

Background: Formation of disulfide bonds between exposed cysteines decreases activity of T7 DNA polymerase.
Results: Only Cys-275 and Cys-313 on the thioredoxin binding domain of T7 DNA polymerase are solvent exposed.
Conclusion: Cys-313 is critical for the interaction of T7 DNA polymerase with its processivity factor, thioredoxin.
Significance: A single cysteine in the thioredoxin binding domain of T7 DNA polymerase is important for activity.
Keywords: Bacteriophage, Cysteine-mediated Cross-linking, DNA Polymerase, DNA Replication, Thioredoxin, DNA Synthesis, Phage T7
Abstract
Gene 5 protein (gp5) of bacteriophage T7 is a non-processive DNA polymerase. It achieves processivity by binding to Escherichia coli thioredoxin (trx). gp5/trx complex binds tightly to a primer-DNA template enabling the polymerization of hundreds of nucleotides per binding event. gp5 contains 10 cysteines. Under non-reducing condition, exposed cysteines form intermolecular disulfide linkages resulting in the loss of polymerase activity. No disulfide linkage is detected when Cys-275 and Cys-313 are replaced with serines. Cys-275 and Cys-313 are located on loop A and loop B of the thioredoxin binding domain, respectively. Replacement of either cysteine with serine (gp5-C275S, gp5-C313S) drastically decreases polymerase activity of gp5 on dA350/dT25. On this primer-template gp5/trx in which Cys-313 or Cys-275 is replaced with serine have 50 and 90%, respectively, of the polymerase activity observed with wild-type gp5/trx. With single-stranded M13 DNA as a template gp5-C275S/trx retains 60% of the polymerase activity of wild-type gp5/trx. In contrast, gp5-C313S/trx has only one-tenth of the polymerase activity of wild-type gp5/trx on M13 DNA. Both wild-type gp5/trx and gp5-C275S/trx catalyze the synthesis of the entire complementary strand of M13 DNA, whereas gp5-C313S/trx has difficulty in synthesizing DNA through sites of secondary structure. gp5-C313S fails to form a functional complex with trx as measured by the apparent binding affinity as well as by the lack of a physical interaction with thioredoxin during hydroxyapatite-phosphate chromatography. Small angle x-ray scattering reveals an elongated conformation of gp5-C313S in comparison to a compact and spherical conformation of wild-type gp5.
Introduction
The use of sliding clamps by replicative DNA polymerases to achieve high processivity is conserved throughout evolution (1–3). Moreover, these sliding clamps provide platforms onto which other replication proteins can assemble. For example, DNA polymerase III of Escherichia coli must have the β-clamp, a ring-shaped protein preassembled on the DNA before the polymerase can productively bind to the primer-template. The polymerase, then tethered to the β-clamp, can polymerize thousands of nucleotides without dissociation from the DNA (4). Bacteriophage T4 DNA polymerase also requires a sliding clamp for processivity as well as a docking site for other proteins (5, 6). Similarly, in eukaryotes, the proliferating cell nuclear antigen forms a ring-shaped structure around DNA and increases the processivity of the polymerase (1). In most instances, the binding of the sliding clamp to DNA requires the assistance of a clamp loader.
In contrast to the sliding clamps, several viruses have evolved different mechanisms to achieve processivity of the polymerase. The DNA polymerase encoded by bacteriophage φ29, for example, effectively mediates processive strand-displacement synthesis on duplex DNA without accessory proteins (7). Similarly, the DNA polymerase of bacteriophage T5 alone polymerizes hundreds of nucleotides per binding event (8). The DNA polymerase (gp5)4 of bacteriophage T7, the subject of this study, is a non-processive polymerase and requires a processivity factor (9–11). However, its structure and assembly onto the DNA differ from the sliding clamps discussed above. T7 gp5 dissociates from a primer-template after incorporation of less than 50 nucleotides. To achieve high processivity, gp5 forms a 1:1 complex with a host protein, thioredoxin (trx), with an apparent dissociation constant KD of 5 nm (9). The binding of trx to gp5 allows the polymerase to incorporate hundreds of nucleotides per polymerization cycle without dissociating from DNA (9–11).
gp5 is classified as a member of the polymerase I family based on sequence homology (12, 13). Comparison of the sequence of T7 gp5 to E. coli DNA polymerase I reveals the existence of a 76-amino acid segment in gp5 that is absent from E. coli DNA polymerase I. Genetic and biochemical studies (14, 15) have shown that this 76-amino acid segment is responsible for the interaction with trx; deletion of this domain abolishes the ability of the polymerase to bind trx and consequently decreases the rate of polymerization of nucleotides due to the loss of processivity. Insertion of this region into DNA polymerase I of E. coli confers a trx-dependent processivity of the polymerase, indicating that this segment itself is sufficient for binding of trx (16). The 76-amino acid region is, therefore, also known as the thioredoxin binding domain (TBD). The crystal structure of gp5/trx (17, 18) captured in a polymerization mode shows the TBD as a flexible loop extended between helices H and H1 of the thumb (Fig. 1). The thumb along with the palm and fingers forms a DNA binding groove with the palm forming the base of a cleft and the fingers and thumb forming the cleft sides (Fig. 1). In this structure trx appears to bind to the extended loop of gp5 and is rotated slightly up and away from the cleft in which the primer-DNA template lies.
FIGURE 1.

Structure of T7 gp5/trx. The x-ray crystallographic structure of gp5/trx in complex with a primer-DNA template and a deoxynucleoside 5′-triphosphate was captured in a polymerization mode at 2.2 Å resolution (18). Primer-DNA template is shown in cyan. gp5 is present in blue with the three subdomains: palm, finger, and thumb. The three subdomains form a DNA binding groove leading to the active site containing the deoxynucleoside 5′-triphosphate. The processivity factor, trx depicted in gray, is bound to gp5 at a flexible loop of 76-amino acids, designated the TBD. The TBD, shown in orange, extends from the thumb. Inset, enlargement of the TBD and trx interface. In this structure, Cys-275 and Cys-313 on the TBD of gp5 and the active sites cysteines Cys-32 and Cys-35 of trx are highlighted in green. Thr-327 is highlighted in pink, whereas basic residues are highlighted in red.
The precise molecular mechanism by which trx mediates processivity of gp5 is not well understood. Trx is a versatile protein originally isolated as a hydrogen donor for ribonucleotide reductase (19–21). The activity of trx in this role relies on two-redox active cysteines in an exposed active loop of a conserved sequence 32CGPC35. The two cysteines can form a disulfide linkage and can participate in reversible oxidation-reduction reactions with other proteins. However, the reducing power of the active site cysteines is not required in the polymerase catalytic reaction as in the case for trx in the other reactions in which it participates (19–21). Both Cys-32 and Cys-35 can be replaced with a residue that abolishes the ability of trx to undergo oxidation-reduction reactions, but these altered trx can form functional gp5/trx complexes in vitro (22, 23). The altered trx do, however, have a reduced binding affinity for gp5, suggesting that the active cysteine sites play a structural role. Indeed, the crystal structure of gp5/trx reveals that one of the two cysteines, Cys-32, hydrogen bonds with Thr-327 (highlighted in pink) in the TBD (Fig. 1).
The TBD of gp5 contains a number of basic residues located in two loops, designated loop A and B (14). These basic residues are in position to contact the phosphodiester backbone of the DNA in the binding cleft of gp5 (Fig. 1). It has been previously shown that the gp5/trx complex remains tightly bound and slides along the DNA while diffusing, whereas gp5 without trx diffuses along the DNA using a hopping mechanism (24). Thus, binding of trx to gp5 may expose a number of basic residues, and the increased affinity of the gp5/trx complex for duplex DNA may be the result of increased electrostatic interactions. It is likely that in the crystal structure shown in Fig. 1, gp5/trx has been captured in a non-processive mode. In a processive mode the thumb and bound trx are postulated to swing down onto the duplex region of the primer-template to prevent the DNA from dissociation before the next polymerization cycle. Suppressor analysis of a genetically altered trx supports this hypothesis (25). Amino acids in gp5 that restore the ability of the altered trx to confer processivity on the polymerase reside within the TBD, whereas another is located within the exonuclease domain. The latter site is interesting because it raises the possibility that the extended loop of the TBD might swing down and dock on the lip of the crevice located within the exonuclease domain, thus encircling gp5/trx around the DNA (26). Moreover, trx does indeed sit over the DNA as shown by footprinting data (27). Loops A and B in the TBD also serve as docking sites for the acidic C termini of the T7 DNA helicase and the T7 ssDNA-binding protein, both essential components of the T7 replisome. The ability of gp5 to bind to this acidic tail of the helicase provides a mechanism for gp5/trx to be captured by the helicase in the event it transiently dissociates from the DNA, thus increasing the processivity of leading strand synthesis (28, 29).
gp5 has 10 cysteine residues distributed throughout the protein sequence; in contrast, E. coli DNA polymerase I of the same family has only one cysteine. Of the 10 cysteines, Cys-275 and Cys-313 are located on loop A and B, respectively, of the TBD (Fig. 1). The crystal structure suggests these two cysteines are exposed to the solvent and protein-protein binding surface (17, 18). The unique location of these two cysteines encouraged us to study their potential role in protein-protein interaction as well as polymerase function of gp5. We have replaced each of exposed cysteines with serine and examined the effect of the mutation on the function of gp5 both in vitro and in vivo. In this study we describe yet another role of trx: the binding of trx to gp5 sequesters an otherwise solvent-exposed cysteine that can form intermolecular disulfide bonds.
EXPERIMENTAL PROCEDURES
Bacterial Strains, Plasmids, Bacteriophage T7, and DNA Templates
E. coli A307 (HrfC, trxA−) lacking the trx coding gene and T7Δ5 phage lacking gene 5 were constructed by Stanley Tabor (Harvard Medical School). M13 mp18 phage purchased from Novagene was used to purify M13 ssDNA using a Lambda Maxi kit (Qiagen Inc.). Plasmids pGP5-3 expressing wild-type T7 gene 5 and pTrx-3 expressing E. coli trxA were generated from parent vector pT7-1 (30) in which the expression of gene 5 and trxA were under the control of a T7 promoter. Similarly, plasmid pTrxA expressing trxA derived from pET-24a backbone (Novagene) was used for a complementation assay. 25-nt oligo(dT) to 350-nt poly(dA) were purchased from Amersham Biosciences. Oligonucleotides used for site-directed mutagenesis and primers for DNA polymerase assay were purchased from Integrated DNA Technologies Inc.
Mutagenesis of T7 Gene 5 and Trx
Plasmids pGP5-C275S, pGP5-C313S, and pGP-5C275S/C313S were constructed using QuikChange II XL site-directed mutagenesis kit (Stratagene). The parent plasmid used for mutagenesis of gene 5 was pGP5-3. The presence of the desired mutation was confirmed by DNA sequencing.
Overproduction and Purification of Proteins
Wild-type-gp5, gp5-C275S, gp5-C313S, and gp5-C275S/C313S were overproduced in E. coli A307 (DE3) (lacking gene trxA) using plasmids pGP5-3 and purified to homogeneity following the procedures described previously (31). Wild-type trx was overproduced in E. coli BL21(DE3) using plasmid pTrx-3 and purified as previously described (10).
Formation of Disulfide Bonds between Exposed Cysteines of gp5
Non-reduced conditions for gp5 were generated by passing 30 μg of protein through a G-25 column (GE Healthcare) by centrifugation at 3000 rpm for 2 min at 4 °C to remove dithiothreitol (DTT) present in the solution. Samples were collected and incubated on ice for 30 min with or without the addition of 1 mm DTT. Samples were then electrophoresed on pre-casted 4–20% SDS-PAGE (Bio-Rad) either under reduced or non-reduced conditions by using protein loading buffer containing or lacking 1 mm DTT, respectively.
Complementation of T7Δ5 Phage Growth
Complementation assays to examine the ability of altered gp5, gp5-C275S, and gp5-C313S to support growth of phage T7Δ5 (lacking gene 5) were carried out as described previously (31). Briefly, 100 μl of 7-fold serial dilutions (10−7) of T7Δ5 phage were mixed with a 400 μl of cell culture of E. coli A307 (trxA−) carrying both pGP5-3 (ampicillin resistance) and pTrxA (kanamycin resistance) plasmids at A600 = 1 and 3 ml of soft agar (1% Tryptone, 0.5% yeast extract, 1% NaCl, and 0.7% agar). Mixtures were plated on LB plates (1% Tryptone, 0.5% yeast extract, 1% NaCl, and 1.5% agar) containing appropriate antibiotics. The expression of gene 5 and trxA were under the control of a T7 promoter that was induced upon the expression of T7 RNA polymerase after T7Δ5 phage infection. The plates were then incubated at 37 °C for 6 h. The efficiency of plating was determined by dividing the number of plaque-forming units obtained with each altered gp5 against the number of plaques obtained with wild-type gp5.
DNA Polymerase Assays
The circular DNA template used for DNA synthesis was prepared by annealing a 24-mer oligonucleotide (M13 primer −47) to M13 mp18 ssDNA at a molar ratio of 1:1. The homopolymeric DNA template was prepared by annealing a 25-nt oligo(dT) to 350-nt poly(dA) (dT25/dA350). The DNA polymerase assay for the primed M13 ssDNA contained 40 mm Tris-HCl, pH 7.5, 50 mm NaCl, 10 mm MgCl2, 5 mm DTT, 250 μm each of dGTP, dATP, and dCTP, [3H]dTTP (10 cpm/pmol), 20 nm primed M13 ssDNA, and 5 nm gp5 with a 40-fold excess of trx. The reactions were carried out at 37 °C and stopped by the addition of EDTA to a final concentration of 25 mm. Aliquots were spotted on DE81 ion exchange filters. Unincorporated radiolabeled nucleotides were removed by washing the filters with three successive 5-min washes in 300 mm ammonium formate (pH 8.0). Filters were dried, and the amount of [3H]dTMP incorporated into DNA was determined by liquid scintillation counting.
Reactions to monitor polymerase activity on linear DNA substrate were similar to those for primed M13 ssDNA except that dT25/dA350 was added at a concentration of 200 nm, resulting in a 40-fold molar excess of DNA over polymerase. The reactions were incubated at 25 °C.
Processivity Assay
Processivity assays were carried out on a circular M13 ssDNA primed with a 5′ 32P-labeled 24-nt primer (M13 primer −47). Primers for processivity assays were labeled with [γ-32P] ATP at the 5′ end using T4 polynucleotide kinase (New England Biolabs). The labeling reaction was incubated at 37 °C for 30 min followed by heating at 65 °C for 10 min. M13 ssDNA was primed by annealing with the labeled 24-nt primer in 50 mm Tris-HCl, pH 7.5, and 50 mm NaCl. Primed M13 DNA was purified using S-400 spin columns (Amersham Biosciences).
The DNA polymerase reaction (100 μl) contained 50 mm Tris-HCl, pH 7.5, 0.1 mm DTT, 50 mm NaCl, 10 mm MgCl2, 250 μm each of four dNTPs, 5 nm wild-type or altered gp5, 200 nm trx, and 40 nm 32P-labeled primed M13 ssDNA. Reaction mixtures were incubated at 37 °C. At the indicated times aliquots (20 μl) were removed, and the reaction was terminated by EDTA (25 mm). The reaction products were separated by electrophoresis on a 0.6% alkaline agarose gel. The gel was dried and exposed to a phosphorimaging plate followed by scanning with a Fuji BAS 1000 Bio-Imaging Analyzer.
Gel-shift Assays of gp5-DNA Binding
100 nm 32P-labeled primed linear DNA primer/template and increasing amount of proteins (gp5 wt or gp5-C313S: 0–10 μm) were incubated for 30 min in ice in binding buffer containing Tris-HCl, pH 7.5, 5 mm DTT, and 50 mm potassium glutamate. Reactions (10 μl) were resolved by electrophoresis on 10% acrylamide gels in 0.5 × Tris borate EDTA buffer at 50 V for 3 h. The gel was dried and exposed to a phosphorimaging plate and analyzed using a Fuji BAS 1000 Bio-Imaging Analyzer.
Analysis of Solvent-accessible Surface Area of Cysteines of gp5
The relative surface accessibility for all cysteines of gp5 was calculated using Naccess (32). Naccess calculates the solvent-accessible surface that is defined by rolling a probe with the size of a water molecule around a Van der Waals surface.
Hydroxyapatite-Phosphate Chromatography
Hydroxyapatite chromatography in phosphate buffer provides an effective method to separate free gp5 from gp5/trx (11). A mixture of gp5 (5 μm) and trx (50 μm) were incubated at 4 °C overnight in buffer containing 20 mm potassium phosphate, pH 7.4, 1 mm EDTA, 2 mm sodium citrate, and 10% glycerol. The mixture was then dialyzed twice against 500 ml of the same buffer. The dialyzed protein was loaded onto a prepacked column of hydroxylapatite (Bio-Rad) at a flow rate of 1 ml/min using a FPLC system (AKTA, GE Healthcare). The column was then washed with 100 ml of the same buffer. Bound proteins were eluted with a 100-ml linear gradient (20–250 mm) of potassium phosphate, pH 7.4, containing 2 mm sodium citrate, 1 mm DTT, and 10% glycerol. Peaks of protein elution were monitored at A280.
Small Angle X-ray Scattering (SAXS)
SAXS analysis of wild-type gp5 and gp5-C313S was measured at the National Synchrotron Light Source (Upton, NY) on beamline X-9 as previously described (27, 33). Measurements were performed in a buffer containing 20 mm potassium phosphate, pH 7.4, 300 mm NaCl, 1 mm DTT, and 1 mg/ml of gp5 at 15 °C. The magnitude of the scattering vector (q) is defined as,
 |
where 2θ is the scattering angle. The experimental SAXS data for all samples showed a linear behavior (see Fig. 8) in the low q, Guinier region, indicating that gp5 did not undergo aggregation. Radii of gyration (Rg)were derived from data in the qRg < 1.3 region using the Guinier approximation,
 |
The scattering curve reflects structural characteristics in reciprocal space. The software GNOM (34) was used to translate the scattering profiles into real space by Fourier transformation, resulting in the pairwise distance distribution function p(r). The p(r) function is proportional to the particle electron density and reflects the distances between pairs of scattering points within the macromolecule, permitting determination of the maximum intermolecular diameter (Dmax). The overall three-dimensional shape of gp5-C313S was restored as described previously (27).
FIGURE 8.
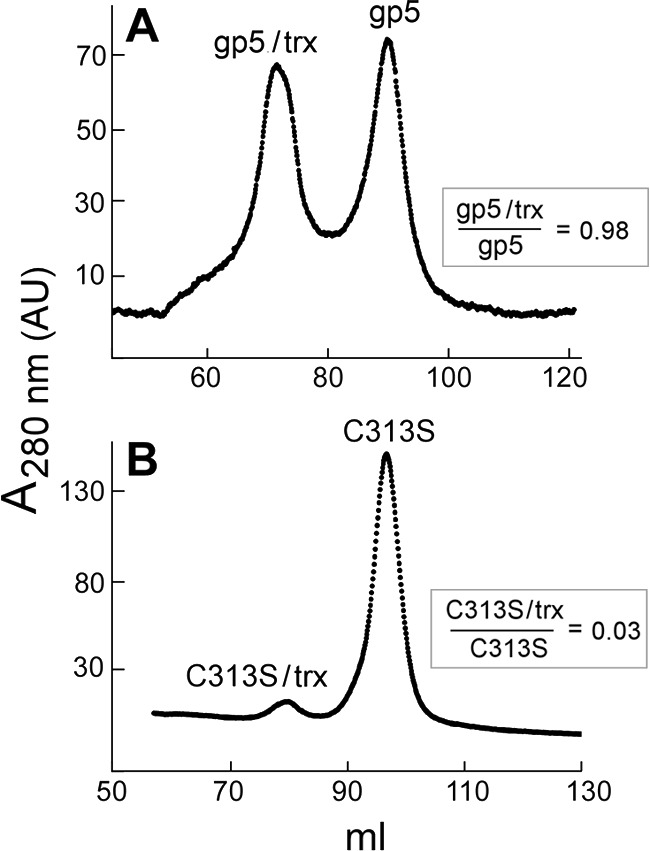
Physical interaction of wild-type gp5 and gp5-C313S with trx. Separation of wt gp5/trx from free wt gp5 was performed on a hydroxyapatite chromatography in phosphate buffer as described under “Experimental Procedures.” Wild-type gp5 or gp5-C313S (5 μm) was premixed with trx (50 μm) and dialyzed against 500 ml of a buffer containing 10 mm potassium phosphate, pH 7.4, 1 mm DTT, 2 mm sodium citrate, and 10% glycerol. Dialyzed proteins were loaded onto the hydroxyapatite column using a FPLC system. Proteins were eluted with a 100-ml gradient from 10 to 250 mm potassium phosphate, pH 7.4. A, a sample containing wild-type gp5 and trx is shown. B, a sample containing gp5-C313S and trx is shown.
RESULTS
Exposed Cysteines of gp5 Form Intermolecular Disulfide Bonds under Non-reduced Conditions
T7 gp5 has 10 cysteines distributed throughout the protein sequence. When gp5 is stored at 4 °C in the absence of a reducing agent such as DTT, there is a progressive loss in activity (> 40% after overnight incubation). A likely explanation for this phenomenon is the formation of disulfide bonds (S-S) between exposed cysteines, resulting in protein aggregation. We examined the solvent accessibility of 10 cysteines of gp5/trx using Naccess (32). Results reveal that only Cys-275 and Cys-313 that are located in loop A and loop B, respectively, of the TBD are potentially exposed to the solution. Cys-275 and Cys-313 have 43.7 and 24.2%, respectively, of the atom area exposed to the solvent (Table 1, in bold). Other cysteines have no or limited exposure to the protein surface (<2.1%).
TABLE 1.
Solvent accessibility of cysteine residues in T7 gp5/trx
The relative surface accessibility for all cysteines of gp5/trx complex was calculated using the program Naccess (32). Naccess calculates the solvent-accessible surface that is defined by rolling a probe with the size of a water molecule around a Van der Waals surface of a protein from a PDB file. The values represent the surface accessibility of each cysteine based on the crystal structure of T7 gp5/trx (PDB code 1T8E) (18). “Side chain” refers to the fraction of amino acid side chain that is exposed to the solvent, “Main chain” refers to the fraction of main chain (not including the side chain) that is exposed to the solvent, and “All atoms” refers to the entire surface area of the amino acid that is exposed to the solvent.
| Residue no. | All atoms | Side chain | Main chain |
|---|---|---|---|
| % | % | % | |
| Cys-20 | 0 | 0 | 0 |
| Cys-88 | 0.4 | 0.2 | 1 |
| Cys-275 | 43.7 | 50 | 27.2 |
| Cys-313 | 24.2 | 25.6 | 20.4 |
| Cys-451 | 0.5 | 0.6 | 0 |
| Cys-483 | 0 | 0 | 0 |
| Cys-622 | 0.4 | 0.6 | 0 |
| Cys-660 | 0 | 0 | 0 |
| Cys-688 | 2.1 | 0.7 | 5.6 |
| Cys-703 | 1.5 | 0 | 5.5 |
| Cys-32a | 0.1 | 0 | 0.4 |
| Cys-35a | 2.6 | 0.3 | 8.8 |
a Cysteine in redox-active site of trx in complex with T7 gp5.
We introduced site-specific mutations into the pGP5-3 plasmid to change the codon TGT for Cys-275 and Cys-313 of gp5 to AGT and the codon for serine to obtain the altered proteins gp5-C275S and gp5-C313S, respectively. gp5-C275S/C313S, in which both of these cysteines are changed to serine, was also constructed. We purified wild-type gp5, gp5-C275S, gp5-C313S, and gp5-C275S/C313S and examined them for their ability to form disulfide bonds under non-reducing conditions (see “Experimental Procedures”). After incubation for 30 min in the absence or presence of DTT, the proteins were analyzed by SDS-PAGE. The results shown in Fig. 2A demonstrate that in the presence of the reducing agent DTT, both wild-type and altered gp5 migrated as a single band corresponding to monomers of gp5 (lanes 1–4). However, in the absence of DTT along with monomer, a significant portion of wild-type gp5 migrated at positions corresponding to dimers and trimers. In addition to monomers, dimers are also detected in the case of gp5-C275S (lane 6) and gp5-C313S (lane 7). No oligomeric forms were detected when both cysteines were replaced with serines as seen in lane 8 containing gp5-C275S/C313S. The results confirm that only Cys-275 and Cys-313 of gp5 are solvent-exposed and can form intermolecular disulfide bonds, most likely the cause of the loss of activity of gp5. Based on these results, DTT is added to storage buffer and to routine enzymatic reactions involving gp5 to achieve full enzyme activity.
FIGURE 2.

SDS-PAGE analysis of gp5 under reduced and non-reduced conditions. The reducing agent (DTT) is removed from the protein sample using a G-25 column (see “Experimental Procedures”). A, lanes 1–4, non-reduced protein samples were preincubated with 1 mm DTT followed by mixing with the loading buffer containing 5 mm DTT. Lanes 5–8, non-reduced protein samples were mixed with the loading buffer lacking DTT to create non-reduced conditions during gel electrophoresis. Lanes 1 and 5, wild-type gp5; lanes 2 and 6, gp5-C275S; lanes 3 and 7, gp5-C313S; lanes 4 and 8, gp5-C275S/C313S. Gel electrophoresis was carried out on 4 - 20% linear gradient SDS-polyacrylamide gel. B, shown is the effect of trx on the disulfide bond formation. Polymerase was mixed with 50-fold excess of trx. Samples were then prepared and analyzed as described for panel A for non-reduced samples.
In the presence of trx, neither wild-type gp5 nor the three altered gp5s form intermolecular disulfide linkages (Fig. 2B, lanes 9–12). This result suggests that trx binds to the TBD over the region containing Cys-275 and Cys-313, thus burying them in the protein-protein interface. These results taken together are important in that they imply that binding of trx to TBD of gp5 protect it from the activity loss due to the formation of the intermolecular disulfide bonds.
DNA Polymerase Activity in the Absence of Trx
T7 gp5 alone has low processivity. It dissociates from a primer-DNA template after incorporation of less than 50 nucleotides (9, 11). We first examined the effect of the replacement of Cys-257 and Cys-313 with a serine on DNA synthesis catalyzed by gp5 in the absence of trx. In this assay we used the homopolymer primer-DNA template, dT25/dA350 because it lacks secondary structure that impedes the non-processive gp5 (23). As shown in Fig. 3, despite low processivity, the amount of [3H]dTMP incorporated into DNA is proportional to the amount of gp5 present. Replacement of either Cys-275 or Cys-313 with serine resulted in a dramatic decrease in the activity of gp5. The initial rate of DNA synthesis catalyzed by gp5-C275S, gp5-C313S, or gp5-C275S/C313S is ∼15-fold lower than that observed with wild-type gp5. We compared the binding affinity of wild-type and mutant gp5 to DNA template. Because both cysteines are located within the TBD in the thumb of gp5, the alterations unlikely lead to a defect in the catalytic core of the polymerase that is distal to the TBD. We carried out gel-shift assays to compare the binding affinity of wt gp5 and gp5-C313S to primer-DNA template. Surprisingly, gp5-C313S and wt gp5 showed comparable binding affinity to DNA (Fig. 4). The results suggest that the reduction in polymerase activity of the altered gp5 is due to an improper functional conformation created by the mutations rather than a reduction in physical binding to DNA.
FIGURE 3.

DNA polymerase activity of gp5 in the absence of trx. Standard reaction mixtures (20 μl) containing 200 nm dA350/dT25 DNA substrate, 250 μm each of dATP, dCTP, dGTP, and [3H]dTTP (10 cpm/pmol), and increasing amounts of the indicated gp5 in the standard reaction mixture containing DTT were incubated at 25 °C for 2 min. The reactions were stopped by the addition of 25 mm EDTA, and the amount of [3H]dTMP incorporated into DNA was determined as described under “Experimental Procedures.” ■, wt gp5; ○, gp5-C275S; ▴, gp5-C313S; ♦, gp5-C275S/C313S.
FIGURE 4.
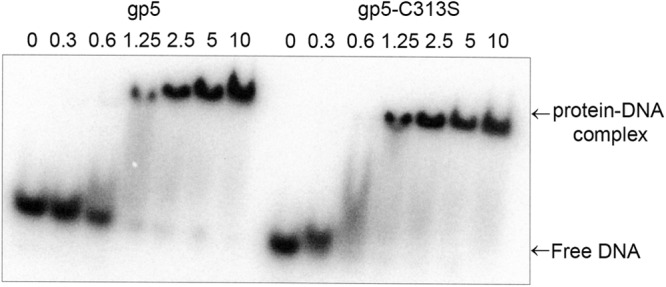
Mobility gel-shift assay of binding of gp5 wt and gp5-C313S to primer-DNA template. Binding and gel analysis was performed as described under “Experimental Procedures.” The picture shows gel retardation of 32P-labeled linear primer-DNA template as a function of increasing concentrations of protein (gp5 wt or gp5-C313S: 0–10 μm) as indicated.
DNA Polymerase Activity in the Presence of Trx
The addition of trx to reaction mixtures containing gp5 has a dramatic effect on the rate of DNA synthesis (Fig. 5). The addition of trx nearly restores DNA synthesis of gp5-C275S to that observed with wild-type gp5/trx using the relatively short homopolymeric primer/template (Fig. 5A). Wild-type gp5/trx and gp5-C275S/trx both incorporate nucleotides at an initial rate of ∼75,000 pmol/μg/min. gp5-C313S/trx and gp5-C275S/C313S/trx have initial rates ∼50% that observed with wild-type gp5/trx. The results indicate that alteration of two cysteines in the TBD do not indeed impair the catalytic core of gp5.
FIGURE 5.
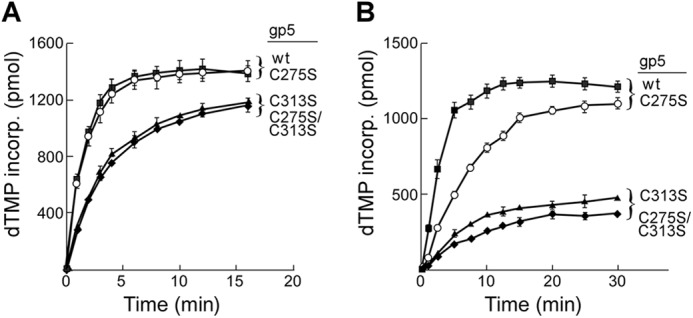
DNA polymerase activity of gp5 in the presence of trx. DNA synthesis was determined by measuring the amount of [3H]dTMP incorporated into DNA over time as described under “Experimental Procedures.” A, DNA polymerase activity on a homopolymer primer-template is shown. Reactions (200 μl) contained 50 mm Tris-HCl, pH 7.5, 5 mm DTT, 50 mm NaCl, 10 mm MgCl2, 250 μm each of dGTP, dATP, dCTP, and [3H]dTTP (10 cpm/pmol), 200 nm dA350/dT25 substrate, 5 nm each of the indicated gp5s, and 250 nm trx. Reaction mixtures were incubated at 25 °C. Aliquots of 20 μl were removed at the indicated times, and the reaction was stopped by the addition of 25 mm EDTA. B, DNA polymerase activity on primed M13 ssDNA is shown. Reactions were similar to those described in A except that dA350/dT25 was replaced by 20 nm M13ss DNA primed with 24-nt primer. Additionally, reactions involving primed M13 ssDNA were incubated at 37 °C. ■, wt gp5; ○, gp5-C275S; ▴, gp5-C313S; ♦, gp5-C275S/C313S.
The dT25/dA350 primer-template is too short to provide meaningful information on the processivity of these gp5/trx complexes. Therefore, we have compared the effect of trx on DNA synthesis mediated by wild-type gp5 and the altered gp5 on primed M13 ssDNA. M13 ssDNA has sites of secondary structure along its ∼7000 nucleotide length that can slow gp5/trx during DNA synthesis (31, 35). Wild-type gp5/trx has a processivity of hundreds of nucleotides per binding event on primed M13 ssDNA (9) and that processivity is reflected in the high rate of DNA synthesis observed on M13 ssDNA (Fig. 5B). The initial rate of DNA synthesis catalyzed by gp5-C275S/trx is more than 50% that observed with wild-type gp5/trx (Fig. 5B) and, as shown below, like wild-type, gp5/trx is able to copy most of the M13 ssDNA after 30 min of incubation. In contrast, gp5-C313S/trx and gp5-C275S/C313S/trx demonstrated significantly reduced rates of synthesis on the long DNA, ∼10% that observed with wild-type gp5/trx. Moreover, the extent of DNA synthesis catalyzed by gp5-C313S/trx and gp5-C275S/C313S/trx over 30 min is considerably lower than that obtained with wild-type gp5/trx.
To determine definitively if the reduced activity observed with gp5-C313S and gp5-C275S/C313S/trx is a result of reduced processivity, we compared their processivity to that of wild-type gp5/trx. In this assay the product obtained during synthesis on M13 ssDNA with a 5′-32P-labeled primer wherein the primer-template is in an 8-fold molar excess over gp5/trx is measured. Under these conditions at short times the synthesis measured is the result of a single binding event. As shown in Fig. 5, wild-type gp5-/trx replicates an entire ssM13 DNA molecule (∼7 kb) within 5 min of incubation as judged by the sizes of the replication products on an alkaline agarose gel. However, a number of strong pause sites are also observed. gp5-C275S/trx replicates the entire M13 DNA molecule within 15 min, but the amount of primer fully extended is far less than that found with wild-type gp5/trx. In sharp contrast, gp5-C313S/trx is far less processive (Fig. 6). This altered polymerase synthesizes only short DNA products less than 1 kb in length; no fully extended products were detected after 20 min of incubation. The results show that gp5-C313S/trx has low processivity compared with wild-type gp5/trx.
FIGURE 6.

Processivity of wild-type gp5, gp5-C275S, and gp5-C313S. Processivity assays were carried out on a M13 ssDNA primed with a 5′ 32P-labeled 24-nt primer in the presence of trx as described under “Experimental Procedures.” DNA synthesis reactions (100 μl) contained 50 mm Tris-HCl, pH 7.5, 5 mm DTT, 50 mm NaCl, 10 mm MgCl2, 250 μm each of dGTP, dATP, dCTP, and dTTP, 5 nm gp5, 250 nm trx, and 40 nm 32P-labeled primed M13 ssDNA. Reaction mixtures were incubated at 37 °C. At the indicated times, aliquots (20 μl) were removed, and reactions were stopped by the addition of 25 mm EDTA. The reaction products were separated on a 0.6% alkaline agarose gel.
Binding Affinity of gp5 to Trx
Trx binds tightly to gp5 (KD 5 nm) and increases the affinity of the DNA polymerase for a primer-DNA template that in turn leads to a dramatic stimulation of the polymerase activity (9, 11). Single molecule biophysical assays show a 10–50-fold increase in processivity of gp5 upon binding of trx (36). We examined the ability of wild-type gp5 and the altered gp5 to form active complexes with trx by measuring the kinetic of incorporation of [3H]dTMP into DNA using dT25/dA350 primer-template as previous described (31). In these experiments (Fig. 7A) activity is measured as a function of trx concentration. The observed equilibrium binding constants (Kobs) that were obtained from Scatchard plots of this data are presented in Fig. 7B. The observed equilibrium binding constant Kobs for gp5-C275S is 17 nm, not too different from that observed (10 nm) for wild-type gp5. In contrast, the Kobs constants obtained with gp5-C313S and gp5-C275S/C313S were 6- and 10-fold higher, respectively, than that for wild-type gp5.
FIGURE 7.
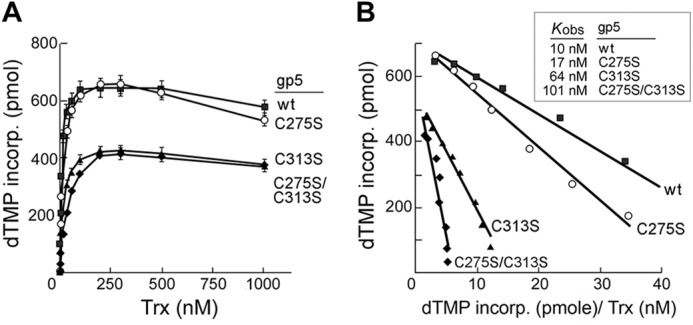
Binding affinity of wild-type gp5 and altered gp5 to trx. A, dependence of trx concentration on initial rate of DNA polymerase activity of gp5/trx is shown. Reactions (20 μl) contained 5 nm concentrations of the indicated gp5, 50 mm Tris-HCl, pH 7.5, 5 mm DTT, 50 mm NaCl, 10 mm MgCl2, 250 μm each of dGTP, dATP, dCTP, and [3H]TTP (10 cpm/pmol), 200 nm dA350/dT25 substrate. Increasing amounts of trx was added to each reaction as indicated. Reactions were incubated at 25 °C. The amount of DNA synthesis for each reaction was determined by measuring the amount of [3H]dTMP incorporated over 1 min as described under “Experimental Procedures.” B, shown is the determination of the observed equilibrium binding constant, Kobs. The data shown in A were used to generate Scatchard plots of initial rates versus ratio of initial rates to added concentration of trx. Kobs is equal to the negative slope of the corresponding curve. ■, wt gp5; ○, gp5-C275S; ▴, gp5-C313S; ♦, gp5-C275S/C313S.
Physical Interaction of gp5 with Trx
Hydroxyapatite chromatography in phosphate buffer can be used effectively to resolve gp5 from gp5/trx (11). We, therefore, used this method to examine the binding of wild-type gp5 and gp5-C313S to trx. The elution profile of a premixed sample of wild-type gp5 with trx is shown in Fig. 8A. Protein eluted in two peaks containing essentially the same amount of protein. Analysis of peak samples on SDS-PAGE show that the earlier-eluting peak contains wt gp5 and trx at a ratio of 1:1, whereas the later-eluting peak contains only wt gp5. Results suggest that wt gp5 bound tightly to trx to form the stable complexes that bound to HA column and co-eluted. The mole ratio of trx to gp5 used in this experiment was 5 to 1, and we found a significant amount of free wt gp5 remained. Probably a higher mole ratio of trx to gp5 is required to saturate free gp5. In contrast, when gp5-C313S and trx were premixed and then chromatographed on hydroxyapatite-phosphate, essentially all (>95%) of gp5-C313S eluted at the position corresponding to free gp5-C313S; only a small portion (<5%) of gp5-C313S eluted at the position corresponding to gp5-C313S/trx complexes (Fig. 8B). The results show that gp5-C313S fails to form a stable complex with trx on hydroxyapatite-phosphate.
SAXS Analysis of gp5
SAXS allows the determination of the solution envelope structure of a protein at low resolution. The experimental SAXS patterns of gp5 and gp5 in complex with trx were reported previously (27). We used SAXS to examine the conformational state of gp5-C313S in comparison with wild-type gp5. The small angle scattering curves and their corresponding Guinier plots (Fig. 9A, q2 = 0.0001–0.0004 Å−2) show clear differences between wild-type gp5 and gp5-C313S. Linearity of the very small angle portion of the spectrum (S2 ≤ 0.0004 Å−2) in the Guinier plot indicates that both proteins are stable and do not aggregate. An upward deviation from linearity in the wider-angle portion of the spectrum (>0.002 Å−2) of gp5-C313S (Fig. 9A, inset) represents a pronounced aberration from internal symmetry of the particles due to conformational change.
FIGURE 9.
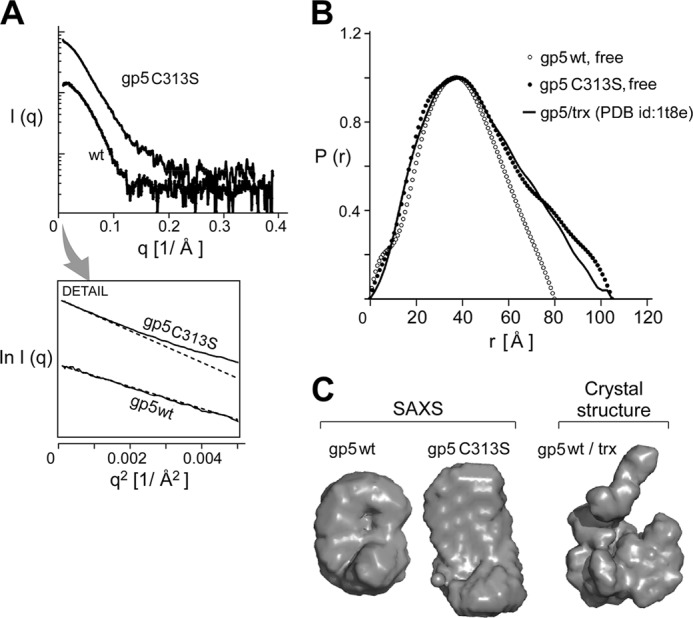
SAXS analysis and low resolution solution structure of wild-type gp5 and gp5-C313S. A, x-ray scattering profiles of wild-type gp5 and gp5-C313S are shown. Inset, small angle data and Guinier plots of wild-type gp5 and gp5-C313S are shown. B, shown is distance distribution function p(r) of wild-type gp5 (gray) and gp5-C313S (black) with Dmax values of 80 and 104 Å, respectively. p(r) was obtained using the computer program GNOM (34) and served as an alternative measure for Rg and the maximum dimension of the measured particle. C, the corresponding ab initio model of gp5 was obtained using the computer program GASBOR (37).
We analyzed the small angle region of the scattering profile using the Guinier approximation (Equation 2 under “Experimental Procedures”) and the software GNOM (34). The analysis of the small angle data of wild-type gp5 and gp5-C313S gives a hydrated radius of gyration (Rg) value of 31 and 34 Å, respectively (Table 2), indicating a different conformation for wild-type gp5 and gp5-C313S. The distance distribution function (p(r)) curve measures the distribution of pairwise distances (centers of mass) within the volume of the particle. The p(r) of wild-type gp5 has a maximum particle size (Dmax) of 80 Å and displays a maximum at 40 Å (Fig. 9B) (27). gp5-C313S presents a wider distribution of the mass indicated by Dmax of 104 Å (Table 2), suggesting an elongated structure. The program GASBOR (37) was used to analyze the scattering profile and to reconstruct low resolution ab initio structural models of gp5-C313S. The theoretical scattering profiles of 20 independently reconstructed models reliably account for the experimental scattering result. The final averaged structural model indicates that wild-type gp5 is more spherical and compact than gp5-C313S (Fig. 9C).
TABLE 2.
Overall parameters for small angle x-ray scattering of gp5 and gp5-C313S
Rg radius of gyration; Dmax maximum dimension of particle.
| Species | Method | Rg | Dmax |
|---|---|---|---|
| Å | Å | ||
| wt gp5 | Guiniera | 31.5b | |
| GNOMc | 30.8b | 80b | |
| gp5/trx | CRYSOLd | 34b | 105b |
| Guinier | 33.3b | ||
| GNOM | 34.9b | 104b | |
| gp5-C313S | Guinier | 34.2 | 104 |
| GNOM | 36.53 | 104 |
a Determined by linear fitting to the Guinier region.
b Values were from a previous study (27).
c Determined using the software GNOM. Results of the course search were refined to obtain a smooth p(r).
Complementation of Phage T7Δ5 Growth by gp5-C313S, gp5-C275S, and gp5-C275S/C313S
As gp5-C313S is defective in interaction with trx, it seemed likely that its ability to support T7 DNA replication in vivo would also be affected. We examined the ability of the altered gp5s to support phage T7Δ5 (lacking gene 5) growth. In this assay, the viability of a phage T7Δ5 depends on a functional gp5 expressed from a plasmid (pGP5-3) harbored in E. coli A307 lacking the trx gene. Host E. coli A307 also carries pTrxA plasmid that expresses trx. Expression of both gp5 and trx were induced upon infection of T7Δ5 (see “Experimental Procedures”). The ability of the altered gp5 to complement T7Δ5 growth was measured by the plating efficiency and plaque size. As shown in Fig. 10, although all the altered gp5 could support phage T7Δ5 and produce visible plaques, none complemented as efficiently as wild-type gp5. gp5-C275S could support T7Δ5 growth with essentially the same plating efficiency as wild-type gp5. However, the size of plaques with gp5-C275S is only one-third those obtained with wild-type gp5 after 6 h of incubation at 37 °C. In sharp contrast, T7Δ5 growth complemented by gp5-C313S not only had lower plating efficiency (0.64) but also yielded tiny plaques (Fig. 10C). gp5 harboring substitutions of both Cys-275 and Cys-313 further decreased plating efficiency (0.47) and also yielded plaques of tiny size (Fig. 10D). Thus the in vivo results are in agreement with the biochemical studies.
FIGURE 10.
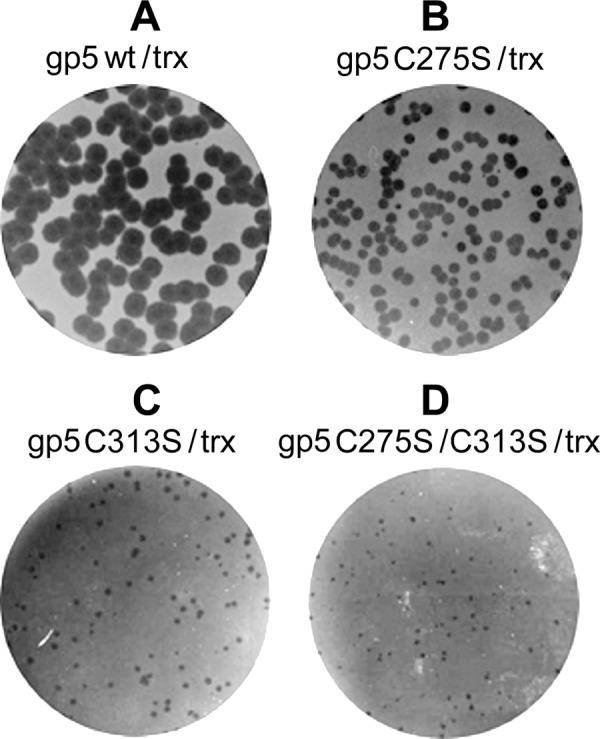
Complementation of phage T7Δ5 growth by gp5. Complementation assays were performed as described under “Experimental Procedures.” In each panel E. coli A307 (trxA−) carrying a pair of plasmids expressing the different species of gp5 and wild-type trx was infected by phage T7Δ5 (lacking gene 5). After incubation at 37 °C for 6 h, phage plaques were photographed. Different gp5 species are wt gp5 (A), gp5-C275S (B), gp5-C313S (C), and gp5-C275S/C313S (D).
DISCUSSION
Gene 5 of bacteriophage T7 encodes a DNA polymerase of low processivity. gp5 alone stalls and dissociates from the primer-template after incorporation of less than 50 nt (9–11). To achieve high processivity, gp5 uses a host protein, trx. The binding of trx to gp5 increases the affinity of gp5 to a primer-DNA template allowing gp5/trx to incorporate hundreds of nucleotides per binding event without dissociating from the DNA (9–11). T7 gp5 is a member of polymerase I family (12, 13). However, in contrast to the large fragment (Klenow) of E. coli polymerase I that has only one cysteine (38), T7 gp5 has 10 cysteines distributed throughout the amino acid sequences. Cys-275 and Cys-313 that are located in loop A and loop B, respectively (15), of the TBD are the subject of the present study. We found that the polymerase activity of gp5 decreases significantly when it is stored under non-reduced conditions. Under non-reduced condition, gp5 forms dimers and trimers as determined on SDS-PAGE. When either Cys-275 or Cys-313 is replaced by a serine, only dimers are formed. Moreover, no oligomeric forms are detected with gp5 having both cysteines replaced with serines. The results suggest that only Cys-275 and Cys-313 are exposed to solvent and that the loss of polymerase activity of gp5 under non-reduced condition is due to intermolecular disulfide linkages (-S-S-) between these two cysteines. Interestingly, neither wild-type nor the altered gp5 form such intermolecular disulfide bonds in the presence of trx. The result suggests that Cys-275 and Cys-313 are buried between the interacting surface of gp5 and trx, thus eliminating the intermolecular disulfide bonds between exposed cysteines of gp5.
Trx has two active site cysteines, Cys-32 and Cys-35. The three-dimensional structure of gp5/trx reveals those two active sites cysteines are not in proximity to Cys-275 and Cys-313 of gp5. In this structure Cys-275 and Cys-313 are located on the tips of two positive charged flexible loop A and B, respectively, of the TBD, and the two loops stretch away from trx (Fig. 1), whereas Cys-32 of trx is exposed on the protein-protein surface and probably hydrogen-bonds with Thr-327 in the thumb of gp5. As predicted from the crystal structure, we did not detect a stable disulfide linkage (gp5-S-S-trx) between the active site cysteines of trx with the exposed cysteines of gp5. No disulfide bond was found even when one of active site cysteines (Cys-35) of trx was replaced with a serine to avoid self-disulfide linkage (data not shown). Additionally, replacement of Thr-327 of gp5 with a cysteine results in stable disulfide linkage formation between the sulfhydryl group of Cys-32 of trx and Cys-327 of altered gp5 (31).
The replacement of either Cys-275 or Cys-313 with a serine, another nucleophilic residue lacking a thiol group, drastically decreased the polymerase activity of gp5 in the absence of trx on DNA templates lacking secondary structure. The presence of trx restores polymerase activity of gp5-C275S to almost identical levels of that observed with wild-type gp5/trx; gp5-C313S and gp5-C275S/C313S are also markedly stimulated by trx but are only 50% as active as wild-type gp5/trx. Trx also greatly stimulates polymerase activity of gp5-C275S as it does wild-type gp5 on primed M13 ssDNA templates. gp5-C313S/trx and gp5-C275S/C313S/trx on the other hand have 10-fold less polymerase activity on the M13 template. A large number of past studies have demonstrated that the stimulation of polymerase activity by trx reflects an increase in processivity that eliminates the rate-limiting steps of dissociation and re-association of gp5 to the primer (29, 35). Indeed, processivity assays show that whereas both wild-type gp5/trx and gp5-C275S/trx fully replicate the M13 molecules, gp5-C313S/trx has difficulty in polymerizing nucleotides through sites of secondary structure. Comparison of Kobs indicates that gp5-C275S has a similar binding affinity to trx as wild-type gp5, whereas gp5-C313S and gp5-C275S/C313S have 6- and 10-fold lower affinity, respectively, for trx as compared with wild-type gp5. In support of this weaker association, gp5-C313S and trx dissociated during chromatography of gp5-C313S/trx on hydroxyapatite-phosphate, whereas wt gp5/trx did not. Hydroxyapatite in phosphate buffer is assumed to mimic the phosphodiester backbone of DNA and has been successfully used to separate gp5/trx complexes from free gp5 (11). The results taken together suggest that Cys-275 and Cys-313 are part of the TBD surface involved in the TBD-trx interaction.
Both Cys-313 and Cys-275 are located on the tips of loop A and B of the TBD. These loops are highly positively charged and have been postulated to interact with the DNA template as well as with the acidic C-terminal tail of the helicase (15, 24). We recently examined the flexibility of gp5 using normal mode analysis (27). The results showed that the TBD of gp5 possesses higher flexibility than the other regions of the polymerase. Binding of trx increases the interaction surface of gp5 to DNA, allowing the basic residues on loops A and B to electrostatically contact the phosphodiester backbone of DNA. Mutations that remove the charges of loops A and B affect the ability of the polymerase to bind to trx as well as decrease polymerase activity (14, 15). Therefore, a proper positioning of the basic residues of loops A and B is critical for this electrostatic interaction. Biochemical data in this study, on the other hand, suggest that the exposed cysteines, in particular Cys-313 on loop B, are required to maintain the correct conformation of the TBD for the binding of gp5 to trx as well as DNA. Serine differs structurally from cysteine by having a hydroxyl at the position of the thiol group found in cysteine. Nevertheless, the differences in the chemistry between sulfur and oxygen could result in a critical difference in the protein structure. Hydrogen bonding of cysteine is relatively weaker than that of serine in the aspect of accepting or donating a hydrogen bond. Therefore, it is likely that the effect of C313S replacement on activity of gp5 is a result of an increase in hydrophilicity of the amino acid side chain rather than significant change in the size. In support of this interpretation, the SAXS analysis suggests that the replacement of Cys-313 by a serine results in a change of the structure of gp5; wild-type gp5 has a compact and spherical conformation in comparison with the elongated conformation obtained with gp5-C313S. Thus, it is likely that the replacement of Cys-313 with serine disrupts the conformation of the TBD, a compact structure that is accessible by trx. Finally, although the data support a structural role of the exposed Cys-313 in the TBD-trx interaction, a more indirect role for this cysteine cannot be excluded at this point.
Acknowledgments
Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the United States Department of Energy, Office of Basic Energy Sciences, under Contract DE-AC02-98CH10886.
This work was supported, in whole or in part, by National Institutes of Health Grant GM54397.
- gp5
- T7 gene 5 protein
- trx
- thioredoxin
- TBD
- thioredoxin binding domain
- nt
- nucleotide(s)
- SAXS
- small angle x-ray scattering.
REFERENCES
- 1. Indiani C., O'Donnell M. (2006) The replication clamp-loading machine at work in the three domains of life. Nat. Rev. Mol. Cell Biol. 7, 751–761 [DOI] [PubMed] [Google Scholar]
- 2. Stukenberg P. T., Studwell-Vaughan P. S., O'Donnell M. (1991) Mechanism of the sliding beta-clamp of DNA polymerase III holoenzyme. J. Biol. Chem. 266, 11328–11334 [PubMed] [Google Scholar]
- 3. Lee Y. S., Kennedy W. D., Yin Y. W. (2009) Structural insight into processive human mitochondrial DNA synthesis and disease-related polymerase mutations. Cell 139, 312–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yao N., Leu F. P., Anjelkovic J., Turner J., O'Donnell M. (2000) DNA structure requirements for the Escherichia coli gamma complex clamp loader and DNA polymerase III holoenzyme. J. Biol. Chem. 275, 11440–11450 [DOI] [PubMed] [Google Scholar]
- 5. Yang J., Zhuang Z., Roccasecca R. M., Trakselis M. A., Benkovic S. J. (2004) The dynamic processivity of the T4 DNA polymerase during replication. Proc. Natl. Acad. Sci. U.S.A. 101, 8289–8294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mueser T. C., Hinerman J. M., Devos J. M., Boyer R. A., Williams K. J. (2010) Structural analysis of bacteriophage T4 DNA replication. A review in the Virology Journal series on bacteriophage T4 and its relatives. Virol. J. 7, 359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Camacho A., Salas M. (2010) DNA bending and looping in the transcriptional control of bacteriophage φ29. FEMS Microbiol. Rev. 34, 828–841 [DOI] [PubMed] [Google Scholar]
- 8. Andraos N., Tabor S., Richardson C. C. (2004) The highly processive DNA polymerase of bacteriophage T5. Role of the unique N and C termini. J. Biol. Chem. 279, 50609–50618 [DOI] [PubMed] [Google Scholar]
- 9. Huber H. E., Tabor S., Richardson C. C. (1987) Escherichia coli thioredoxin stabilizes complexes of bacteriophage T7 DNA polymerase and primed templates. J. Biol. Chem. 262, 16224–16232 [PubMed] [Google Scholar]
- 10. Modrich P., Richardson C. C. (1975) Bacteriophage T7 deoxyribonucleic acid replication in vitro. A protein of Escherichia coli required for bacteriophage T7 DNA polymerase activity. J. Biol. Chem. 250, 5508–5514 [PubMed] [Google Scholar]
- 11. Tabor S., Huber H. E., Richardson C. C. (1987) Escherichia coli thioredoxin confers processivity on the DNA polymerase activity of the gene 5 protein of bacteriophage T7. J. Biol. Chem. 262, 16212–16223 [PubMed] [Google Scholar]
- 12. Braithwaite D. K., Ito J. (1993) Compilation, alignment, and phylogenetic relationships of DNA polymerases. Nucleic Acids Res. 21, 787–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ollis D. L., Kline C., Steitz T. A. (1985) Domain of E. coli DNA polymerase I showing sequence homology to T7 DNA polymerase. Nature 313, 818–819 [DOI] [PubMed] [Google Scholar]
- 14. Yang X. M., Richardson C. C. (1997) Amino acid changes in a unique sequence of bacteriophage T7 DNA polymerase alter the processivity of nucleotide polymerization. J. Biol. Chem. 272, 6599–6606 [DOI] [PubMed] [Google Scholar]
- 15. Hamdan S. M., Marintcheva B., Cook T., Lee S. J., Tabor S., Richardson C. C. (2005) A unique loop in T7 DNA polymerase mediates the binding of helicase-primase, DNA-binding protein, and processivity factor. Proc. Natl. Acad. Sci. U.S.A. 102, 5096–5101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bedford E., Tabor S., Richardson C. C. (1997) The thioredoxin binding domain of bacteriophage T7 DNA polymerase confers processivity on Escherichia coli DNA polymerase I. Proc. Natl. Acad. Sci. U.S.A. 94, 479–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Doublié S., Tabor S., Long A. M., Richardson C. C., Ellenberger T. (1998) Crystal structure of a bacteriophage T7 DNA replication complex at 2.2 Å resolution. Nature 391, 251–258 [DOI] [PubMed] [Google Scholar]
- 18. Brieba L. G., Eichman B. F., Kokoska R. J., Doublié S., Kunkel T. A., Ellenberger T. (2004) Structural basis for the dual coding potential of 8-oxoguanosine by a high fidelity DNA polymerase. EMBO J. 23, 3452–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eklund H., Gleason F. K., Holmgren A. (1991) Structural and functional relations among thioredoxins of different species. Proteins 11, 13–28 [DOI] [PubMed] [Google Scholar]
- 20. Holmgren A. (1985) Thioredoxin. Annu. Rev. Biochem. 54, 237–271 [DOI] [PubMed] [Google Scholar]
- 21. Kallis G. B., Holmgren A. (1980) Differential reactivity of the functional sulfhydryl groups of cysteine 32 and cysteine 35 present in the reduced form of thioredoxin from Escherichia coli. J. Biol. Chem. 255, 10261–10265 [PubMed] [Google Scholar]
- 22. Huber H. E., Russel M., Model P., Richardson C. C. (1986) Interaction of mutant thioredoxins of Escherichia coli with the gene 5 protein of phage T7. The redox capacity of thioredoxin is not required for stimulation of DNA polymerase activity. J. Biol. Chem. 261, 15006–15012 [PubMed] [Google Scholar]
- 23. Adler S., Modrich P. (1983) T7-induced DNA polymerase. Requirement for thioredoxin sulfhydryl groups. J. Biol. Chem. 258, 6956–6962 [PubMed] [Google Scholar]
- 24. Etson C. M., Hamdan S. M., Richardson C. C., van Oijen A. M. (2010) Thioredoxin suppresses microscopic hopping of T7 DNA polymerase on duplex DNA. Proc. Natl. Acad. Sci. U.S.A. 107, 1900–1905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Himawan J. S., Richardson C. C. (1996) Amino acid residues critical for the interaction between bacteriophage T7 DNA polymerase and Escherichia coli thioredoxin. J. Biol. Chem. 271, 19999–20008 [DOI] [PubMed] [Google Scholar]
- 26. Kelman Z., Hurwitz J., O'Donnell M. (1998) Processivity of DNA polymerases. Two mechanisms, one goal. Structure 6, 121–125 [DOI] [PubMed] [Google Scholar]
- 27. Akabayov B., Akabayov S. R., Lee S. J., Tabor S., Kulczyk A. W., Richardson C. C. (2010) Conformational dynamics of bacteriophage T7 DNA polymerase and its processivity factor, Escherichia coli thioredoxin. Proc. Natl. Acad. Sci. U.S.A. 107, 15033–15038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee S. J., Marintcheva B., Hamdan S. M., Richardson C. C. (2006) The C-terminal residues of bacteriophage T7 gene 4 helicase-primase coordinate helicase and DNA polymerase activities. J. Biol. Chem. 281, 25841–25849 [DOI] [PubMed] [Google Scholar]
- 29. Hamdan S. M., Johnson D. E., Tanner N. A., Lee J. B., Qimron U., Tabor S., van Oijen A. M., Richardson C. C. (2007) Dynamic DNA helicase-DNA polymerase interactions assure processive replication fork movement. Mol. Cell 27, 539–549 [DOI] [PubMed] [Google Scholar]
- 30. Tabor S., Richardson C. C. (1985) A bacteriophage T7 RNA polymerase/promoter system for controlled exclusive expression of specific genes. Proc. Natl. Acad. Sci. U.S.A. 82, 1074–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Johnson D. E., Richardson C. C. (2003) A covalent linkage between the gene 5 DNA polymerase of bacteriophage T7 and Escherichia coli thioredoxin, the processivity factor. Fate of thioredoxin during DNA synthesis. J. Biol. Chem. 278, 23762–23772 [DOI] [PubMed] [Google Scholar]
- 32. Hubbard J. J., Thornton J. H. (1993) NACCESS, computer program. Department of Biochemistry and Molecular Biology, University College, London [Google Scholar]
- 33. Allaire M., Yang L. (2011) Biomolecular solution X-ray scattering at the National Synchrotron Light Source. J. Synchrotron Radiat. 18, 41–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Svergun D. (1992) Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J. Appl. Crystallogr. 25, 8 [Google Scholar]
- 35. Myers T. W., Romano L. J. (1988) Mechanism of stimulation of T7 DNA polymerase by Escherichia coli single-stranded DNA-binding protein (SSB). J. Biol. Chem. 263, 17006–17015 [PubMed] [Google Scholar]
- 36. Wuite G. J., Smith S. B., Young M., Keller D., Bustamante C. (2000) Single-molecule studies of the effect of template tension on T7 DNA polymerase activity. Nature 404, 103–106 [DOI] [PubMed] [Google Scholar]
- 37. Svergun D. I., Petoukhov M. V., Koch M. H. (2001) Determination of domain structure of proteins from x-ray solution scattering. Biophys. J. 80, 2946–2953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jovin T. M., Englund P. T., Kornberg A. (1969) Enzymatic synthesis of deoxyribonucleic acid. XXVII. Chemical modifications of deoxyribonucleic acid polymerase. J. Biol. Chem. 244, 3009–3018 [PubMed] [Google Scholar]
- 39. Svergun D. I., Barberato C., Koch M. H. J. (1995) CRYSOL. A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr.. 28, 768–773 [Google Scholar]