activation and binds to sevenless and Sos proteins in vitro. Cell 73, 169–177 [DOI] [PubMed] [Google Scholar]
- 30. Aronheim A., Engelberg D., Li N., al-Alawi N., Schlessinger J., Karin M. (1994) Membrane targeting of the nucleotide exchange factor Sos is sufficient for activating the Ras signaling pathway. Cell 78, 949–961 [DOI] [PubMed] [Google Scholar]
- 31. Lowenstein E. J., Daly R. J., Batzer A. G., Li W., Margolis B., Lammers R., Ullrich A., Skolnik E. Y., Bar-Sagi D., Schlessinger J. (1992) The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell 70, 431–442 [DOI] [PubMed] [Google Scholar]
- 32. Rozakis-Adcock M., Fernley R., Wade J., Pawson T., Bowtell D. (1993) The SH2 and SH3 domains of mammalian Grb2 couple the EGF receptor to the Ras activator mSos1. Nature 363, 83–85 [DOI] [PubMed] [Google Scholar]
- 33. Kim Y. S., Burns A. L., Goldsmith P. K., Heppner C., Park S. Y., Chandrasekharappa S. C., Collins F. S., Spiegel A. M., Marx S. J. (1999) Stable overexpression of MEN1 suppresses tumorigenicity of RAS. Oncogene 18, 5936–5942 [DOI] [PubMed] [Google Scholar]
- 34. Gao S. B., Hua X., Jin G. H. (2008) Menin regulates endocrine diseases by controlling histone modification and gene transcription. Ann. Endocrinol. (Paris) 69, 426–432 [DOI] [PubMed] [Google Scholar]
- 35. Jin S., Mao H., Schnepp R. W., Sykes S. M., Silva A. C., D'Andrea A. D., Hua X. (2003) Menin associates with FANCD2, a protein involved in repair of DNA damage. Cancer Res. 63, 4204–4210 [PubMed] [Google Scholar]
- 36. Guan K. L., Jenkins C. W., Li Y., Nichols M. A., Wu X., O'Keefe C. L., Matera A. G., Xiong Y. (1994) Growth suppression by p18, a p16INK4/MTS1- and p14INK4B/MTS2-related CDK6 inhibitor, correlates with wild-type pRb function. Genes Dev. 8, 2939–2952 [DOI] [PubMed] [Google Scholar]
- 37. Hirai H., Roussel M. F., Kato J. Y., Ashmun R. A., Sherr C. J. (1995) Novel INK4 proteins, p19 and p18, are specific inhibitors of the cyclin D-dependent kinases CDK4 and CDK6. Mol. Cell Biol. 15, 2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Karnik S. K., Chen H., McLean G. W., Heit J. J., Gu X., Zhang A. Y., Fontaine M., Yen M. H., Kim S. K. (2007) Menin controls growth of pancreatic beta-cells in pregnant mice and promotes gestational diabetes mellitus. Science 318, 806–809 [DOI] [PubMed] [Google Scholar]
- 39. Jackson E. L., Willis N., Mercer K., Bronson R. T., Crowley D., Montoya R., Jacks T., Tuveson D. A. (2001) Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 15, 3243–3248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Risch A., Plass C. (2008) Lung cancer epigenetics and genetics. Int. J. Cancer 123, 1–7 [DOI] [PubMed] [Google Scholar]
- 41. Rhee I., Bachman K. E., Park B. H., Jair K. W., Yen R. W., Schuebel K. E., Cui H., Feinberg A. P., Lengauer C., Kinzler K. W., Baylin S. B., Vogelstein B. (2002) DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416, 552–556 [DOI] [PubMed] [Google Scholar]
- 42. Kwon O., Jeong S. J., Kim S. O., He L., Lee H. G., Jang K. L., Osada H., Jung M., Kim B. Y., Ahn J. S. (2010) Modulation of E-cadherin expression by K-Ras. Involvement of DNA methyltransferase-3b. Carcinogenesis 31, 1194–1201 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y., Ozawa A., Zaman S., Prasad N. B., Chandrasekharappa S. C., Agarwal S. K., Marx S. J. (2011) The tumor suppressor protein menin inhibits AKT activation by regulating its cellular localization. Cancer Res. 71, 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Milne T. A., Hughes C. M., Lloyd R., Yang Z., Rozenblatt-Rosen O., Dou Y., Schnepp R. W., Krankel C., Livolsi V. A., Gibbs D., Hua X., Roeder R. G., Meyerson M., Hess J. L. (2005) Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. U.S.A. 102, 749–754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Karnik S. K., Hughes C. M., Gu X., Rozenblatt-Rosen O., McLean G. W., Xiong Y., Meyerson M., Kim S. K. (2005) Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. U.S.A. 102, 14659–14664 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.